

Abstract
Objective: To describe the phenotypic features of an ethnically homogenous group of patients with Fanconi–Bickel syndrome harboring the p.R310X mutation.
Methods: The study group consisted of eight patients from a single Bedouin family with clinically and molecularly diagnosed Fanconi–Bickel syndrome who had been followed at the same tertiary medical center for 8 years or more. All were homozygous for the p.R310X mutation. The medical files were reviewed for presenting signs and symptoms, laboratory and imaging findings, treatment regimens, and disease severity over time.
Results: Seven patients were diagnosed at our center before age 1 year, and one transferred from another center at age 16 years. Most patients presented with failure to thrive and/or hepatomegaly. All had short stature and doll-like facies. Most had biochemical abnormalities. Evaluation of the long-term findings revealed a wide spectrum of disease severity according to the following parameters: growth patterns, maximal electrolyte replacement therapy, skeletal and renal complications, frequency of follow-up visits, and hospitalizations for disease exacerbations. There was no apparent association of the clinical picture at presentation and later disease severity.
Conclusion: Fanconi–Bickel syndrome has a broad phenotypic variability in patients harboring the same homozygous p.R301X mutation. This finding might be explained by genetic elements such as modifier genes and epigenetic factors, as well as the effects of still-undetermined environmental and nutritional factors.
Introduction
Fanconi–Bickel syndrome (FBS, OMIM 227810) is a rare autosomal recessive disease caused by a deficiency of glucose transporter protein 2 (GLUT2) (Santer et al. 1998), a member of the facilitative glucose transporter family. GLUT passively mediates the bidirectional transport of monosaccharides, mainly d-glucose and d-galactose, in hepatocytes, pancreatic β cells, renal tubular cells, and enterocytes (Santer et al. 2002a). The gene encoding GLUT2 (SLC2A2) is located on chromosome 3q26.1–q26.3 (OMIM 138160). Of the 59 or more known mutations that are responsible for FBS, p.R301X, a nonsense mutation at nucleotide c.1213 of exon 7, is the most common (http://www.biobase-international.com).
Signs and symptoms of FBS begin in infancy and include failure to thrive (FTT), hepatomegaly, hypophosphatemic rickets, and short stature (Santer et al. 2002b). Biochemically, the disease is characterized by a general renal proximal tubular defect (glycosuria, bicarbonate wasting, aminoaciduria, renal tubular acidosis, hyperphosphaturia) and carbohydrate abnormalities that include postprandial hyperglycemia, fasting hypoglycemia, and hypergalactosemia (Odièvre et al. 2002). The diagnosis is suspected on the basis of the clinical and laboratory findings and confirmed by molecular genetic testing. Management consists of nutritional supplement with uncooked cornstarch and fluid and electrolyte replacement therapy (Lee et al. 1995).
FBS is generally considered a well-defined clinical condition (Odièvre et al. 2002), although there are reports of atypical clinical signs or an unusually mild disease course (Aperia et al. 1981; Berry et al. 1995; Grünert et al. 2012). Long-term follow-up studies have focused mainly on specific disease characteristics, such as renal (Manz et al. 1987) and hepatic complications (Saltik-Temizel et al. 2005). In a study of the genotype–phenotype correlation in FBS, Santer et al. (2002b) investigated the various possible presentations of FBS due to null versus missense mutations.
The aim of the present study was to describe the presenting and long-term features of FBS and to assess phenotypic variability in an ethnically homogenous group of patients all harboring the p.R301X mutation.
Patients and Methods
The Bedouin are an ethnic Arabian, traditionally nomadic, group divided into tribes and clans with a complex consanguinity. The cohort for the present study consisted of seven patients (four male) of a single family (Fig. 1) and a male patient born to third-degree consanguineous parents from the same Bedouin clan, all homozygous for the p.R301X mutation. The patients had been followed at the Day Hospitalization Unit of Schneider Children's Medical Center of Israel for more than 8 years. For the present study, the medical files were reviewed for data on presenting symptoms and signs, laboratory and imaging findings, and treatment regimens. Disease severity was evaluated by the following parameters: growth patterns, mean electrolyte replacement therapy, skeletal and renal complications, frequency of follow-up visits, and hospitalizations for disease exacerbation. In addition the parents of the minors and the adult patient were assessed for the severity of the disease on a scale from 1 (mild) to 3 (severe).
Fig. 1.

