

Abstract
Background
The mechanism(s) by which androgen receptor (AR) splice variants contribute to castration-resistant prostate cancer (CRPC) is still lacking.
Methods
Expressions of Epithelial-to-Mesenchymal Transition (EMT) and stem cell markers were molecularly tested using prostate cancer (PCa) cells transfected with AR and AR3 (also known as AR-V7) plasmids or siRNA, and also cultured cells under androgen deprivation therapy (ADT) condition. Cell migration, clonogenicity, sphere forming capacity was assessed using PCa cells under all experimental conditions and 3, 3′-diindolylmethane (DIM; BR-DIM) treatment. Human PCa samples from BR-DIM untreated or treated patients were also used for assessing the expression of AR3 and stem cell markers.
Results
Overexpression of AR led to the induction of EMT phenotype, while overexpression of AR3 not only induced EMT but also led to the expression of stem cell signature genes. More importantly, ADT enhanced the expression of AR and AR3 concomitant with up-regulated expression of EMT and stem cell marker genes. Dihydrotestosterone (DHT) treatment decreased the expression of AR and AR3, and reversed the expression of these EMT and stem cell marker genes. BR-DIM administered to PCa patients prior to radical prostatectomy inhibited the expression of cancer stem cell markers consistent with inhibition of self-renewal of PCa cells after BR-DIM treatment.
Conclusion
AR variants could contribute to PCa progression through induction of EMT and acquisition of stem cell characteristics, which could be attenuated by BR-DIM, suggesting that BR-DIM could become a promising agent for the prevention of CRPC and/or for the treatment of PCa
Keywords: Androgen Receptor splice variants; Cancer stem cells; self-renewal; 3, 3′-diindolylmethane
Introduction
Prostate cancer (PCa) initiation and progression is well known to be dependent on androgen receptor (AR) signaling. Despite initial favorable response to androgen deprivation therapy (ADT), most of the patients progress to a more aggressive castration-resistant prostate cancer (CRPC) and ultimately lead to metastasis, which is the primary cause of death of patients initially diagnosed with PCa. Therefore, it is critical to understand the underlying molecular mechanism(s) for the development of CRPC. Recent studies have shown that CRPC cells expressing Nkx3-1 (CARNs), a known regulator of prostate epithelial differentiation, exhibited the expression of stem cell signature genes [1]. These cells showed expression of luminal markers such as cytokeratin-18 and AR, and negative for basal cell marker p63, and also display self-renewal capacity in vivo. Furthermore, deletion of the PTEN tumor suppressor gene in CARNs led to the development of carcinoma after regeneration [1]. These results suggest that androgen deprivation could in part be responsible for PCa progression after ADT through the acquisition of stem cell signatures, mediated by the activation of oncogenes or inactivation of tumor repressors in AR-positive PCa cells. The question is why the cells expressing AR could survive after ADT and then initiate the processes of tumor progression and metastasis, the answer to such a question is currently lacking.
Recent studies have shown that the expression of AR splice variants lacking ligand-binding domain was found to be increased in androgen-independent PCa cell lines, CRPC and metastasis [2-7]. These AR splice variants are constitutively active and localized in the nuclear compartment, and their transcriptional activity is not regulated by androgens or anti-androgens [5, 8, 9]. Human AR gene is structurally composed of eight exons and encodes a multi-domain protein, including an NH2 terminal transactivation domain (NTD, encoded by exon 1), a central DNA binding domain (DBD, encoded by exon 2-3), a hinge region (encoded by exon 4), and a COOH terminal ligand-binding domain (LBD, encoded by exon 4-8). AR3 is encoded by exon 1-3 and 3b [10-12], which contains intact NTD and DBD but lacks the hinge region and LBD, and has been found to be associated with CRPC and higher risk of tumor recurrence [12]. These results suggest that PCa cells expressing AR splice variants might be the cells that are responsible for the development of CRPC phenotype after ADT, which in turn could be responsible for tumor progression, recurrence and metastasis.
Tumor recurrence and metastasis are believed to be associated with the acquisition of stem cell signatures and Epithelial-to-Mesenchymal Transition (EMT) phenotype [13]. ADT has been shown to be linked with the acquisition of EMT phenotype [14-16]. Increasing evidences have demonstrated that EMT phenotypic cells are also the source for cancer stem cells [17-20]. Interestingly, recent studies have shown that AR splice variants (constitutively active) are associated with castration resistance phenotype in PCa [3, 5, 6, 10]. Thus, AR variants might contribute to the induction of EMT and the acquisition of stem cell signatures in PCa after ADT. We have recently shown that EMT markers are expressed in bone metastatic human PCa specimens [21]. These findings suggest that the inhibition in the expression of AR variants and/or inactivation of their function is urgently needed for the elimination of EMT phenotypic cells and cancer stem cells (CSCs) for the prevention of tumor progression and/or treatment of aggressive and metastatic PCa. To that end, studies have shown that 3,3′-diindolylmethane (DIM obtained from BioResponse and termed as BR-DIM), which is typically found in cruciferous vegetables, is a very potent agent for the inhibition of PCa cell growth, which is in part mediated by alterations in multiple cellular signaling pathways [22-25], including inactivation of NF-κB. Studies have also shown that Enzalutamide (anti-androgen) resistance could be in part mediated through NF-κB, whose activation maybe attributed to increased expression of AR and AR variants [26].
In the current study, we found that BR-DIM significantly down-regulated the expression of AR and AR3, and caused down-regulation in the expression of AR target gene, PSA, in both androgen-dependent LNCaP cells as well as androgen-independent 22RV1 cells. PSA has been reported to promote cell proliferation, migration, invasion and metastasis of PCa cells through several mechanisms [27, 28]. As a proof-of-concept, here we provide evidence that administration of BR-DIM to PCa patients prior to radical prostatectomy led to reduced expression of AR and AR3 in the tumor specimens, and these results were consistent with our in vitro experimental findings. Thus, BR-DIM could become a promising natural agent for overcoming resistance to ADT, which will lead to achieve better treatment outcome of PCa patients.
Materials and methods
Cell lines and culture condition
PZ-HPV-7, RWPE-1, LNCaP, DU145, PC3, VCaP, 22RV1, and MDA-PCa-2b cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). PZ-HPV-7 and RWPE-1 cells were maintained in Keratinocyte Serum Free Medium (K-SFM, Invitrogen, Carlsbad, CA) supplemented with 0.05 mg/ml bovine pituitary extract (BPE) and 5 ng/ml human recombinant epidermal growth factor (EGF), 50 units/ml Penicillin, and 50 μg/ml Streptomycin. LNCaP, C4-2B, DU145, PC3, VCaP, 22RV1 and MDA-PCa-2b were maintained in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), 10 μmol/L Hepes, 50 units/ml Penicillin and 50 μg/ml Streptomycin. All cells were maintained in a 5% CO2-humidified atmosphere at 37°C.
Reagents and antibodies
Antibodies against AR (441), ZEB1, vimentin and Fibronectin were purchased from Santa Cruz (Dallas, Texas). AR3 (AR-V7) was obtained from A&G Pharmaceutical, Inc (Columbia, MD). Antibodies against PSA, Lin28B, Nanog were purchased from Cell Signaling (Danvers, MA), N-cadherin was obtained from BD Biosciences (Bedford, MA). Antibody to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was purchased from Affinity BioReagents (Golden, CO). Goat anti-mouse or anti-rabbit IgG (H + L)-HRP conjugates were obtained from Bio-Rad (Reinach, BL). BR-DIM, a formulated DIM, manufactured by BioResponse (Boulder, CO, abbreviated as BR-DIM), was kindly provided by Dr. Michael Zeligs, and was dissolved in DMSO and stored at -20°C in multiple aliquots for in vitro study.
