

Abstract
Alternating hemiplegia of childhood (AHC) is a rare disorder caused by heterozygous mutations in ATP1A3. AHC is associated with early-onset plegic and tonic/dystonic attacks and permanent neurologic deficits. Attacks tend to persist through life. Flunarizine therapy occasionally reduces the severity, duration and frequency of attacks. A ketogenic diet/modified Atkins diet (KD/MAD) can attenuate paroxysmal movement disorders associated with GLUT1 deficiency syndrome (GLUT1DS), but there are no reports on the effect of KD/MAD in AHC. We describe the case of a young girl with AHC who had tonic/dystonic and plegic attacks, mostly triggered by exercise, together with mild permanent dystonia and mental retardation. Her family had a history of dominant (three affected generations) paroxysmal exercise-induced dystonia. A history of plegic attacks that ceased after childhood was retraced from the medical records of the three affected adults, leading to the diagnosis of familial AHC due to ATP1A3 p.Asp923Asn mutation (Roubergue et al 2013). KD/MAD was considered for the proband when she was 3½ years old, following initial misdiagnosis of GLUT1DS. MAD, a KD variant, was chosen because it is easier to manage than KD and is similarly effective to KD in most GLUT1DS patients. MAD resulted in complete disappearance of the attacks during 15 months of follow-up. Conclusions: A modified Atkins diet had a sustained beneficial effect on attacks associated with AHC. Although preliminary, this observation suggests that a ketogenic diet might be a therapeutic option for paroxysmal disorders in some patients with alternating hemiplegia of childhood.
Introduction
Alternating hemiplegia of childhood (AHC) (OMIM #614820) is a rare disorder characterized by episodes of alternating hemiplegia/quadriplegia and tonic/dystonic attacks associated with permanent neurological deficits (Sweney et al 2009; Panagiotakaki et al 2010). Paroxysmal events typically start before age 18 months and are often precipitated by specific triggers such as physical exertion (Panagiotakaki et al 2010; Sweney et al 2009). Most cases are sporadic. The main genetic cause of AHC was recently identified as heterozygous mutations in ATP1A3, the gene encoding the neuron-specific Na+/K+-ATPase α3 subunit (Heinzen et al 2012; Rosewich et al 2012). Mutations in this gene had previously been shown to cause rapid-onset dystonia parkinsonism (RDP) (de Carvalho Aguiar et al 2004).
Current treatments for AHC are disappointing. Flunarizine, a calcium channel blocker, reduces the frequency, duration and severity of attacks in some patients (Panagiotakaki et al 2010; Mikati et al 2000).
The ketogenic diet (KD), or its variant the modified Atkins diet (MAD), is used to treat some metabolic paroxysmal movement disorders such as those encountered in glucose transporter type 1 deficiency syndrome (GLUT1DS) (De Vivo et al 1991; Ito et al 2011). The effect of such diets on the paroxysmal disorders associated with AHC is not known. Here we tested MAD in a child with AHC, in whom paroxysmal attacks were the predominant disease manifestation.
Patient and Methods
The index case and her family have been described briefly elsewhere (Roubergue et al 2013). Their history and neurological and genetic findings are summarized in Fig. 1. The treatment response of their paroxysmal features is reported in Table 1.
Fig. 1.
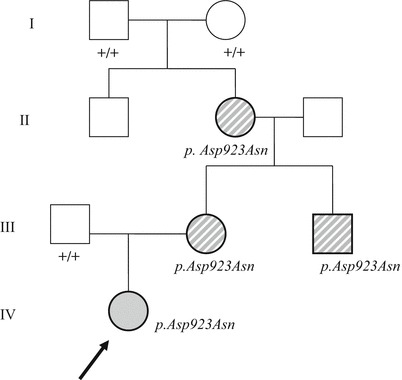
Family pedigree. Uniform grey shading: individual with plegic and tonic/dystonic episodes. Diagonal shading: adults with isolated dystonic episodes (mainly exercise-induced), and no plegic episodes after childhood. Shapes surrounded by a thick line: individuals with permanent mild dystonia/chorea and mild cognitive deficits. +/+: individuals with a normal genotype. p.Asp923Asn: ATP1A3 heterozygous mutation. The arrow indicates the index patient
Table 1.