The pedigree of the Bedouin sibship Note: III.2 and IV.11, III.11 and IV 10, IV. 9, and IV.13 are the same individuals. The intrafamilial relation of patient 4 could not be tracked
Growth was evaluated in the pre-pubertal stage (age 5–7 years) by calculating the standard deviation score (Z-score) for mean height for age and sex, as defined by the guidelines of the World Health Organization (WHO) Global Database on Child Growth and Malnutrition (de Onis and Blössner 2003). Hyperglycemia and hypoglycemia were defined as glucose >170 mg/dL and <50 mg/dL, respectively. Biochemical urinary parameters were taken by urine spots; amino acids excretion was measured qualitatively and phosphaturia at presentation/diagnosis was estimated by calculating the tubular reabsorption of phosphate (TRP). The presence of rickets or osteopenia/osteoporosis was evaluated after diagnosis by plain bone X-rays of the distal femur, proximal tibia, distal radius, and distal ulna. The presence of nephropathy and nephrocalcinosis was evaluated by repeated urine analyses and sonography.
Results
Patients
Seven patients were referred to our medical center before age 1 year; 3 were 1 month old or younger. The remaining patient (no. 6) was diagnosed at age 3 years at another hospital, where he was managed until age 16 years. In all cases, the diagnostic investigation was prompted by patient symptoms and signs or by incidental physical findings on routine general checkup. None of the patients was diagnosed prenatally. All were managed by corn starch supplement and fluid and electrolyte replacement therapy.
Detailed Patient Descriptions
Patient 1 presented at age 11 months with FTT, doll-like facies, hepatomegaly, developmental delay, rachitic rosary, and bowed legs. Laboratory work-up at presentation revealed remarkable fluctuations in blood glucose levels, metabolic acidosis, hypocalcemia, hypophosphatemia, hypouricemia, and hypokalemia in addition to massive glucosuria, hyperphosphaturia, and generalized aminoaciduria compatible with proximal tubulopathy. Over the course of follow-up, early radiologic abnormalities of rickets and osteopenia improved considerably. At no time did the patient require hospitalization. Relatively low amounts of phosphorus and potassium supplementation were needed.
Patient 2 presented at age 7 months with FTT, doll-like facies, hepatomegaly, rachitic rosary, and enlarged wrists and ankles. Laboratory work-up at presentation revealed significant fluctuations in blood glucose levels, hypoglycemia, postprandial hyperglycemia, mild metabolic acidosis, hypophosphatemia, hypouricemia, massive glucosuria, excessive phosphate losses, and generalized aminoaciduria. Skeletal evaluation during follow-up revealed severely stunted growth, bilateral genu varum, rickets, and osteopenia; nephrocalcinosis developed at age 5 years. High amounts of electrolyte supplementation were necessary. During follow-up, the patient was hospitalized nine times for metabolic acidosis, hypokalemia, hypophosphatemia, and three events of hypocalcemic tetany.
Patient 3 presented at age 6 months with FTT, doll-like facies, and hepatomegaly. Laboratory work-up at presentation revealed remarkable fluctuations in blood glucose levels, hypokalemia, hypouricemia, massive glucosuria, hyperphosphaturia, and generalized aminoaciduria. Skeletal evaluation over the course of follow-up showed short stature, bilateral genu varum, and osteopenia. Low amounts of phosphorus replacement therapy were needed. During follow-up, the patient was hospitalized once for metabolic acidosis during acute gastroenteritis.
Patient 4 presented at age 1 month with symptomatic hypoglycemia, prolonged diarrhea, FTT, hepatomegaly, hypotonia, and rickets. Laboratory work-up at presentation showed significant fluctuations in blood glucose levels, hypoglycemia, postprandial hyperglycemia, hypocalcemia, hypophosphatemia, hypouricemia, hypokalemia, and hyponatremia. His urine studies revealed massive glucosuria, hyperphosphaturia, and generalized aminoaciduria compatible with proximal tubulopathy. X-rays showed signs of rickets. Skeletal evaluation during follow-up revealed severely stunted growth and rickets. The patient had recurrent nocturnal symptomatic hypoglycemic episodes that necessitated gastrostomy for continuous night feedings until age 3 years. He had hypercalciuria and developed nephrocalcinosis at age 6 years. He required high amounts of electrolyte replacement therapy. During follow-up, he was hospitalized 8 times for electrolyte imbalance and once, in the pediatric intensive care unit (PICU), for hyponatremia. This patient was initially misdiagnosed with insulin-dependent diabetes mellitus; FBS was diagnosed at age 5 months.
Patient 5 presented at age 1 month with recurrent vomiting, FTT, hypotonia, and hepatomegaly. Laboratory work-up at presentation revealed significant fluctuations in blood glucose levels, postprandial hyperglycemia, hypocalcemia, hypophosphatemia, and hypouricemia; urine studies showed massive glucosuria, excessive phosphate, and generalized aminoaciduria compatible with proximal tubulopathy. Skeletal evaluation during follow-up revealed severely stunted growth, rickets, and osteopenia. Over the course of follow-up he had hypercalciuria, and developed nephrocalcinosis at age 4.5 years. High amounts of phosphorus replacement therapy were needed. During follow-up, he was hospitalized nine times for diarrhea, prerenal azotemia, recurrent pneumonia, and symptomatic hypocalcemia, and twice, in the PICU, for hypocalcemia.