Plasmids and transfection
p5HBhAR-A plasmid expressing full length AR and p5HBhAR-1-2-3-CE3 expressing AR3 were kindly provided by Scott M. Dehm (Masonic Cancer Center, University of Minnesota, Minneapolis, MN). The pCMV5 plasmid was purchased from ATCC (Manassas, VA). LNCaP, PC3 and DU145 Cells were transfected with p5HBhAR-A plasmid (AR), p5HBhAR-1-2-3-CE3 (AR3) or pCMV5 plasmid using ExGen 500 in vitro Transfection Reagent (Thermo Scientific Fermentas, Pittsburgh, PA). After transfection and incubation for different time, the cells were prepared for making cell lysates and isolation of RNA, or for cell migration and clonogenic assays.
Patients and prostate tissue specimen collection
Retrospective archival pre-treatment PCa tissues and matched adjacent normal tissues were collected from patients who underwent routine radical prostatectomy from 2004-2011 at Karmanos Cancer Institute (KCI), and obtained from Biospecimen Core of Karmanos Cancer Institute after obtaining institutional review board approval, and the PCa tissue specimens from our clinical trial of BR-DIM (B-DIM, http://clinicaltrials.gov/show/NCT00888654) prior to radical prostatectomy of newly diagnosed PCa patients were also obtained after receiving approval from Wayne State University Institutional Review Board and written informed consents obtained from all study subjects. All tissues were retrieved from the formalin-fixed paraffin-embedded (FFPE) tissue blocks, and from which 10 micron sections were cut. The clinical characteristics of patients were obtained from the hospital database as shown in our previously published paper [23].
Clonogenic assay
LNCaP cells transfected with p5HBhAR-A plasmid expressing full length AR, p5HBhAR-1-2-3-CE3 plasmid expressing AR3 and control pCMV5 plasmid, and then incubated for 24 h. The cells were collected after trypsinization, and re-suspended in the complete medium for clonogenic assay performed as previously described by our laboratory [23].
Sphere-forming assay
Sphere-forming assay was used to determine the self-renewal capacity in vitro. Single cell suspensions of LNCaP, C4-2B and 22RV1 cells, or PC3 and DU145 cells transfected with p5HBhAR-A plasmids expressing full length AR and p5HBhAR-1-2-3-CE3 plasmids expressing AR3 as well as pCMV5 plasmids were seeded in 6-well plates with ultra-low attachment surface (Corning, Lowell, MA) at 2000 cells/well in DMEM/F12 (Invitrogen) supplemented with B27 and N2 (Invitrogen). Sphere-forming assay was performed as previously described by our laboratory [18].
Western blot analysis
Total cell lysates were prepared from transfected cells, sphere-forming cells and BR-DIM treated cells by lysing the cells in RIPA buffer. Protein concentration was measured using BCA protein assay kit (Pierce, Rockford, IL). Western blot assay was performed as previously described [25, 29].
Real-time RT-PCR
The total RNA from cells was prepared using the RNeasy Mini Kit (Qiagen, Valencia, CA), and the DNA was removed through incubation of RNA extract with RNase-free NDase from an RNase-free NDase kit (Qiagen). 1μg of total RNA for each sample was reverse transcribed into cDNA using a High Capacity RNA-to-cDNA Kit (Applied biosystems, Fostor, CA) following the manufacturer's instruction. To measure the levels of mRNA in PCa patient tissues, the total RNA was isolated from formalin-fixed paraffin-embedded (FFPE) tissues using miRNeasy FFPE Kit (Qiagen) and the DNA was removed by treating the samples using RNase-free NDase according to the manufacturer's instruction. Real time PCR was conducted as previously described [23, 30]. The ΔΔCT method was used for calculation of the relative expression. The relative amount of mRNA was normalized to the expression of beta-actin or GAPDH.
Wound healing assay
PC3 and DU145 cells were seeded in 6-well plates and incubated for 24 h, and then transfected with plasmids expressing AR and AR3 or empty vector. After 48h incubation, the cells reached ∼80-90% confluence as a monolayer. The wound was made by scratching the monolayer with a 200 μl pipette tip across the center of the well. After scratching, gently washing the well twice with complete medium to remove the detached cells and then adding fresh complete medium into the wells. The images were captured for cell monolayer on a microscope. The cells were incubated for another 24h and then washed twice with PBS. The images of cells were taken using a microscope.
Immunohistochemistry
For the immunohistochemical staining, FFPE tumor tissue sections were used and incubated with specific primary antibody against Lin28B, followed by incubation with anti-rabbit secondary antibody and 3, 3′-diaminobenzidine (DAB). Sections were visualized under an Olympus microscope (Olympus, Japan) and images were captured by the camera linked to a computer.
Statistical methods
The data are presented as the mean and standard deviation (SD) in the bar graphs. Comparisons of the continuous variables between two independent groups were made using two tailed student's t test. Comparisons between two independent groups from patient samples were made using the Wilcoxon rank sum test. Spearman correlations were used to describe the strength of linear relationship between two variables. All statistical tests were two sided at significance level of 0.05.
Results
Expressions of AR and AR3 in PCa tissue specimens and PCa cell lines
Activated AR signaling has been commonly found in PCa patients, especially in castration-resistant prostate cancer (CRPC) patients. In this study, we found that the expression of AR was found to be increased in patients' tumor tissues with Gleason grade 6, but there were no significant differences in patients' tumor tissues with Gleason grade ≥7 (Fig-1A). Whereas the expression of AR3 was significantly up-regulated in patients' tumor tissues, especially in patients' tumor tissues with Gleason grade ≥8 compared with adjacent normal tissues (Fig-1CB). Consistent with the results from patient tissues, the mRNA expression of AR and AR3 was found to be increased in PCa cell lines (LNCaP, C4-2B, MDA-PCa-2B, VCaP and 22RV1 cells) compared with immortalized normal prostate epithelial cell lines (RWPE-1 and PZ-HPV-7 cells; Fig-1C). The results from Western blots also showed increased expression of AR and AR3 as well as AR signaling target gene PSA in PCa cell lines compared with normal prostate epithelial cell lines (Fig-1D, left panel). Interestingly, the expression of stem cell markers including Nanog and Oct4 as well as mesenchymal marker such as fibronectin (FN) was up-regulated in PCa cell lines compared with normal prostate epithelial cell lines (Fig-1D, right panel). These results suggest that the expression of AR, especially AR3 is strongly associated with aggressive PCa in patients and in PCa cell lines.
Fig-1. AR and AR3 expression in PCa tissue specimens and PCa cell lines.
