

Abstract
ATP synthase or complex V (cV) of the oxidative phosphorylation system is responsible for the production of ATP, dissipating the electrochemical gradient generated by the mitochondrial respiratory chain. In addition to maternally transmitted cV dysfunction caused by mutations in mtDNA genes (MT-ATP6 or MT-ATP8), encoding cV subunits, recessive mutations in the nuclear TMEM70 are the most frequent cause of ATP synthase deficiency.
We report on a cohort of ten Italian patients presenting with neonatal lactic acidosis, respiratory distress, hypotonia, cardiomyopathy and psychomotor delay and harbouring mutations in TMEM70, including the common splice mutation and four novel variants. TMEM70 protein was virtually absent in all tested TMEM70 patients’ specimens.
The exact function of TMEM70 is not known, but it is considered to impact on cV assembly since TMEM70 mutations have been associated with isolated cV activity reduction. We detected a clear cV biochemical defect in TMEM70 patients’ fibroblasts, whereas the assay was not reliable in frozen muscle. Nevertheless, the evaluation of the amount of holocomplexes in patients with TMEM70 mutations showed a nearly absent cV in muscles and a strong decrease of cV with accumulation of sub-assembly species in fibroblasts. In our cohort we found not only cV deficiencies but also impairment of other OXPHOS complexes. By ultrastructural analysis of muscle tissue from one patient with isolated cV deficiency, we found a severely impaired mitochondrial morphology with loss of the cristae. These findings indicate that cV impairment could indirectly alter other respiratory chain complex activities by disrupting the mitochondrial cristae structure.
Introduction
The mitochondrial oxidative phosphorylation (OXPHOS) system consists of five multi-subunit complexes, acting in concert to generate energy in the form of ATP molecules. Genetic OXPHOS disorders result from mutations in either mitochondrial DNA (mtDNA) or nuclear genes encoding structural subunits of the OXPHOS complexes or factors involved in their synthesis, assembly and function. Complex V or mitochondrial ATP synthase consists of 16 different polypeptides, two of which, ATPase 6 and ATPase 8, being encoded by mtDNA (Holt et al. 1990; Rahman et al. 1996). Most isolated cases of ATP synthase deficiency are caused by mutations in the mitochondrial genes MT-ATP6 (MIM516060) and MT-ATP8 (MIM516070) and are associated with different clinical phenotypes, including maternally inherited Leigh syndrome (MILS) (Rahman et al. 1996), adult-onset NARP (neuropathy, ataxia and retinitis pigmentosa), Leber hereditary optic neuropathy and hypertrophic cardiomyopathy. Disease-causing mutations in patients with isolated cV deficiency have been identified in only four nuclear genes, two encoding assembly factors: ATPAF2 and TMEM70, and two structural subunits—ATP5E encoding the epsilon subunit of the F1 domain (Mayr et al. 2010) and ATP5A1 encoding the alpha subunit of the F1 complex (Jonckheere et al. 2013). Whilst mutations in ATPAF2 (MIM604273), ATP5A1 (MIM615228) and ATP5E (MIM614053) have been detected in single patients, TMEM70 (MIM614052) mutations are a frequent cause of autosomal recessive ATP synthase deficiency, usually associated with a distinctive phenotype consisting of neonatal-onset hypertrophic cardiomyopathy, facial dysmorphisms, severe lactic acidosis and 3-methylglutaconic aciduria (3-MGA) (Cízková et al. 2008). Several patients share a common Roma descent, being homozygous for a founder c.317-2A>G mutation which causes aberrant splicing with marked loss of TMEM70 transcript (Honzík et al. 2010). Few other mutations have been described, mainly in Arab-Muslim families (Spiegel et al. 2011).
Herein, we report the clinical, biochemical and genetic characteristics of ten patients presenting with neonatal severe hypertrophic cardiomyopathy and carrying mutations in TMEM70, including four novel changes.
Patients and Methods
Patients
Written informed consent was obtained from patients’ parents, in agreement with the Declaration of Helsinki upon approval of the Ethical Committees of the Foundation IRCCS Istituto Neurologico “C.Besta”, Milan, Italy.