Patient 6 presented at age one month with FTT, doll-like facies, hepatomegaly, and hypotonia. Laboratory work-up at presentation revealed significant fluctuations in blood glucose levels including hypoglycemia and postprandial hyperglycemia, hypocalcemia, hypophosphatemia, hypokalemia, hypouricemia, massive glucosuria, hyperphosphaturia, and generalized aminoaciduria; X-rays revealed signs of rickets. He was diagnosed at another hospital when he was 3 years old. The patient was hospitalized only once, for pathological fracture. Final height was 154 cm, with mild osteopenia. Since he was under our follow-up he did not require electrolyte supplementation to age 29 years, when he was lost to follow-up. The patient is married and has three healthy children.
Patient 7 presented at age 1 year with hepatomegaly. Laboratory work-up at presentation revealed significant fluctuations in blood glucose levels with postprandial hyperglycemia, mild metabolic acidosis, hypokalemia, hypouricemia, massive glucosuria, hyperphosphaturia, and generalized aminoaciduria. Skeletal evaluation during follow-up showed short stature and radiologic signs of rickets. High amounts of electrolyte replacement therapy were needed. During follow-up, the patient was hospitalized 14 times for bronchopneumonia, acute gastroenteritis, and hypokalemia, and twice, in the PICU, for hypokalemia and metabolic acidosis.
Patient 8 presented at age 1 year with FTT, hepatomegaly, bowed legs, and enlarged wrists and ankles. Laboratory work-up at presentation revealed significant fluctuations in blood glucose levels with postprandial hyperglycemia, metabolic acidosis, hypocalcemia, hypophosphatemia, hypouricemia, massive glucosuria, hyperphosphaturia, and generalized aminoaciduria compatible with proximal tubulopathy; X-rays showed signs of rickets. Skeletal evaluation during follow-up showed severely stunted growth and rickets. Very high amounts of phosphorus supplementation were needed. Nephrocalcinosis developed at age 9 years. During follow-up, the patient was hospitalized six times for bone pain that necessitated intravenous therapy with opiates.
Summary of Findings
Presentation
The main demographic, clinical, laboratory, and radiological findings at presentation and diagnosis are summarized in Table 1. All patients had short stature and doll-like facies. All had prominent fluctuations in blood glucose levels and hypouricemia; most patients had also metabolic acidosis, hypophosphatemia hypocalcemia, and hypokalemia. In addition, all had massive glucosuria, excessive phosphate losses with low TRP and generalized aminoaciduria compatible with proximal tubulopathy; none had microalbuminuria at presentation. Five patients had clinical and/or radiological signs of rickets at presentation.
Table 1.
Main demographic, clinical, laboratory, and radiological findings at first presentation in eight patients with Fanconi–Bickel syndrome with the p.R310X mutation
| Pt. no./year of birth | Sex | Age at presentation/symptoms and signs | Main laboratory findings at diagnosis | Skeletal findings | ||
|---|---|---|---|---|---|---|
| Blood | Urine | Clinical | Radiologic | |||
| 1/2001 | M | 11 months/ FTT Wt 15, length 3 Hepatomegaly (+5) Developmental delay |
Ca 8.7 pH 7.29 HCO3 17.4 K 3.0 Na 132 Phos 2.5 Uric acid 1.5 Glucose 63–157 |
Glucose >1,000 mg/dL General aminoaciduria TRP 83% |
Bowed legs Rachitic rosary |
Rickets Osteopenia |
| 2/2002 | F | 7 months/ FTT Wt <2, length <2 Hepatomegaly (+6) |
Ca 8.4 pH 7.36 HCO3 12.5 K 2.5 Na 135 Phos 1.8 Uric acid 0.8 Glucose 46–202 |
Glucose >1,000 mg/dL General aminoaciduria TRP 68% |
Rachitic rosary Enlarged wrists and ankles |
Rickets Osteopenia |
| 3/2001 | F | 6 months/ FTT Wt 25, length 3 Hepatomegaly (+3) |
Ca 10.3 pH 7.33 HCO3 17 K 2.0 Na 133 Phos 4.4 Uric acid 0.8 Glucose 61–160 |
Glucose >1,000 mg/dL General aminoaciduria TRP 80% |
None | Osteopenia |
| 4/1998 | M | 1 month/ Symptomatic hypoglycemia Prolonged diarrhea Hepatomegaly(+3) |