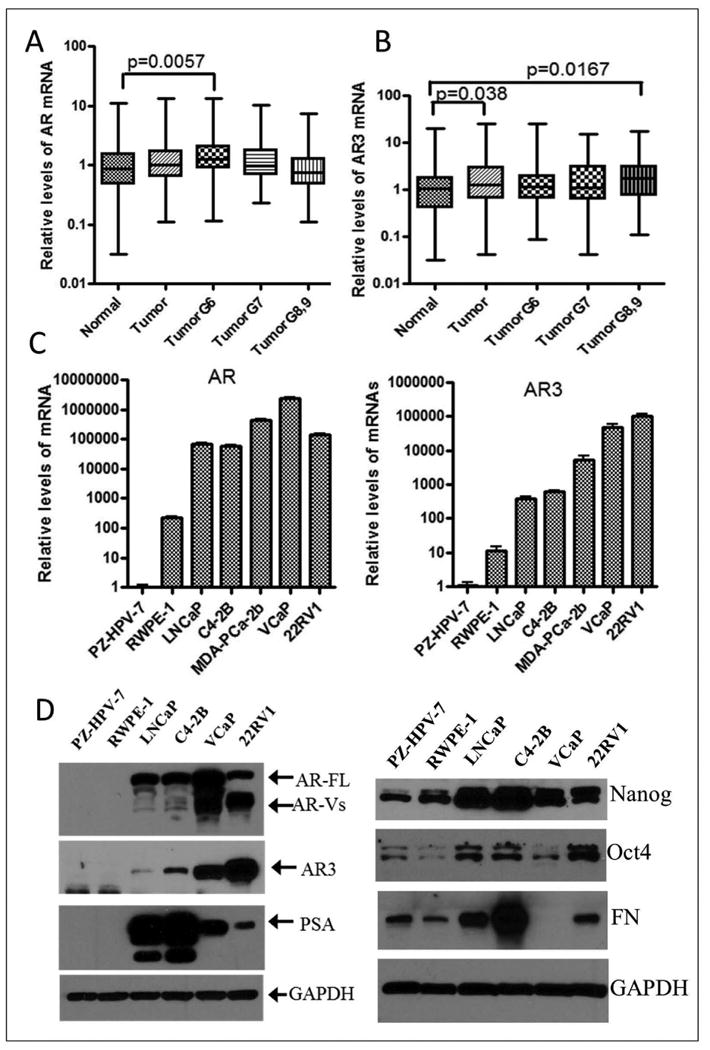
(A) The expression of AR mRNA was increased in PCa tissue specimens with Gleason grade 6, but not in patients' samples with Gleason score ≥7 compared with adjacent normal tissues. Similar data for AR mRNA levels were reported previously [Kong et at. Am J Transl Res 2012; 4(1): 14-23], and also showed different expression pattern between AR and AR3 in PCa tissue specimens (TumorG6: tumor tissues from patients with Gleason grade 6. N = 93 for normal, N=136 for tumor, N = 42 for Tumor G6, N = 45 for Tumor G7, N = 49 for Tumor G8 and 9). (B) The levels of AR3 were found to be significantly up-regulated in PCa tumor tissue specimens and especially in tumor tissues with Gleason grade≥8 compared with adjacent normal tissues. (N = 91 for normal, N=134 for tumor, N = 42 for Tumor G6, N = 44 for Tumor G7, N = 48 for Tumor G8 and 9). (C) Significant up-regulation in the expression of AR and AR3 mRNA in PCa cell lines compared with immortalized normal prostate epithelial cell lines such as RWPE-1 and PZ-HPV-7. (D) Increased protein levels of AR and AR3 as well as stem cell markers such as Nanog and Oct4, and mesenchymal marker fibronectin (FN) in PCa cell lines compared with normal prostate epithelial cell lines. Expression of PSA was found to be increased in PCa cell lines, which was inversely correlated with AR3 expression.
Overexpression of AR and AR3 induced EMT and stem cell signatures
AR positive cell line, LNCaP and AR negative cell line, DU145 cells were transfected with wild-type AR and AR3 plasmids. The results from Western blot showed that overexpression of AR led to increased expression of mesenchymal markers such as fibronectin in LNCaP cells and ZEB1 in DU145 cells (Fig-2AC). Whereas interestingly, overexpression of AR3 not only led to increased expression of fibronectin and ZEB1 in LNCaP and DU145 cells, respectively, but also increased the expression of stem cell markers such as Nanog in LNCaP cells and Lin28B in DU145 cells (Fig-2A), which were consistent with increased mRNA expressions of EMT and stem cell markers in LNCaP and DU145 cells transfected with AR or AR3 plasmids compared with transfected cells with control plasmids (Fig-2B). In contrast, down-regulation of AR3, but not AR through transfection with siRNA reduced the expression of Nanog, Oct4 and ZEB1 in 22RV1 cells (Fig-2C), which normally express high levels of AR3 (Fig-1D left panel). These results demonstrate that overexpression of AR, especially overexpression of AR3 leads to the acquisition of EMT and stem cell phenotype consistent with increased expression of EMT and stem cell marker genes, which is believed to be linked with increased cancer cell aggressiveness (increased tumor cell migration, invasion and tumor growth). As predicted, transfection of AR-negative cells (DU145 and PC3 cells) with AR and AR3 plasmids dramatically promoted cell migration after 24 hours as assessed by wound healing assay (Fig-2D). Moreover, overexpression of AR and AR3 in AR-positive LNCaP cells further promoted clonogenic growth of the cells compared with those cells transfected with control plasmids (Fig-2E). We also found that overexpression of AR3 but not AR increased prostaspheres (sphere-forming capacity of PCa cells) of PC3 (Fig-3A and 3C) and DU145 cells (Fig-3B and 3D) transfected with AR3 plasmids compared with the cells transfected with control plasmids.
Fig-2. Overexpression of AR and AR3 resulted in the induction of EMT and stem cell phenotype in PCa cells.
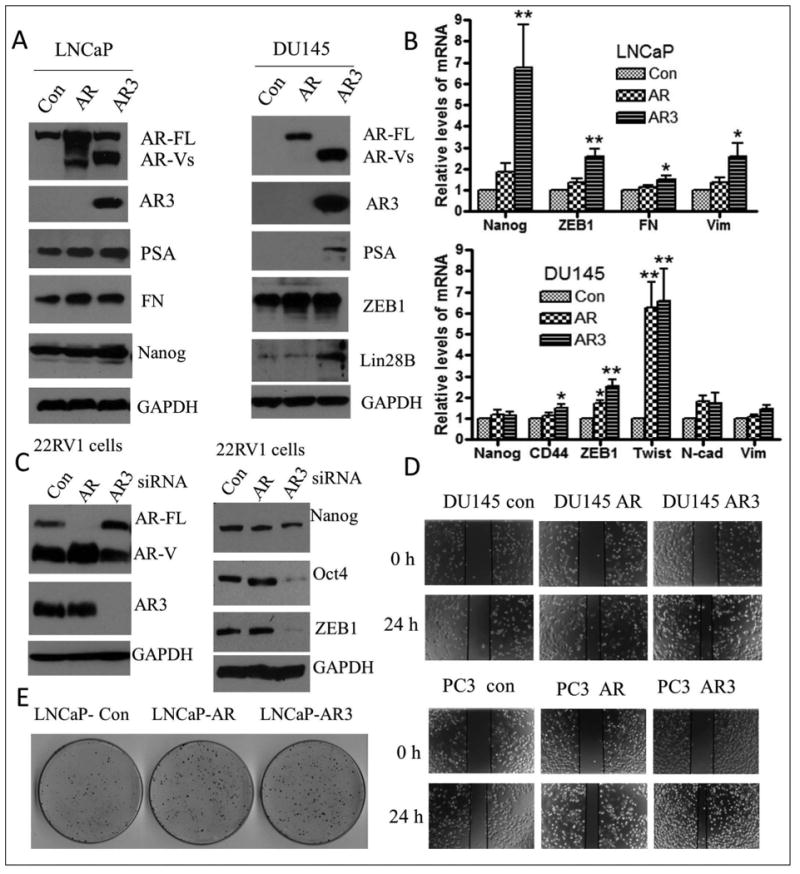
(A) and (B) Transfection of LNCaP and DU145 cells with plasmids expressing AR showed increased expression of mesenchymal markers such as fibronectin (FN), ZEB1 and Twist. Transfection of LNCaP and DU145 cells with plasmid expressing AR3 showed not only increased expression of mesenchymal markers such as fibronectin and ZEB1 but also enhanced expression of stem cell marker genes such as Nanog, Lin28B and CD44 (* p<0.05, ** p<0.01). (C) Down-regulation of AR3 but not AR by transfection of 22RV1 cells with siRNA inhibited the expression of Nanog, Oct4 and ZEB1. (D) Overexpression of AR and AR3 by transfection of DU145 and PC3 with plasmids expressing AR and AR3 promoted cell migration. (E) LNCaP cells transfected with plasmids expressing AR (LNCaP-AR), AR3(LNCaP-AR3) or control vector (LNCaP-con) and incubated for 24 hours, then the transfected cells were collected and seeded in 100 mm dishes at density of 2000 cells. After 2 weeks of incubation, the colonies were stained and photographed.