Treatment responses of attacks in the proband and other affected family members
| Age | Age at onset of Pl/Dyst attacks | Duration of Pl/Dyst attacks | Monthly frequency of Pl/Dyst attacks | Triggers (alleviating factors) of Pl/Dyst attacks | Current therapy, age at outset [previous therapy, age at outset] | Attack-free periods: age of onset, duration | Age at end of Pl /Dyst attacks | |
|---|---|---|---|---|---|---|---|---|
| Proband | 4 years 8 months | 2 years/ 8 months |
Mins to 15 days/mins to 2 days | 2 to 4/2 | Exercise, stress (sleep)/ exercise +++, fever |
MAD, 3 years 5 months | 3 years 5 months/ 3 years 5 months |
|
| Mother | 27 years | 2 years/ 2 months |
NA/ mins to hours |
NA/4 | NA/ exercise +++ (rest, sleep) |
PHT, 8 years [CZP, 13 months]; [FNR, 7 years] |
Age 8 years, 4 months Age 7 years, 6 months |
5 ½ years/ ongoing |
| Maternal uncle | 25 years | 2 years/8 years | NA/ mins to hours |
30 then 4/10 to 20 | Exercise/ exercise +++ |
FNR, 2 years 9 months; PHT, 6 years |
5 years/ ongoing |
|
| Maternal grandmother | 52 years | 3 years/ NA |
5 min to 1 hour/ 30 min to hours |
NA/4 | NA/exercise +++ (rest) |
PB, < 30 years; PHT, 34 years [CZP, < 30 years]; [FNR, 32 years]; [other AEs] |
NA/ ongoing |
Abbreviations: Pl plegic, Dyst dystonic, MAD Modified Atkins Diet, AE antiepileptic drug, CZP carbamazepine, FNR flunarizine, PB Phenobarbital, PHT phenytoin, NA not available, +++ usual trigger
The proband, now 4 years 8 months old, was referred to us at age 2½ years for plegic attacks. The attacks had begun at age 2 years with transient upper-limb paresis. Episodes of monoparesis/plegia, hemiplegia and quadriplegia subsequently occurred 2–4 times monthly, lasting from several minutes to 15 days. They were triggered by prolonged crying, nervousness or physical exertion. They disappeared during night-time sleep and, when long lasting, recurred within half an hour after awakening.
Previously, the child had had episodes of paroxysmal dystonia, mostly induced by prolonged physical exertion. At age 8 months, she had wrist dystonia induced by walking on all fours and, from age 20 months, had orofacial-laryngeal dystonia induced mainly by playing with toys. She had an attack of generalized dystonia at age 11 months. The attacks lasted from 30–60 min to 2–3 days and occurred about twice a month.
The family had a history of dominant exercise-induced paroxysmal dystonia (Fig. 1, Table 1). Plegic attacks during childhood, of which the affected adult members of the family were unaware, were retraced from the medical records.
Glycorrhachia was 2.65 mmol/l in the proband. Genetic analysis of DYT1, GLUT1/SLC2A1, PDHE1 alpha, ATP1A2 and CACNA1A, as well as karyotyping and the Illumina CGH array were negative in the proband or in one or several symptomatic family members. Analysis of ATP1A3 revealed the heterozygous substitution c.2767G>A (p.Asp923Asn) in all four symptomatic family members (Roubergue et al 2013). The mutation had appeared de novo in the maternal grandmother and then segregated over three generations (Fig. 1).
Procedure
The modified Atkins diet, a KD variant, was considered before the final diagnosis of AHC was made, as the patient’s clinical phenotype resembled that of mild GLUT1DS, in which KD/MAD was known to be effective.
MAD was introduced at age 3 years 5 months, using the protocol recommended by Kossoff and Dorward (2008), i.e. starting without a fasting period, with no calorie, fluid or protein restriction, and with an initial upper limit of 10 g of carbohydrates per day. Fat intake was encouraged. MAD was initiated with 1,250 cal (88 cal/kg) daily, provided by 93 g of fat, 94 g of protein and 10 g of carbohydrates. The ketogenic ratio (ratio of g of fat to g of protein plus g of carbohydrate) was 0.9:1, compared to 4:1 in the classic KD. Carbohydrate intake was increased to 20 g after 4 months, and then to 30 g daily after a further 6 months. The duration of the diet was 15 months at the last follow-up.
Results
After MAD initiation, the dystonic and plegic attacks disappeared completely during the 15 months of follow-up (Table1). This prompted the family to continue the diet even after the child started school. Interictal neurological status was unchanged, with persistent mild permanent dystonia.