Ca 7.8 pH 7.30 HCO3 16.0 K 2.4 Na 127 Phos 2.4 Uric acid 0.7 Glucose 35–271 |
Glucose >1,000 mg/dL General aminoaciduria TRP 57% |
Enlarged wrists and ankles | Rickets |
| 5/2002 | M | 1 month/ FTT Wt <3, length <3 Recurrent vomiting Hypotonia Hepatomegaly (+2) |
Ca 8.7 pH 7.32 HCO3 18.0 K 4.1 Na 140 Phos 2.4 Uric acid 0.8 Glucose 50–190 |
Glucose >1,000 mg/dL General aminoaciduria TRP 64% |
None | None |
| 6/1980 | M | 1 month/ FTT Wt 5, length 3 Hepatomegaly (+5) Hypotonia |
Ca 7.8 pH 7.36 HCO3 24.6 K 3.3 Na 138 Phos 2.3 Uric acid 1.1 Glucose 46–202 |
Glucose >1,000 mg/dL General aminoaciduria TRP 79% |
None | Rickets |
| 7/1997 | M | 1 year/ Hepatomegaly (+4) |
Ca 10.8 pH 7.33 HCO3 19.8 K 3.2 Na 135 Phos 3.5 Uric acid 1.8 Glucose 61–210 |
Glucose >1,000 mg/dL General aminoaciduria TRP 81% |
None | None |
| 8/1999 | F | 1 year/ FTT Wt 5, length <3 Hepatomegaly (+4) |
Ca 7.1 HCO3 18 pH 7.28 K 3.8 Na 136 Phos 2.0 Uric acid 1.3 Glucose 53–190 |
Glucose >1,000 mg/dL General aminoaciduria TRP 80% |
Bowed legs Enlarged wrists and ankles |
Rickets |
Note: Patients 4 and 6 were diagnosed at ages 5 months and 3 years, respectively
Normal biochemical values in blood: Hypoglycemia <50 mg/dL, hyperglycemia >170 mg/dL, Ca 9.0–11.0 mg/dL, HCO3 22–26 mmol/L, pH 7.35–7.45, K 3.5–5.0 mEq/L, Na 135–145 mEq/L, uric acid 2.0–7.0 mg/dL, phosphorus 3.8–6.5 mg/dL. Normal biochemical values in urine: TRP (Total reabsorption phosphate): >85%. Calcium/creatinine ratios by age: 0–6 months. < 0.8, 7–12 months. <0.6, >2 years <0.21
FTT—failure to thrive (growth below the 5rd percentile or a change in growth that has crossed two major growth percentiles in a short time); numbers of weight and length percentiles are presented
Hepatomegaly (centimeters below midline costal margin)
Follow-up
The duration of follow-up ranged from 8.7 to 14 years (average 11.4 years). The following biochemical abnormalities were found in all patients during follow-up: postprandial hyperglycemic events, asymptomatic fasting ketotic hypoglycemia, hypouricemia, hyponatremia, hypokalemia; most had hypocalcemia and metabolic acidosis. All had massive glucosuria, generalized aminoaciduria, hypercalciuria, and hyperphosphaturia. Tables 2 and 3 summarize the main clinical, laboratory, and radiological findings during the course of the disease; subjective disease-severity assessment is presented in Table 3; Figs. 2a, b show the males and females' height curves, respectively. Longitudinal follow-up revealed variability in disease severity. Two patients (nos.1 and 3) needed small amounts of replacement therapy, and patient 6 (referred to our center at age 16 years) needed no replacement therapy when he was under our follow-up. Two patients (nos. 2 and 4) had exceptionally severe biochemical abnormalities: patient 2 had three hypocalcemic tetanic events, and patient 4 had recurrent nocturnal symptomatic hypoglycemia that necessitated gastrostomy for continuous night feedings until age 3 years.
Table 2.
Clinical findings in eight patients with Fanconi–Bickel syndrome with the p.R310X mutation during follow-up
| Pt. no. | F-U duration (year) | Annual visits (average) | No. hospitalizations/diagnoses | No. ICU admissions/diagnoses | Ht. SDSa |
|---|---|---|---|---|---|
| 1 | 11.7 | 5.6 | None | None | −2.78 |
| 2 | 9.3 | 9.6 | 9/Hypocalcemia with/without tetany Metabolic acidosis Hypophosphatemia |
1/Hypocalcemic tetany |
−4.33 |
| 3 | 10.5 | 4.3 | 1/Metabolic acidosis during acute gastroenteritis | None | −2.70 |
| 4 | 14 | 12.5 | 8/Hyponatremia Hypokalemia Hypophosphatemia |
1/Hyponatremia | −3.89 |
| 5 | 10 | 7.5 | 9/Diarrhea Acute prerenal azotemia Recurrent pneumonia Hypocalcemia |
2/Hypocalcemia | −5.32 |
| 6 | 13 | 2.6 | 1/Pathological fracture, It tibia | None | |
| 7 | 14 | 6.1 | 14/Bronchopneumonia Acute gastroenteritis Hypokalemia |
2/Hypokalemia Metabolic acidosis |
−3.18 |
| 8 | 8.7 | 6.6 | 6/Bone pain Hypophosphatemia |
None | −3.77 |
aHeight standard deviation score (Z-score) for mean height for age and gender
Table 3.