Fig-3. Overexpression of AR3 increased sphere-forming (prostasphere-forming) capacity.
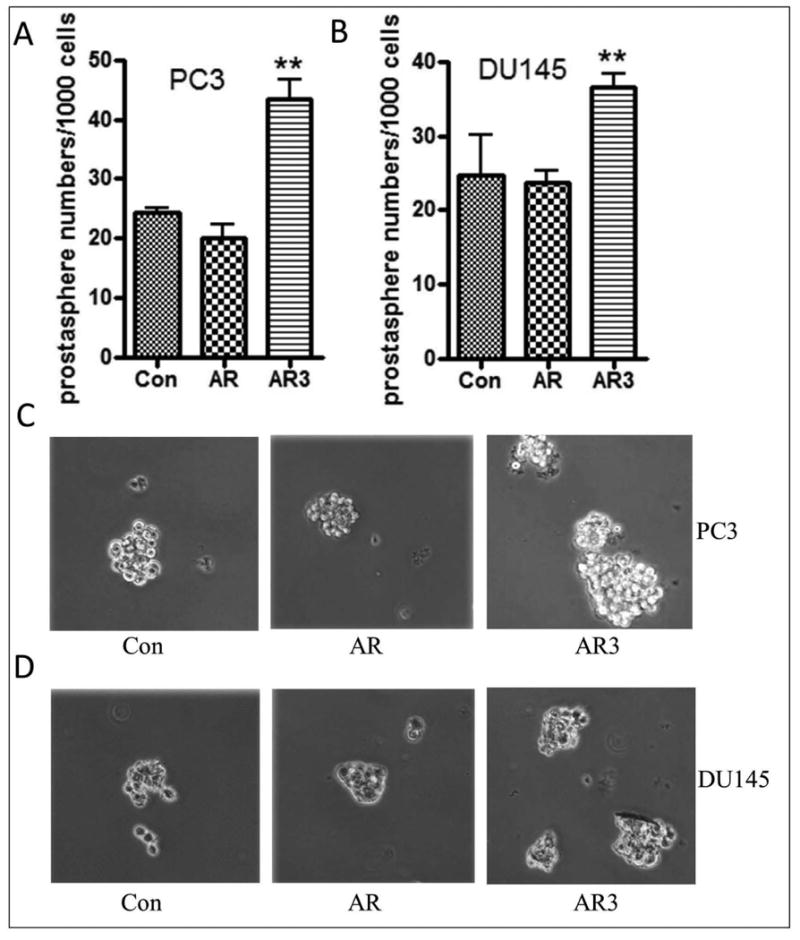
(A) and (C) PC3 cells transfected with AR3 but not AR showed increased prostasphere-forming ability. (B) and (D) DU145 cells overexpressing AR3 but not AR showed increased prostasphere numbers and Size (**, p<0.01).
Androgen deprivation was linked with the acquisition of EMT and stem cell features
Androgen deprivation therapy (ADT) becomes ineffective even though most cancer cells in metastatic CRPC (mCRPC), especially bone metastasis are still AR positive [31, 32]. The molecular mechanisms for the promotion of more aggressive tumor growth after ADT remained widely elusive. In this study, we found that C4-2B cells cultured in charcoal stripped fetal bovine serum (CS-FBS) after 2 weeks showed increased expression of AR and AR3, and decreased expression of PSA (Fig-4CA, left panel). Interestingly, androgen deprivation enhanced the expression of pluripotent stem cell factors including Lin28B, Nanog, and Sox2 as well as increased expression of EMT markers such as ZEB1, Twist, N-cadherin and vimentin (Fig-4A, middle and right panels); whereas, dihydrotestosterone (DHT) treatment reversed the expression of these EMT and stem cell markers in C4-2B cells (Fig-4BC, upper panel). Moreover, DHT treatment significantly inhibited the expression of AR and AR3 whereas increased the expression of PSA concomitant with inhibition of EMT and stem cell factors in VCaP cells (Fig-4CB, lower panel), which were consistent with the results from Western blots showing that androgen deprivation for 2 weeks led to the up-regulation of AR and AR3, and increased expression of Nanog after 4 weeks in C4-2B cells (Fig-4C). Whereas DHT treatment inhibited AR and AR3 expression concomitant with inhibition of Lin28B, vimentin and N-cadherin protein in C4-2B and VCaP cells (Fig-4D). These results suggest that androgen deprivation is in part responsible for the induction of EMT and the expression of stem cell marker genes in PCa cells, which confers cancer cells to become more aggressive after ADT, and eventually leads to metastatic disease.
Fig-4. EMT and stem cell signatures were found in PCa cells grew in androgen deprivation condition.
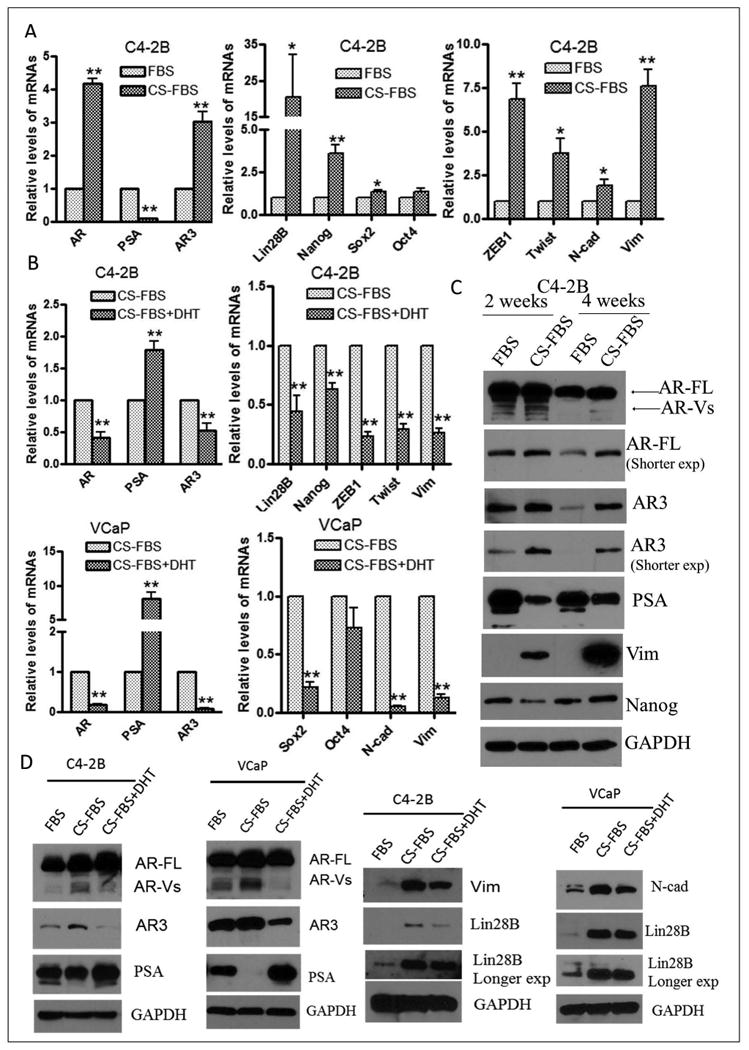
(A) C4-2B cells grew in charcoal stripped fetal bovine serum (CS-FBS), at least 2 weeks, showed increased expression of AR and AR3 mRNA, inhibited the expression of PSA, and also up-regulated the expression of stem cell marker genes such as Lin28B, Nanog and Sox2, and EMT markers such as ZEB1, Twist, N-cadherin and vimentin. (B) C4-2B (upper panel) and VCaP cells (lower panel) grew in CS-FBS for 3 days and then treated with or without 10 nM DHT and incubated for another 24 hours. DHT treatment inhibited mRNA expression of AR and AR3, concomitant with decreased mRNA expression of Lin28B, Nanog, ZEB1, Twist and vimentin in C4-2B cells, and depressed mRNA expression of Sox2, N-cadherin and vimentin in VCaP cells. (C) C4-2B cells grew in CS-FBS for, at least 2 weeks, promoted protein expression of AR, AR3 and mesenchymal marker such as vimentin, whereas C4-2B cells grew in CS-FBS for 4 weeks showed increased protein expression of stem cell marker such as Nanog. (D) DHT treatment for 24 hours reversed protein expression of AR, AR3, PSA, Lin28B, vimentin and N-cadherin in C4-2B and VCaP cells (*p<0.05; **p<0.01).