A total of ten patients (five boys and five girls) from eight apparently unrelated families were examined. The clinical characteristics and biochemical investigations carried out in the patients are summarized in Table 1. All our patients showed hypertrophic cardiomyopathy as the main clinical feature, associated with high lactate levels, psychomotor delay, respiratory distress and muscular hypotonia. All showed intrauterine growth restriction (IUGR) and most of them had an emergency delivery because of oligohydramnios, indicating antenatal disease onset. Dysmorphic features were observed in all subjects of our cohort; none of the boys presented hypospadias, often reported in TMEM70-mutant patients (Spiegel et al. 2011; Torraco et al. 2012). One patient (P3) showed subacute intestinal obstruction, as reported elsewhere (Spiegel et al. 2011). Although there was no consistent brain MRI pattern, lesions included bulbar and cerebellar atrophy (P1, P2), pseudocysts in frontal (P3) or occipital lobe (P1), signs of incomplete brain development (P5, P6 and P7) and in one case a severe haemorrhagic lesion in the parieto-occipital lobe (P8). Six patients died during the first year of life because of respiratory distress consequent to cardiomyopathy. Four cases are still alive, with the oldest patient, P1, being 9 years old. Biomarkers in body fluids reported in TMEM70-mutant cases, such as 3-MGA, organic acids in urine and hyperammonaemia (Wortmann et al. 2009; Honzik et al. 2012; Wortmann et al. 2013), were detected in some, but not all, of our patients (Table 1).
Table 1.
Clinical, genetic and biochemical features of individuals with TMEM70 mutations
| P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 | P10 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Origin | Italian | Italian | Italian | Roma | Macedonian | Roma | Italian | Italian | Italian | Italian |
| Consanguinity | No | No | No | Yes | No | Yes | No | No | No | No |
| Sex | F | F | F | F | M | M | M | M | F | M |
| Onset | 2 years | 1 year | 8 months | Neonatal | Neonatal | Neonatal | Neonatal | Birth | Prenatal | Prenatal |
| Age/age of death | Alive (9 years) | Died (1 year) | Alive (3 years) | Died (2 years) | Alive (4 years) | Died (4 years) | Alive (1 years) | Died (1 months) | Died (48 h) | Died (3 months) |
| Delivery weeks | 37 | 40 | 33 | 35 + 6 | 39 | 36 | 36 + 6 | 33 | 37 | 34 |
| Birth weight | Reduced | 2,460 g | Reduced | 1,680 g | 2,620 g | 2,000 g | 2,140 g | 1,975 g | 2,650 g | 1,855 g |
| Oligohydramnios | Yes | Yes | Yes | No | Yes | N.a. | No | Yes | Yes | Yes |
| IUGR | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Apnoea/distress | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Hypotonia | Yes | Yes | No | Yes | Yes | Yes | Yes | Yes | N.a. | Yes |
| Hypertrophic cardiomyopathy | Yes | Yes | Yes | Yes (NCC) | Yes | Yes (NCC) | Yes | Yes | Yes | Yes |
| Psychomotor delay | Yes | Yes | Yes | Yes | Yes | Yes | Yes | N.a. | N.a. | Yes |
| Dysmorphic features | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Lactic acidosis | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | N.a. | Yes |
| Hyperammonaemia | Yes | Yes | No | No | No | Yes | Yes | No | No | No |
| 3-MGA mmol/Mol creatinine | Normal | High | Normal | High | High | High | High | Normal | N.d. | Normal |
| Brain imaging | Occipital cysti, cerebellar hypoplasia | Bulbar hypoplasia | Frontal pseudocyst, vermis hypotrophy, WM alterations | Cerebellar hypoplasia, thin corpus callosum, lactate peak | Incomplete hippocampal inversion, WM alterations | Incomplete hippocampal inversion, WM alterations | Abnormal cortical development lactate peak, WM alterations | Haemorrhagic lesion in the parietal occipital lobe | N.d. | N.d. |