The diet was well tolerated. The child lost 1.5 kg in the early phase but regained it after her caloric intake was increased. Urine ketosis varied from 1+ to 4+. Mild hypercholesterolemia, present before MAD initiation (5.1 mmol/l), increased early after MAD initiation (6.2 mmol/l) and had returned close to the pretreatment level when controlled at 5 months (5.4 mmol/l; N = 1.8–4.6 mmol/l). Five months after MAD initiation, she was found to have elevated calciuria (urine calcium/creatinine ratio 2.91, N = 0.22–0.50) and low carnitinemia (free carnitine 13.7 μmol/l, N = 24–63; total L carnitine 23.7 μmol/l, N = 35–84; free to total carnitine ratio 0.58, N = 0.70–0.90).
Discussion
We describe the case of a child with a mild form of typical AHC. Initial misdiagnosis of GLUT1DS deficiency led to treatment with a modified Atkins diet, resulting in complete cessation of her dystonic and plegic attacks. This beneficial effect was sustained for more than 15 months. Although based on a single observation, our findings suggest that KD might be a therapeutic option in AHC.
Attacks associated with AHC usually persist throughout life (Bourgeois et al 1993; Panagiotakaki et al 2010; Sweney et al 2009). At best, current drug therapies reduce the severity, duration and frequency of attacks. Flunarizine is the most consistently effective drug, as shown in large cohorts of patients (Casaer 1987; Sweney et al 2009; Panagiotakaki et al 2010).
The cessation of both types of attack in our patient supports the role of the modified Atkins diet in the observed improvement. We found only three other reports of AHC patients whose attacks ceased completely after treatment, which always consisted of flunarizine; they included the first patient to be treated with flunarizine, whose publication ultimately led to current use of this drug to prevent attacks in AHC patients (Casaer and Azou 1984; Silver and Andermann 1993; Mikati et al 2000). Follow-up was reported in only one of these three cases and, as in our patient, lasted more than 1 year.
We cannot rule out the possibility that the link between MAD initiation and the cessation of attacks might have been fortuitous in our patient. In particular, the childhood plegic attacks experienced by her family’s three affected adults gradually ceased, leaving only isolated tonic/dystonic attacks. Isolated cessation of either plegic or tonic/dystonic attacks has previously been reported in a small number of AHC patients (Bourgeois et al 1993; Mikati et al 2000; Panagiotakaki et al 2010). In addition, lengthy attack-free periods (up to 2 years) have been reported in very few patients (Bourgeois et al 1993; Salmon and Wilson 1984). Such periods were experienced by our patient’s mother after treatment with flunarizine and phenytoin (6 and 4 months, respectively). Yet these attack-free periods were far shorter than that experienced by our patient after MAD initiation (currently 15 months).
We considered the ketogenic diet following initial misdiagnosis of GLUT1DS with dominant paroxysmal exercise-induced dystonia (PEID) and plegic attacks. PEID occurs in some AHC individuals (Sweney et al 2009), but autosomal dominant PEID is not a known phenotype of AHC. By contrast, autosomal dominant PEID is a core feature of GLUT1DS type 2 (OMIM # 612126), the mild form of GLUT1DS, and may be associated with episodes of hemiplegia, quadriplegia, mild mental retardation and permanent neurological symptoms (Suls et al 2008; Weber et al 2008; Rotstein et al 2009; Pons et al 2010). The long duration (up to several days) of some attacks in our patient was not typical of paroxysmal episodes in GLUT1DS (Suls et al 2008; Weber et al 2008). However, the absence of frank hypoglycorrhachia in our patient, and the apparent absence of SLCA2/GLUT1 mutation, did not rule out GLUT1DS. Indeed, glycorrhachia in the lower range of normal has been reported in GLUT1DS, and only 70–80% of patients harbour a mutation in the SLC2A/GLUT1 gene (Klepper 2012, 2013).
KD is the treatment of choice for patients with GLUT1DS (De Vivo et al 1991; Suls et al 2008; Klepper 2012). KD may also be considered for SLCA2/GLUT1-negative patients, even in the absence of frank hypoglycorrhachia (as in our proband), and an immediate response to the diet would support the diagnosis of GLUT1DS (Klepper 2012, 2013). We opted for a modified Atkins diet, a KD variant used in GLUT1DS (Ito et al 2011; Leen et al 2013), because it is easier to manage (the proband’s mother was mentally impaired), and more palatable than KD.
The mechanism underlying the therapeutic effect of KD is unknown. Ketones provide an alternative brain fuel that may compensate for defective cerebral glucose metabolism (De Vivo et al 1991) as observed in GLUT1DS (Pascual et al 2007; Ito et al 2011). KD might act similarly in AHC. Indeed, cerebral glucose hypometabolism is observed in AHC patients, as well as in AHC model mice and, interestingly, in the only rapid-onset-dystonia-parkinsonism patient studied by means of F-18 fluorodeoxyglucose positron emission tomography, who harboured precisely the same ATP1A3 mutation (p.Asp923Asn) as the family described here (Sasaki et al 2009; Kirshenbaum et al 2013; Anselm et al 2009). KD might also reduce neuronal excitability (for reviews see Lutas and Yellen 2013; Stafstrom and Rho 2012; Dhamija et al 2013), thereby correcting the altered membrane excitability due to ATP1A3 dysfunction and thus reducing the frequency and/or severity of the paroxysmal features of AHC.