Up-regulation of AR3 in PCa tissue specimens, which was positively correlated with increased expression of Lin28B
The mRNA levels of Lin28B were measured by real time RT-PCR using RNA extracted from FFPE PCa tissue specimens in comparison with adjacent normal tissues. We found that Lin28B expression was significantly higher in tumor tissues with Gleason grade ≥7 compared with adjacent normal tissues (Fig-5A and 5B). More importantly, AR3 expression was positively correlated with the expression Lin28B in PCa tissue specimens with Gleason grade 8 and 9 (Fig-5C), suggesting that AR3 could contribute to the regulation of Lin28B expression in PCa, and especially could be associated with the acquisition of cancer cell “stemness” that may be responsible for the development of CRPC and acquisition of therapeutic resistance. Therefore, elimination of stem cells by repression of stem cell markers or inactivation of AR and AR3 could become a promising strategy for the prevention of CRPC development and/or for the treatment of patients diagnosed with PCa, especially for the treatment of mCRPC with better treatment outcome. Interestingly, 3,3′-diindolylmethane (DIM obtained from BioResponse and termed as BR-DIM) was able to significantly inhibit the expression of Lin28B in patients' tumor specimens (obtained from our BR-DIM clinical trial as described under “Material and Methods” section) compared with untreated patients' tumor tissue samples (Fig-5). Further proof-of-concept results came from in vitro studies as documented below.
Fig-5. Lin28B was increased in PCa tissue specimens, which was positively correlated with AR3 expression, and abrogated by BR-DIM treatment in a phase II clinical trial.
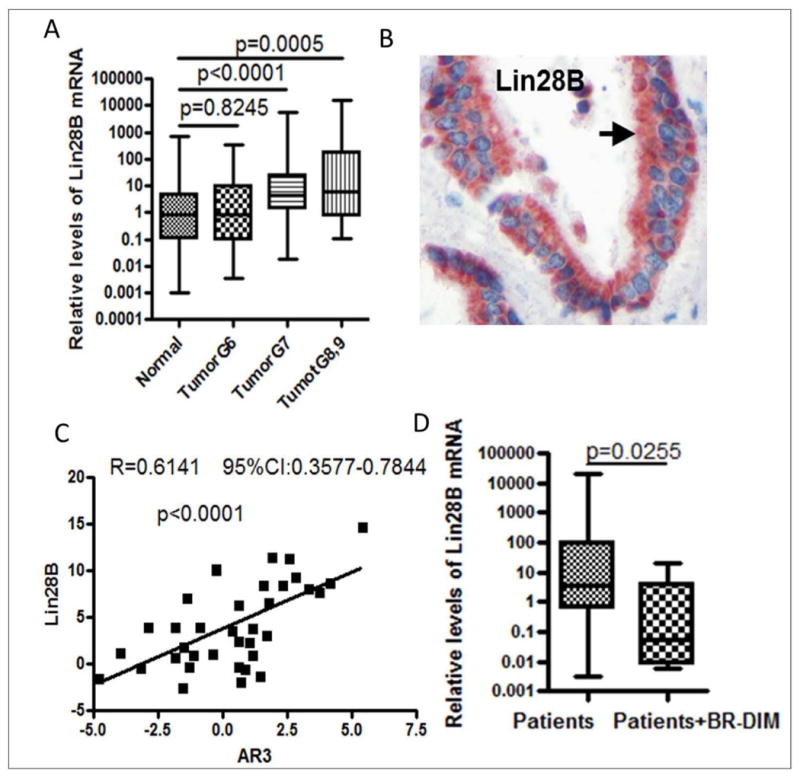
Total RNA was obtained from formalin-fixed paraffin-embedded (FFPE) tissue specimens of PCa patients and BR-DIM clinical trial samples with matched tumor Gleason grade, tumor stage and patient age as control group. BR-DIM was given to patients for 2-4 weeks prior to surgery. (A) The levels of Lin28B mRNA were found to be significantly increased in patients' tumors with higher Gleason grade (Gleason score ≥7) compared with adjacent normal tissues (N = 75 for normal, N = 38 for Tumor G6, N = 46 for Tumor G7, N = 34 for Tumor G8 and 9). (B) Immunohistochemical staining showed increased expression of Lin28B localized in the cytoplasm. (C) Lin28B expression was positively correlated with expression of AR3 in patients' tumors with Gleason grade ≥8 (N = 36). (D) BR-DIM treatment led to decreased expression of Lin28B (N = 67 for patients, N = 7 for patients with BR-DIM treatment).
BR-DIM inhibited the expression of AR and AR3, resulting in the repression of “stemness” in PCa cells
Sphere-forming capacity is the hallmark of stem cell characteristics. The prostaspheres (sphere-forming ability of PCa cells) derived from LNCaP cells showed increased expression of stem cell marker genes including Lin28B, Nanog, Oct4 and Sox2 (Fig-6A). Although there was no significant difference in the expression of AR and AR3 between prostaspheres from LNCaP (an androgen-responsive cell line), and the parental LNCaP cells (Fig-6A), the expression of AR and AR3 was found to be higher in prostaspheres derived from 22RV1 cells (an androgen-independent cell line), compared with the parental 22RV1 cells. Expressions of stem cell marker genes including Lin28B, Nanog, Sox2, CD44, and EMT markers such as ZEB1 and vimentin were also increased in prostaspheres derived from 22RV1 cells (Fig-6B). Interestingly, BR-DIM treatment for 4 days inhibited the expression of AR variants and PSA (Fig-6C), and also inhibited the expression of Nanog (Fig-6D) in LNCaP and LNCaP-derived prostasphere cells. We also found that BR-DIM treatment for 4 days repressed the expression of AR, PSA and AR3 mRNA concomitant with the inhibition in the expression of stem cell marker genes (Lin28B, Nanog, Oct4, Sox2 and CD44) in 22RV1 prostasphere cells (Fig-6E). These results are consistent with the results from Western blot analysis showing that BR-DIM treatment down-regulates the expression of AR, AR3 and PSA proteins in 22Rv1 cells and prostaspheres derived from 22RV1 cells (Fig-6F). Furthermore, we treated LNCaP and C4-2B cells with BR-DIM for a shorter period of time such as 1, 2 or 3 days, and we found that BR-DIM treatment led to reduced expression of AR, AR variants, PSA and fibronectin but not Nanog as early as 24 hours (Fig-7A). Moreover, BR-DIM treatment not only inhibited the expression of AR, AR variants, PSA and EMT marker fibronectin, but also repressed the expression of stem cell markers such as Nanog (Fig-7B) in both LNCaP and C4-2B cell lines after 2 days or 3 days, which was consistent with the results showing that BR-DIM treatment repressed the prostasphere-forming capacity as documented by reduced numbers and size of prostaspheres (Fig-7C) and cell migration capacity (Fig-7D) of C4-2B cells compared with untreated cells. These results suggest that BR-DIM treatment could eliminate stem cell characteristics, and thus could prevent tumor recurrence and metastasis through inactivation of AR signaling. Collectively, our results suggest that BR-DIM might become a promising therapeutic drug for the prevention of tumor progression to CRPC and/or for the treatment of PCa.