Laboratory and radiological findings of 8 patients with Fanconi–Bickel syndrome with the p.R310X mutation during follow-up
| Pt. no. | Main laboratory findings | Mean electrolyte replacement (kg/day) | Skeletal findings | Nephrocalcinosis (age at detection) | Disease severity score | ||
|---|---|---|---|---|---|---|---|
| Blood | Urine | Clinical | Radiologic | ||||
| 1 | Ca 8.7–10.2 HCO3 17.0–23.5 K 3.0–4.6 Na 132–137 Phos 2.5–4.6 Uric acid 1.3–2.0 Alk phos (8–10): 333–336. (11–13): 330–482 PTH 30.7–155.0 |
Ca/creat 0.26–0.95 Glucosuria >1,000 mg/dL General aminoaciduria Microalbumin (9 years) 460 |
Phos 70 mg K 0.8 meq |
Valgus knee | Mild osteopenia | No | 1 |
| 2 | Ca 8.4–10.1 HCO3 12.5–23.7 K 2.5–5.2 Na 130–139 Phos 1.8–7.6 Uric acid 0.8 -1.0 Alk phos (3–5): 317–554 (8–10): 504–697 PTH 10.6-68.8 |
Ca/creat 0.13–0.6 Glucosuria >1,000 mg/dL General aminoaciduria Microalbumin (5 years) 1,133 |
Phos 110 mg K 6.6 meq |
Bilateral genu varum |
Rickets Osteopenia |
Yes (5 years) | 2 |
| 3 | Ca 8.7–11.0 HCO3 19.1–27.1 K 2.0–4.7 Na 133–143 Phos 3.3–9.5 Uric acid 0.6–1.8 Alk phos (3–5): 280–522 (8–10): 376–4,400 PTH 14.4–85 |
Ca/creat 0.14–4.5 Glucosuria >1,000 mg/dL General aminoaciduria Microalbumin (6 years) 563 |
Phos 25 mg | Bilateral genu varum |
Osteopenia | No | 1 |
| 4 | Ca 7.8–9.7 HCO3 14.0–25.3 K 2.4–4.4 Na 127–139 Phos 1.9–6.2 Uric acid 0.7–2.0 Alk phos (3–5): 600–1,400 (8–10): 388–1,280 (11–13): 505–986 PTH 12.7–224.3 |
Ca/creat 0.22–2.38 Glucosuria >1,000 mg/dL General amino aciduria Microalbumin (6 years) 302 |
Phos 98 mg K 4.4 meq |
Bilateral genu varum |
Rickets Osteopenia |
Yes (6 years) | 3 |
| 5 | Ca 6.5–10.7 HCO3 13.4–28.6 K 2.6–5.9 Na 131–145 Phos 2.2–11.6 Uric acid 0.6–1.5 Alk phos (3–5): 342–677.0 (8–10): 373–1,159 PTH 60.9–191.0 |
Ca/creat 0.15–5.0 Glucosuria >1,000 mg/dL General aminoaciduria Microalbumin (4 years) 124 |
HCO3 0.3 meq Phos 120 mg K 2.1 meq |
Rickets Osteopenia |
Yes (4.5 years) | 2 | |
| 6 | Ca 7.8–11.0 HCO3 24.6 K 3.3–4.8 Na 132–144 Phos 2.3–3.6 Uric acid 1.0–1.5 Alk phos (20–30): 150–255 PTH 30.0–40.5 |
Ca/creat 0.34–2.9 Glucosuria >1,000 mg/dL General aminoaciduria Microalbumin (24 years) 60 |
Mild osteopenia | No | 1 | ||
| 7 | Ca 9.87–11.0 HCO3 17.0–22.0 K 3.2–4.7 Na 134–143 Phos 4.3–5.4 Uric acid 1.5–1.8 Alk phos (3–5): 397–4,100 (8–10): 368–440 (11–13): 437–747 PTH 15.3–67.3 |
Ca/creat 0.22–0.23 Glucosuria >1,000 mg/dL General aminoaciduria Microalbumin (11 years) 293 |
HCO3 0.5 meq Phosphor 85 mg K 1.7 meq |
Rickets | No | 2 | |
| 8 | Ca 6.8–10.6 HCO3 11.0–21.4 K 2.6–4.9 Na 132–139 Phos 1.4–5.5 Uric acid 0.5–1.4 Alk phos (8–10): 327–1,210 (11–13): 380–740 PTH 68.7–425.0 |
Ca/creat 0.16–1.0 Glucosuria >1,000 mg/dL General aminoaciduria Microalbumin (9 years) 1,207 |
HCO3 3.2 meq Phos 180 mg K 2.8 meq/kg |
Bone pains Bilat. genu varum |
Rickets Osteoporosis |
Yes (9 years) | 3 |
Note: Normal biochemical values in blood: Ca 9.0–11.0 mg/dL, HCO3 22–26 mMol/L, K 3.5–5.0 mEq/L, Na 135–145 mEq/L
Phosphorus values by age in years: 1–3: 3.8–6.5 mg/dL, 4–11: 3.7–5.6 mg/dL, 12–15: 2.9–5.5 mg/dL, 16–19: 2.7–4.7 mg/dL, Uric acid 2.0–7.0 mg/dL, Hypoglycemia <50 mg/dL, hyperglycemia >170 mg/dL
PTH (parathyroid hormone) 15.0–65 ng/L
For alkaline phosphatase, because norms change with growth, measurements are presented by age (indicated in parentheses). Normal values: 3-5/8–10 years 37–448 U/L, 11–13 years 74–390 U/L, 20–30 years 37–270 U/L
Normal biochemical values in urine: microalbuminuria (age at diagnosis in years) 0–17 mcg/mgCr, calcium/creatinine ratios by age: 0–6 months, <0.8, 7–12 months, <0.6, >2 years <0.21
Disease severity scores as assessed subjectively by the parents of the minors and the adult patient on a scale from 1 (mild) to 3 (severe)
Fig. 2.