Synopsis
This is the first report on the effect of a ketogenic diet in a patient with alternating hemiplegia of childhood.
Compliance with Ethics and Guidelines
Conflict of Interest
Anne Roubergue, Bertrand Philibert, Agnès Gautier, Alice Kuster, Karine Markowicz, Thierry Billette de Villemeur, Sandrine Vuillaumier-Barrot, and Diane Doummar declare that they have no conflict of interest.
Sophie Nicole has received research grants from the French association against alternating hemiplegia (AFHA).
Emmanuel Roze declares he is the recipient of a grant “poste d’accueil” AP-HP/CNRS. He received research support from INSERM (COSSEC), AP-HP (DRC-PHRC), Fondation pour la Recherche sur le Cerveau (FRC), the Dystonia Coalition (Pilot project), Ipsen, and Merz-Pharma, Novartis, Teva, Lundbeck, Orkyn; served on scientific advisory boards for Orkyn, Ipsen, and Merz-pharma; received speech honorarium from Novartis and Orkyn; received travel funding from Teva, Novartis, the Dystonia Coalition, the Movement Disorders Society, the World Federation of Neurology Association of Parkinsonism and Related Disorders, International Federation of Clinical Neurophysiology
Informed Consent
No ethical approval was required for this work.
The child’s parents have given their consent for the publication of this case study.
Animal Rights
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Details of the Contributions of Individual Authors
Planning: AR, BP, AG, AK, KM, TBV, SV, SN, ER, DD
Conduct: AR, TBV, DD
Reporting of the work described in the article: AR, BP, AG, AK, KM, DD
Drafting/revising: AR, ER, DD
Footnotes
Competing interests: None declared
Contributor Information
Anne Roubergue, Email: an_schlum@yahoo.fr.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Anselm IA, Sweadner KJ, Gollamudi S, Ozelius LJ, Darras BT. Rapid-onset dystonia-parkinsonism in a child with a novel atp1a3 gene mutation. Neurology. 2009;73:400–401. doi: 10.1212/WNL.0b013e3181b04acd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois M, Aicardi J, Goutières F. Alternating hemiplegia of childhood. J Pediatr. 1993;122:673–679. doi: 10.1016/S0022-3476(06)80003-X. [DOI] [PubMed] [Google Scholar]
- Casaer P, Azou M. Flunarizine in alternating hemiplegia in childhood. Lancet. 1984;2:579. doi: 10.1016/S0140-6736(84)90794-3. [DOI] [PubMed] [Google Scholar]
- Casaer P. Flunarizine in alternating hemiplegia in childhood. An international study in 12 children. Neuropediatrics. 1987;18:191–195. doi: 10.1055/s-2008-1052478. [DOI] [PubMed] [Google Scholar]
- de Carvalho Aguiar P, Sweadner KJ, Penniston JT, et al. Mutations in the Na+/K+-ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron. 2004;43:169–175. doi: 10.1016/j.neuron.2004.06.028. [DOI] [PubMed] [Google Scholar]
- De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood–brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med. 1991;325:703–709. doi: 10.1056/NEJM199109053251006. [DOI] [PubMed] [Google Scholar]
- Dhamija R, Eckert S, Wirrell E. Ketogenic diet. Can J Neurol Sci. 2013;40:158–167. doi: 10.1017/S0317167100013676. [DOI] [PubMed] [Google Scholar]
- Heinzen EL, Swoboda KJ, Hitomi Y, et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat Genet. 2012;44:1030–1034. doi: 10.1038/ng.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Oguni H, Ito S, Oguni M, Osawa M. A modified Atkins diet is promising as a treatment for glucose transporter type 1 deficiency syndrome. Dev Med Child Neurol. 2011;53:658–663. doi: 10.1111/j.1469-8749.2011.03961.x. [DOI] [PubMed] [Google Scholar]
- Kirshenbaum GS, Dawson N, Mullins JG et al (2013) Alternating hemiplegia of childhood-related neural and behavioural phenotypes in Na+, K+-ATPase α3 missense mutant mice. PLoS One 3:e60141 [DOI] [PMC free article] [PubMed]