Fig-6. BR-DIM treatment inhibited the expression of AR, AR variants and stem cell markers.
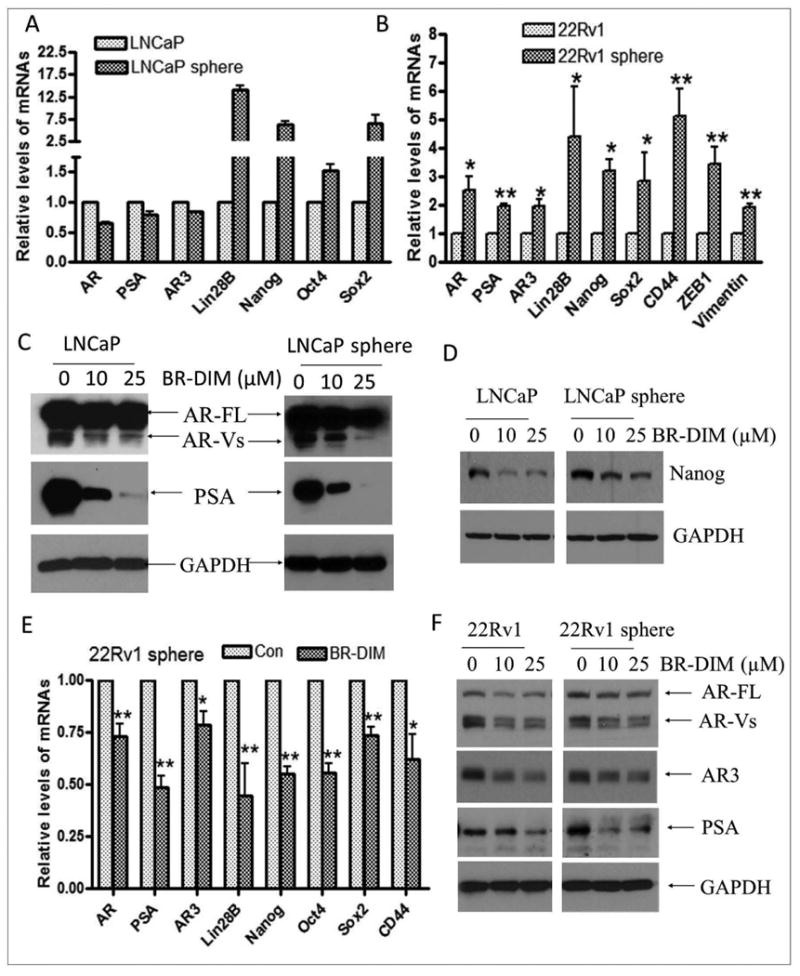
Single cell suspensions of LNCaP and 22RV1 cells were plated in 6-well plates with ultra-low attachment surface at 2000 cells/well and incubated for 3 days. The cells subsequently received fresh medium with or without BR-DIM, and then incubated for another 4 days. Prostasphere cells were collected for preparation of cell lysates or isolation of RNA. (A) Increased expression of Lin28B, Nanog, Oct4 and Sox2 mRNA were found in sphere-forming cells from LNCaP. (B) The mRNA expression of AR, AR3, EMT markers such as ZEB1 and vimentin, and stem cell markers such as Lin28B, Nanog, Sox2 and CD44 were found to be increased in sphere-forming cells from 22RV1 cells. (C) BR-DIM treatment decreased the expression of AR variants (AR-Vs) and PSA in LNCaP and LNCaP sphere-forming cells. (D) BR-DIM treatment inhibited the expression of stem cell marker: Nanog in LNCaP and LNCaP sphere cells. (E) BR-DIM treatment also repressed the mRNA expression of stem cell markers such as Lin28B, Nanog, CD44, Sox2 and Oct4 in 22RV1 sphere cells. (F) Expression of full length AR (AR-FL), AR variants and AR3 was reduced by BR-DIM treatment in 22RV1 cells and 22RV1 sphere-forming cells (*p<0.05; **p<0.01).
Fig-7. BR-DIM treatment inhibited cell migration and prostasphere-forming ability.
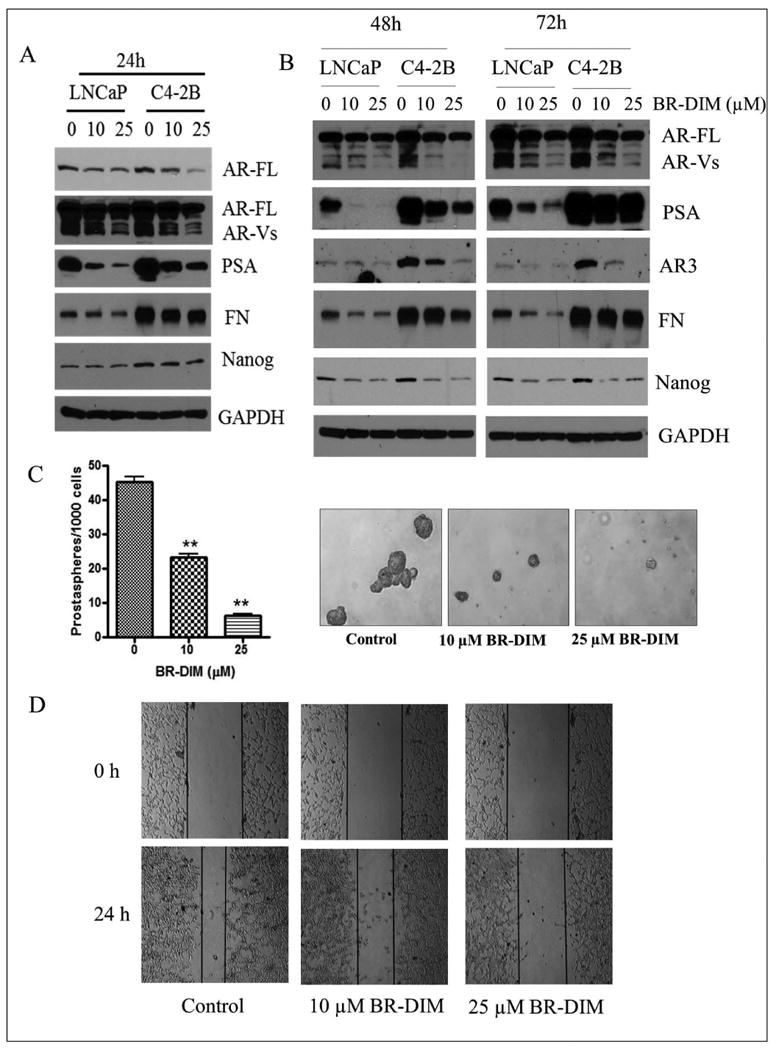
(A) BR-DIM treatment for 24 hours down-regulated the expression of AR, AR variants, AR3 and PSA as well as fibronectin (FN) in LNCaP and C4-2B cells. (B) BR-DIM treatment for 2 days and 3 days inhibited the expression of AR, AR variants, AR3, PSA, fibronectin and Nanog in LNCaP and C4-2B cells. (C) BR-DIM treatment significantly reduced prostasphere numbers and size (**p<0.01). (D) BR-DIM treatment significantly inhibited C4-2B cell migration after 24h.