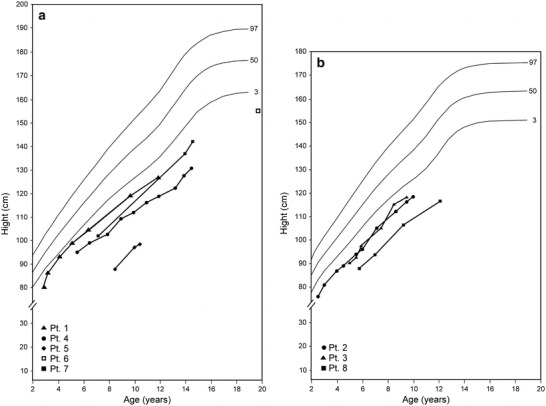
Patient height curves. (a) Males; (b) Females
Clinical and/or radiological bone pathologies (rickets, osteopenia, osteoporosis) occurred in all patients during follow-up. Patient 8 had a severe bilateral genu varum deformity associated with incapacitating pain that warranted intravenous analgesics and surgical orthopedic correction. Four patients (nos. 2, 4, 5, and 8) acquired nephrocalcinosis at ages 4.5–9 years. Microalbuminuria developed in all patients within 3–26 years following diagnosis.
Discussion
The present study describes the early and long-term characteristics of eight patients with FBS from a single Bedouin sibship, all of whom were homozygous for the p.R301X GLUT2 mutation. The findings highlight the typical clinical features at presentation and point to a phenotypic variability over time despite the presence of the identical mutation.
FTT and/or hepatomegaly, prominent fluctuations in blood glucose levels, and proximal tubulopathy were found at presentation or during diagnosis or both in all our patients, similar to other series of FBS (Santer et al. 1998, 2002b). Although there were only mild differences in age at diagnosis, skeletal involvement ranged from none to radiological and/or clinical signs of rickets and osteopenia.
Evaluation of the follow-up findings revealed a spectrum of disease severity as reflected by growth parameters, frequency of follow-up visits, hospitalizations for disease exacerbations, mean amount of electrolyte replacement therapy, and skeletal and renal complications (Fig. 2a, b, Tables 2 and 3). Of note, the variability in disease severity correlated with the subjective disease-severity scores assessment of the parents and the adult patient.
A broad phenotypic expression in FBS has been reported previously, with specific mutations causing atypical clinical manifestations such as intestinal malabsorption without hepatomegaly (Aperia et al. 1981) and glomerular hyperfiltration (Berry et al. 1995). Others described an unusually mild disease course with normal growth and absence of hepatomegaly, renomegaly, or rickets (Grünert et al. 2012).
Few patients with FBS due to homozygosity for the p.R301X mutation have been described. Santer et al. (1998) followed the first patient reported with what is now recognized as typical features of FBS. Like our patient 6, he had not been receiving continuous medications since infancy, and like our patients 1, 3, and 6 he had neither rickets nor nephrocalcinosis on follow-up. Riva et al. (2004) described a neonate who presented with fever, vomiting, mild hepatomegaly, and proximal tubulopathy who was managed with galactose-free diet, frequent small meals, and phosphate and vitamin D supplements. At 1 year, growth was normal, and there were no clinical abnormalities. Of our three patients who presented in the neonatal period, one (patient 5) had also recurrent vomiting and hepatomegaly at presentation but he had a severe disease course. The other two presented with prolonged diarrhea with symptomatic hypoglycemia and rickets (patient 4) and hepatomegaly with hypotonia (patient 6). The former had a severe disease course while patient 6 had a mild disease. Another reported patient with the p.R301X mutation presented with hypoglycemic seizures, FTT, hepatomegaly, tubulopathy, and rickets, in addition to facial dysmorphism, global developmental delay, hyperinsulinism, and refractory hypoglycemia (Hoffman et al. 2007). Interestingly, he inherited the disease via maternal isodisomy of chromosome 3. Patient 4 had recurrent symptomatic hypoglycemic episodes but they were not refractory and they resolved in late infancy. In contrast to the patient of Hoffman et al. (2007) none of our patients had facial dysmorphism and none exhibited severe developmental delay.