- Klepper J. GLUT1 deficiency syndrome in clinical practice. Epilepsy Res. 2012;100:272–277. doi: 10.1016/j.eplepsyres.2011.02.007. [DOI] [PubMed] [Google Scholar]
- Klepper J. Absence of SLC2A1 mutations does not exclude glut1 deficiency syndrome. Neuropediatrics. 2013;44:235–236. doi: 10.1055/s-0033-1336015. [DOI] [PubMed] [Google Scholar]
- Kossoff EH, Dorward JL. The modified Atkins diet. Epilepsia. 2008;49(Suppl 8):37–41. doi: 10.1111/j.1528-1167.2008.01831.x. [DOI] [PubMed] [Google Scholar]
- Leen WG, Mewasingh L, Verbeek MM, Kamsteeg EJ, van de Warrenburg BP, Willemsen MA (2013) Movement disorders in GLUT1 deficiency syndrome respond to the modified Atkins diet. Mov Disord 10:1439–1442 [DOI] [PubMed]
- Lutas A, Yellen G. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013;36:32–40. doi: 10.1016/j.tins.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikati MA, Kramer U, Zupanc ML, Shanahan RJ. Alternating hemiplegia of childhood: clinical manifestations and long-term outcome. Pediatr Neurol. 2000;23:134–141. doi: 10.1016/S0887-8994(00)00157-0. [DOI] [PubMed] [Google Scholar]
- Panagiotakaki E, Gobbi G, Neville B, et al. Evidence of a non-progressive course of alternating hemiplegia of childhood: study of a large cohort of children and adults. Brain. 2010;133:3598–3610. doi: 10.1093/brain/awq295. [DOI] [PubMed] [Google Scholar]
- Pascual JM, Wang D, Hinton V, et al. Brain glucose supply and the syndrome of infantile neuroglycopenia. Arch Neurol. 2007;64:507–513. doi: 10.1001/archneur.64.4.noc60165. [DOI] [PubMed] [Google Scholar]
- Pons R, Collins A, Rotstein M, Engelstad K, De Vivo DC. The spectrum of movement disorders in Glut-1 deficiency. Mov Disord. 2010;25:275–281. doi: 10.1002/mds.22808. [DOI] [PubMed] [Google Scholar]
- Rosewich H, Thiele H, Ohlenbusch A, et al. Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: a whole-exome sequencing gene- identification study. Lancet Neurol. 2012;11:764–773. doi: 10.1016/S1474-4422(12)70182-5. [DOI] [PubMed] [Google Scholar]
- Rotstein M, Doran J, Yang H, Ullner PM, Engelstad K, De Vivo DC. GLUT1 deficiency and alternating hemiplegia of childhood. Neurology. 2009;73:2042–2044. doi: 10.1212/WNL.0b013e3181c55ebf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roubergue A, Roze E, Vuillaumier-Barrot S, et al. The multiple faces of the ATP1A3-related dystonic movement disorder. Mov Dis. 2013;28:1457–1459. doi: 10.1002/mds.25396. [DOI] [PubMed] [Google Scholar]
- Salmon MA, Wilson J. Drug for alternating migraine. Lancet (letter) 1984;2:980. doi: 10.1016/S0140-6736(84)91191-7. [DOI] [Google Scholar]
- Sasaki M, Sakuma H, Fukushima A, Yamada K, Ohnishi T, Matsuda H. Abnormal cerebral glucose metabolism in alternating hemiplegia of childhood. Brain Dev. 2009;31:20–26. doi: 10.1016/j.braindev.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Silver K, Andermann F. Alternating hemiplegia of childhood: a study of 10 patients and results of flunarizine treatment. Neurology. 1993;43:36–41. doi: 10.1212/WNL.43.1_Part_1.36. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Rho JM (2012) The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front Pharmacol 3:59. doi: 10.3389/fphar.2012.00059. eCollection 2012 [DOI] [PMC free article] [PubMed]
- Suls A, Dedeken P, Goffin K, et al. Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain. 2008;131:1831–1844. doi: 10.1093/brain/awn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweney MT, Silver K, Gerard-Blanluet M, et al. Alternating hemiplegia of childhood: early characteristics and evolution of a neurodevelopmental syndrome. Pediatrics. 2009;123:e534–e541. doi: 10.1542/peds.2008-2027. [DOI] [PubMed] [Google Scholar]
- Weber YG, Storch A, Wuttke TV, et al. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest. 2008;118:2157–2168. doi: 10.1172/JCI34438. [DOI] [PMC free article] [PubMed] [Google Scholar]