Discussion
Androgen receptor splice variants (AR-Vs) have been suggested to be mechanistically involved in castration-resistant prostate cancer (CRPC), and it has also been implicated in metastatic CRPC (mCRPC). These AR-Vs lacking a COOH terminal ligand-binding domain (LBD) localize in the nuclear compartment, and are constitutively active. In this study, we showed that AR expression was higher in prostate cancer (PCa) tissue specimens with Gleason grade 6, but not in those patient tissues with Gleason grade ≥7. In contract, one of the AR-Vs, AR3 expression was higher in patients' tumor tissues, especially in patients' tissues with higher Gleason grade. Moreover, AR3 expression but not AR expression was positively correlated with Lin28B expression in patients' tissues with higher Gleason grade. These results suggest that AR3 but not AR is involved in the regulation of stem cell marker gene expression, which are consistent with our results showing that overexpression of AR3 but not AR induced stem cell signatures such as increased expression of Nanog and Lin28B, resulting in the promotion of cell migration and clonogenicity as well as led to enhanced prostasphere formation of PCa cells. Hu et al demonstrated that ligand-dependent full length AR and ligand-independent AR variants mediates different signaling pathway. AR full length signaling regulates the gene expression related to the biosynthesis, metabolism and secretion pathways, whereas AR-Vs up-regulates the expression of cell cycle genes such as UBE2C [33]. Moreover, they found that suppression of AR full length signaling pathway by targeting the AR-LBD via androgen deprivation or treatment with MDV3100 (Enzalutamide) led to increased expression of AR-Vs, especially the expression of AR3 [33], which is consistent with our results showing that androgen deprivation leads to the inhibition in the expression of PSA, and increased the expression of AR and AR3 in PCa cells. Moreover, two PCa cell lines such as VCaP [34] derived from patients with hormone refractory PCa, and 22RV1 cells [35] derived from a xenograft that was serially propagated in mice after castration-induced regression and relapse of the parental, androgen-dependent CWR22 xenograft, showed higher levels of AR3 expression compared with LNCaP and C4-2B cells. These results suggest that the inhibition of AR signaling by targeting the AR-LBD could activate the expression of AR-Vs, especially AR3, which might be responsible for the progression of PCa into CRPC and mCRPC after ADT in most PCa patients.
Although increasing evidence from in vivo and in vitro studies has indicated that androgen deprivation-mediated up-regulation of AR-Vs are involved in the progression of PCa into CRPC [5-7, 12], the mechanisms underlying castration resistance need to be further elucidated in order to design appropriate therapeutic strategy for better treatment of patients with CRPC, especially mCRPC. In the present study, we showed that androgen deprivation resulted in the induction of EMT phenotype and the expression of stem cell marker genes (acquisition of “stemness”) as characterized by up-regulated expression of EMT markers such as ZEB1, N-cadherin and vimentin and stem cell markers such as Lin28 and Nanog, which was found to be associated with increased expression of AR3. DHT treatment of PCa cells reversed these molecular changes, suggesting that the suppression of full length AR signaling-mediated activation of AR3 signaling could contribute to the induction of EMT and “stemness”. This was further confirmed by the results showing that overexpression of AR3 but not AR in PCa cell lines led to increased expression of Lin28B and Nanog, which was associated with increased prostasphere formation and clonogenicity. Overexpression of AR3 also promoted the expression of ZEB1, fibronectin and vimentin, which is linked with increased cell migration. These results are consistent with the findings from Cottard et al showing that overexpression of AR-Vs promotes the expression of N-cadherin, vimentin and ZEB1 [8]. Sun et al showed that androgen deprivation could induce EMT and stem cell-like features in normal mouse prostate tissue and human LuCaP35 prostate tumor explants. They also found that increased mesenchymal signatures in prostate tumors from patients treated with androgen-deprivation therapy [16]. A recent study further suggest that AR3 driven PCa initiation and progression can occur in a transgenic mouse model (AR3Tg) by modulating TGF-beta pathway, which was associated with the elaboration of EMT signatures and increasing prostatic progenitor cell population [11]. These in vivo results confirmed our findings indicating that androgen deprivation-mediated increased expression of AR3 contributes to the development of CRPC and mCRPC by regulating EMT and stem cell pathways. Thus, one could envision that effective drugs for the treatment PCa would be those which can inhibit the acquisition of EMT and “stemness” characteristics and could also eliminate these cells. This indeed could be possible (as discussed below) by targeted inactivation of AR-Vs especially AR3 for the inhibition of tumor growth by eliminating stem cells to prevent tumor recurrence and metastasis after ADT.
BR-DIM has been demonstrated to possess anti-tumor activity in vivo and in vitro by regulating multiple pathways [23-25]. In this study, we showed that BR-DIM not only inhibited the activity of AR canonical signaling pathway as characterized by significant inhibition of PSA expression, but also repressed the expression of AR variant (AR3), resulting in the suppression in the expression of EMT markers including ZEB1, N-cadherin, fibronectin as well as stem cell markers such as Nanog and Lin28B. These results were further associated with the inhibition of sphere-forming capacity of PCa cells. One recent study has shown that the inhibition of AR-FL signaling by targeting AR-LBD via androgen deprivation or treatment of cells with MDV3100 (enzalutamide), which inhibits AR activity by binding to AR-LBD, led to increased expression of AR variants [33], which has been involved in PCa progression to more aggressive CRPC and mCRPC [3, 5, 6, 9, 12, 36, 37]. The results of our study suggest that AR-Vs especially AR3 could contribute to the acquisition of CRPC by regulating EMT and stem cell related pathways. Interestingly, BR-DIM showed inhibition of both AR-FL and/or AR3 in PCa cells in vitro and also in vivo as documented through using PCa tissue specimens obtained from BR-DIM clinical trial. Thus, BR-DIM treatment could not only inhibit tumor growth by blocking the signaling of AR-FL, but could also prevent tumor recurrence and metastasis after conventional ADT and/or enzalutamide (MDV3100) treatment, suggesting that BR-DIM could serve as a promising agent for the prevention of tumor progression and/or treatment of PCa especially CRPC and mCRPC.
Acknowledgments
Special thanks to Scott M. Dehm who kindly provided the p5HBhAR-A plasmid expressing full length AR and p5HBhAR-1-2-3-CE3 expressing AR3 (Masonic Cancer Center, University of Minnesota, Minneapolis, MN). Special thanks to Dr. Gerold Bepler (President and CEO of Karmanos Cancer Institute, Wayne State University School of Medicine, Detroit, Michigan) and Dr. Anthony Shields (Professor & Associate Center Director for Clinical Research, Karmanos Cancer Institute, Wayne State University School of Medicine, Detroit, Michigan) for supporting the BR-DIM clinical trial. We would like to thank Drs. Michael Cher*, Isaac Powell*, Clara Hwang**, Nilesh Gupta**, Dhananjay Chitali**, and Mani Menon***, Departments of Urology*, Wayne State University; Departments of Pathology** and Urology***, Henry Ford Health System, Detroit, Michigan for their participation in the BR-DIM clinical trial. Grant support: This work was funded by grants from the National Cancer Institute, NIH (5R01CA108535-06 and 5R01CA083695-08 to FHS).