It is noteworthy that although earlier studies found that disease severity is somewhat ameliorated during the second decade of life (Santer et al. 2002b), we noted no remarkable change in our patients during follow-up from infancy to age 10–14 years.
The bone complications, mainly rickets, in five patients (nos 2, 4, 5, 7, 8) and the development of nephrocalcinosis in most of them may have been due to difficulty in balancing the high tubular phosphorus loss, as reflected by their need for high doses of phosphorus replacement therapy and frequent follow-up visits to monitor electrolyte levels. This led to secondary hyperparathyroidism in most cases as well as to renal calcium–phosphate deposits, with consequent bone mineralization abnormalities and nephrocalcinosis, respectively.
Glomerulopathy is not a common complication of FBS (Santer et al. 1998). It was previously reported in one patient with microalbuminuria, glomerular hyperfiltration, and glomerular mesangial expansion (Berry et al. 2005), and in four patients with reduced glomerular filtration (Manz et al. 1987). It is unclear if microalbuminuria is a manifestation of a glomerular insult. It is possible that in our patients, the microalbuminuria was attributable to excessive fluid intake for chronic dehydration secondary to the osmotic diuresis induced by impaired glucose reabsorption (Clark et al. 2008; Viberti et al. 1982).
Patient compliance with electrolyte replacement therapy and corn starch supplementation is crucial to avoid complications of FBS. Good parental care during follow-up was found for all our patients with severe disease, as reflected by their strict attendance to scheduled visits, frequent telephone consults, and unscheduled visits for minor health problems. At the same time, our sole adult patient (no. 6), who frequently failed to appear for follow-up, had a mild FBS phenotype without significant disease complications or need for replacement therapy. This patient is one of the few reported adults with FBS (Santer et al. 1997; Pena and Charrow 2011), and the third reported fertile male (Von Schnakenburg and Santer 2011).
We speculate that the broad spectrum of disease severity among our patients might be explained by modifier genes, as suggested for other monogenic diseases (Weatherall 2001), or unknown epigenetic factors (Jansen et al. 2010). At the same time, despite the similar lifestyle and place of residence, the disease course in the individual patients could have been modified by external causes, such as insufficient corn starch management or undetermined environmental factors.
In conclusion, the present follow-up study points to the variability in the clinical spectrum of FBS among patients homozygous for the p.R301X mutation. The initial clinical manifestations are not predictive of the long-term severity of the disease.
Take-Home Message
There is a broad spectrum of clinical severity of Fanconi–Bickel syndrome in patients harboring a homozygous p.R301X mutation.
Author Contributions
Elena Fridman, MD, MSc: Guarantor, data analysis and interpretation, drafting of the manuscript.
Avraham Zeharia, MD: Conception and design of the study, intellectual input, revision of the manuscript.
Tal Markus-Eidlitz, MD: Revision of the content.
Yishai Haimi Cohen, MD: Design, analysis, and revision of the manuscript.
All coauthors have seen the final version of the manuscript, confirm that the work has not been published/submitted elsewhere, and agree with the submission.
Details of funding
No funding was received for the study.
Conflict of Interest
Elena Fridman, Avraham Zeharia, Tal Markus-Eidlitz, and Yishai Haimi Cohen declare that they have no conflict of interest.
Ethics
This retrospective analysis of prospectively collected data was approved by the Rabin Medical Center Research Review Board. Any identifying information about patients was omitted after initial data collection; hence, the need for individual-patient informed consent was waived.