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
Reference List
- 1.Wang X, Kruithof-de JM, Economides KD, Walker D, Yu H, Halili MV, Hu YP, Price SM, Abate-Shen C, Shen MM. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461:495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, Hirsch B, Beckman KB, Silverstein KA, Dehm SM. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 2012;31:4759–4767. doi: 10.1038/onc.2011.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang X, Morrissey C, Sun S, Ketchandji M, Nelson PS, True LD, Vakar-Lopez F, Vessella RL, Plymate SR. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One. 2011;6:e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A, Wikstrom P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011;6:e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, Nelson PS, Plymate SR. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cottard F, Asmane I, Erdmann E, Bergerat JP, Kurtz JE, Ceraline J. Constitutively active androgen receptor variants upregulate expression of mesenchymal markers in prostate cancer cells. PLoS One. 2013;8:e63466. doi: 10.1371/journal.pone.0063466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem. 2012;287:19736–19749. doi: 10.1074/jbc.M112.352930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–489. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun F, Chen HG, Li W, Yang X, Wang X, Jiang R, Guo Z, Chen H, Huang J, Borowsky AD, Qiu Y. Androgen Receptor Splice Variant AR3 Promotes Prostate Cancer via Modulating Expression of Autocrine/Paracrine Factors. J Biol Chem. 2014;289:1529–1539. doi: 10.1074/jbc.M113.492140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, Brodie AM, Edwards J, Qiu Y. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kong D, Li Y, Wang Z, Sarkar FH. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition (EMT)-Phenotypic Cells: Are They Cousins or Twins? Cancers (Basel) 2011;3:716–729. doi: 10.3390/cancers30100716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen CL, Mahalingam D, Osmulski P, Jadhav RR, Wang CM, Leach RJ, Chang TC, Weitman SD, Kumar AP, Sun L, Gaczynska ME, Thompson IM, Huang TH. Single-cell analysis of circulating tumor cells identifies cumulative expression patterns of EMT-related genes in metastatic prostate cancer. Prostate. 2013;73:813–826. doi: 10.1002/pros.22625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, Herold CI, Marcom PK, George DJ, Garcia-Blanco MA. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res. 2011;9:997–1007. doi: 10.1158/1541-7786.MCR-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, Chen D, Seo K, Modrusan Z, Gao WQ, Settleman J, Johnson L. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res. 2012;72:527–536. doi: 10.1158/0008-5472.CAN-11-3004. [DOI] [PubMed] [Google Scholar]
- 17.Klarmann GJ, Hurt EM, Mathews LA, Zhang X, Duhagon MA, Mistree T, Thomas SB, Farrar WL. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clin Exp Metastasis. 2009;26:433–446. doi: 10.1007/s10585-009-9242-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong D, Banerjee S, Ahmad A, Li Y, Wang Z, Sethi S, Sarkar FH. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS One. 2010;5:e12445. doi: 10.1371/journal.pone.0012445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, Haluska P, Ingle JN, Hartmann LC, Manjili MH, Radisky DC, Ferrone S, Knutson KL. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009;69:2887–2895. doi: 10.1158/0008-5472.CAN-08-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sethi S, Macoska J, Chen W, Sarkar FH. Molecular signature of epithelial-mesenchymal transition (EMT) in human prostate cancer bone metastasis. Am J Transl Res. 2010;3:90–99. [PMC free article] [PubMed] [Google Scholar]
- 22.Chen D, Banerjee S, Cui QC, Kong D, Sarkar FH, Dou QP. Activation of AMP-activated protein kinase by 3,3′-Diindolylmethane (DIM) is associated with human prostate cancer cell death in vitro and in vivo. PLoS One. 2012;7:e47186. doi: 10.1371/journal.pone.0047186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong D, Heath E, Chen W, Cher ML, Powell I, Heilbrun L, Li Y, Ali S, Sethi S, Hassan O, Hwang C, Gupta N, Chitale D, Sakr WA, Menon M, Sarkar FH. Loss of let-7 up-regulates EZH2 in prostate cancer consistent with the acquisition of cancer stem cell signatures that are attenuated by BR-DIM. PLoS One. 2012;7:e33729. doi: 10.1371/journal.pone.0033729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Kong D, Ahmad A, Bao B, Sarkar FH. Targeting bone remodeling by isoflavone and 3,3′-diindolylmethane in the context of prostate cancer bone metastasis. PLoS One. 2012;7:e33011. doi: 10.1371/journal.pone.0033011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kong D, Banerjee S, Huang W, Li Y, Wang Z, Kim HR, Sarkar FH. Mammalian target of rapamycin repression by 3,3′-diindolylmethane inhibits invasion and angiogenesis in platelet-derived growth factor-D-overexpressing PC3 cells. Cancer Res. 2008;68:1927–1934. doi: 10.1158/0008-5472.CAN-07-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nadiminty N, Tummala R, Liu C, Yang J, Lou W, Evans CP, Gao AC. NF-kappaB2/p52 induces resistance to enzalutamide in prostate cancer: role of androgen receptor and its variants. Mol Cancer Ther. 2013;12:1629–1637. doi: 10.1158/1535-7163.MCT-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen P, Peehl DM, Graves HC, Rosenfeld RG. Biological effects of prostate specific antigen as an insulin-like growth factor binding protein-3 protease. J Endocrinol. 1994;142:407–415. doi: 10.1677/joe.0.1420407. [DOI] [PubMed] [Google Scholar]
- 28.Webber MM, Waghray A, Bello D. Prostate-specific antigen, a serine protease, facilitates human prostate cancer cell invasion. Clin Cancer Res. 1995;1:1089–1094. [PubMed] [Google Scholar]
- 29.Kong D, Wang Z, Sarkar SH, Li Y, Banerjee S, Saliganan A, Kim HR, Cher ML, Sarkar FH. Platelet-derived growth factor-D overexpression contributes to epithelial-mesenchymal transition of PC3 prostate cancer cells. Stem Cells. 2008;26:1425–1435. doi: 10.1634/stemcells.2007-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong D, Li Y, Wang Z, Banerjee S, Ahmad A, Kim HR, Sarkar FH. miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells. 2009;27:1712–1721. doi: 10.1002/stem.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohler JL, Gregory CW, Ford OH, III, Kim D, Weaver CM, Petrusz P, Wilson EM, French FS. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10:440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 32.Agoulnik IU, Weigel NL. Androgen receptor action in hormone-dependent and recurrent prostate cancer. J Cell Biochem. 2006;99:362–372. doi: 10.1002/jcb.20811. [DOI] [PubMed] [Google Scholar]
- 33.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, Edwards J, Isaacs WB, Nelson PS, Bluemn E, Plymate SR, Luo J. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–3462. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korenchuk S, Lehr JE, MClean L, Lee YG, Whitney S, Vessella R, Lin DL, Pienta KJ. VCaP, a cell-based model system of human prostate cancer. In Vivo. 2001;15:163–168. [PubMed] [Google Scholar]
- 35.Sramkoski RM, Pretlow TG, Giaconia JM, Pretlow TP, Schwartz S, Sy MS, Marengo SR, Rhim JS, Zhang D, Jacobberger JW. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell Dev Biol Anim. 1999;35:403–409. doi: 10.1007/s11626-999-0115-4. [DOI] [PubMed] [Google Scholar]
- 36.Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer. 2011;18:R183–R196. doi: 10.1530/ERC-11-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haile S, Sadar MD. Androgen receptor and its splice variants in prostate cancer. Cell Mol Life Sci. 2011;68:3971–3981. doi: 10.1007/s00018-011-0766-7. [DOI] [PMC free article] [PubMed] [Google Scholar]