Footnotes
Competing interests: None declared
Contributor Information
Elena Fridman, Email: slomovel@gmail.com.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Aperia A, Bergqvist G, Linne T, Zetterstorm R. Familial Fanconi syndrome with malabsorption and galactose intolerance normal kinase and transferase activity: a report on two siblings. Acta Paediatr Scand. 1981;70:527–533. doi: 10.1111/j.1651-2227.1981.tb05735.x. [DOI] [PubMed] [Google Scholar]
- Berry GT, Baker L, Kaplan FS, Witzeleben CL. Diabetes-like renal glomerular disease in Fanconi-Bickel syndrome. Pediatr Nephrol. 1995;9:287–291. doi: 10.1007/BF02254185. [DOI] [PubMed] [Google Scholar]
- Berry GT, Baynes JW, Wells-Knecht KJ, Szwergold BS, Santer R. Elements of diabetic nephropathy in a patient with GLUT 2 deficiency. Mol Genet Metab. 2005;86:473–477. doi: 10.1016/j.ymgme.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Clark WF, Kortas C, Suri RS et al (2008) Excessive fluid intake as a novel cause of proteinuria CMAJ 178(2):173–175 [DOI] [PMC free article] [PubMed]
- de Onis M, Blössner M. The World Health Organization global database on child growth and malnutrition: methodology and applications. Int J Epidemiol. 2003;32(4):518–526. doi: 10.1093/ije/dyg099. [DOI] [PubMed] [Google Scholar]
- Grünert SC, Schwab KO, Pohl M, Sass JO, Santer R. Fanconi-Bickel syndrome: GLUT2 mutations associated with a mild phenotype. Mol Genet Metab. 2012;105:433–437. doi: 10.1016/j.ymgme.2011.11.200. [DOI] [PubMed] [Google Scholar]
- Hoffman TL, Blanco E, Lane A, et al. Glucose metabolism and insulin secretion in a patient with ABCC8 mutation and Fanconi–Bickel syndrome caused by maternal isodisomy of chromosome 3. Clin Genet. 2007;71:551–557. doi: 10.1111/j.1399-0004.2007.00802.x. [DOI] [PubMed] [Google Scholar]
- Jansen A, Gemayel R, Verstrepen KJ (2010) Unstable microsatellite repeats facilitate rapid evolution of coding and regulatory sequences. Genome Dyn 7:108–125. [Epub 2012 Jun 25] Review [DOI] [PubMed]
- Lee PJ, Van't Hoff WG, Leonard JV. Catch-up growth in Fanconi-Bickel syndrome with uncooked cornstarch. J Inherit Metab Dis. 1995;18(2):153–156. doi: 10.1007/BF00711753. [DOI] [PubMed] [Google Scholar]
- Manz F, Bickel H, Brodehl J, et al. Fanconi-Bickel syndrome. Pediatr Nephrol. 1987;1:509–518. doi: 10.1007/BF00849262. [DOI] [PubMed] [Google Scholar]
- Odièvre MH, Lombes A, Dessemme P, et al. A secondary respiratory chain defect in a patient with Fanconi Bickel syndrome. J Inherit Metab Dis. 2002;25:379–384. doi: 10.1023/A:1020147716990. [DOI] [PubMed] [Google Scholar]
- Pena L, Charrow J. Fanconi-Bickel syndrome: report of life history and successful pregnancy in an affected patient. Am J Med Genet. 2011;55A:415–417. doi: 10.1002/ajmg.a.33822. [DOI] [PubMed] [Google Scholar]
- Riva S, Ghisalberti C, Parini R, Furlan F, Bettinelli A, Somaschini M. The Fanconi-Bickel syndrome: a case of neonatal onset. J Perinatol. 2004;24:322–323. doi: 10.1038/sj.jp.7211092. [DOI] [PubMed] [Google Scholar]
- Saltik-Temizel IN, Coşkun T, Yüce A, Koçak N. Fanconi-Bickel syndrome in three Turkish patients with different homozygous mutations. Turk J Pediatr. 2005;47(2):167–169. [PubMed] [Google Scholar]
- Santer R, Schneppenheim R, Dombrowski A, Götze H, Steinmann B, Schaub J (1997) Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome. Nature Genet 17:324–326 [Erratum in Nat Genet 1998;18:298] [DOI] [PubMed]
- Santer R, Schneppenheim R, Suter D, Schaub J, Steinmann B. Fanconi-Bickel syndrome – the original patient and his natural history, historical steps leading to the primary defect, and a review of the literature. Eur J Pediatr. 1998;157:783–797. doi: 10.1007/s004310050937. [DOI] [PubMed] [Google Scholar]
- Santer R, Steinmann B, Schaub J. Fanconi-Bickel syndrome–a congenital defect of facilitative glucose transport. Curr Mol Med. 2002;2:213–227. doi: 10.2174/1566524024605743. [DOI] [PubMed] [Google Scholar]
- Santer R, Groth S, Kinner M. The mutation spectrum of the facilitative glucose transporter gene SLC2A2 (GLUT2) in patients with Fanconi–Bickel syndrome. Hum Genet. 2002;110:21–29. doi: 10.1007/s00439-001-0638-6. [DOI] [PubMed] [Google Scholar]
- Viberti GC, Mogensen CE, Keen H, Jacobsen FK, Jarrett RJ, Christensen CK. Urinary excretion of albumin in normal man: the effect of water loading. Scand J Clini Lab Invest. 1982;2:147–157. doi: 10.3109/00365518209168065. [DOI] [PubMed] [Google Scholar]
- Von Schnakenburg C, Santer R. Fanconi- Bickel syndrome and fertility. Am J Med Genet Part A. 2011;155:2607. doi: 10.1002/ajmg.a.34202. [DOI] [PubMed] [Google Scholar]
- Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001;2:245–255. doi: 10.1038/35066048. [DOI] [PubMed] [Google Scholar]