| Mutations | c.317-2A > G; c.349_352del | c.317-2A > G; c.317-2A > G | c.317-2A > G; c.783A > G | c.317-2A > G; c.317-2A > G | c.701A > C; c.701A > C | c.317-2A > G; c.317-2A > G | c.238C > T; c.238C > T | c.317-2A > G; c.317-2A > G | c.317-2A > G; c.317-2A > G | c.317-2A > G; c.317-2A > G |
| Transcript lossa p.Ile117Alafs*36 | Transcript lossa | Transcript lossa p.*261Trpext*17 | Transcript lossa | p.His234Pro | Transcript lossa | p.Arg80* | Transcript lossa | Transcript lossa | Transcript loss* | |
| MRC deficiency (muscle) | I (48 %); V (53 %) | N.d. | Normal | V (23 %); I (67 %); II (48 %); III (55 %) | Normal | Normal | Normal | N.d. | N.d. | I (15 %) |
| MRC deficiency (fibroblasts) | V (24 %) | V (28 %) | V (47 %) | V (35 %) | V (37 %) | V (17 %) | V (25 %) | N.d. | N.d. | Normal |
N.d. not done/not available, N.a. not applicable, IUGR intrauterine growth restriction, 3-MGA 3-methylglutaconic aciduria, WM white matter, MRC mitochondrial respiratory chain, NCC non-compaction cardiomyopathy
aThe intronic variant c.317-2A > G leads to aberrant splicing and loss of TMEM70 transcript
Biochemistry and Morphology
Muscle and skin biopsies were performed after receiving written informed consent. Muscle morphology and histochemistry were performed as previously described (Dubowitz 1985) in all patients but P2, P8 and P9. Electron microscopy of muscle tissue from P3 was performed as described (Jonckheere et al. 2011).
Fibroblasts were grown in 35 mm imaging dishes (iBIDI, Thistle Scientific) to 50–70% confluence. Cells were then incubated with complete DMEM media (Gibco) supplemented with 100 nM MitoTracker Red (Invitrogen) for 25 min at 37° C. After washing with PBS, cells were visualized with a live-imaging system (CARV-II, Crisel) and analysed using the MetaMorph Imaging System (version 7.1.2, Molecular Devices Corporation).
Respiratory chain activities of complexes I to V were measured by spectrophotometric methods (Bugiani et al. 2004) in supernatants of 800 x g muscle homogenates or digitonin-treated cultured skin fibroblasts. The ATPase activity of cV was assessed although this assay in frozen tissues is considered to be poorly reliable (Jonckheere et al. 2012) and a high oligomycin-resistant activity may interfere with the dosage in cultured cells (Barrientos et al. 2009).
Genetic Analyses
DNA was extracted from blood samples obtained from affected patients and available parents. Sequence analyses of the three TMEM70 exons and their flanking splice junction consensus sequences were performed using the BigDye Terminator Kit (Applied Biosystems, Foster City, California, USA) and the ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). For P10, whole-exome next-generation sequencing (WES) and variant filtering were performed as described (Haack et al. 2012). The complete sequencing of mtDNA was performed on DNA extracted from muscle homogenate, as previously described (Greaves et al. 2006).
Immunoblot Analyses
Protein gel electrophoresis (GE) and blotting analyses were performed in P1, P3 and P8 skeletal muscle, in P8 cardiac autopsy muscle and in P1, P2, P3, P4, P5 and P7 fibroblasts.
For SDS-GE, samples containing 50 μg protein were separated by 12% SDS–polyacrylamide gels, transferred to nitrocellulose membranes (Tiranti et al. 1999) and incubated with antibodies against TMEM70 (Proteintech) and GAPDH (Millipore).
The analysis of the assembled respiratory complexes was performed by using Blue Native Gel Electrophoresis (BNGE). Samples obtained from 25 mg of skeletal/cardiac muscle biopsy or from 2 × 106 fibroblasts (Nijtmans et al. 2002) were loaded and run into a 5–13 % gradient non-denaturing 1-dimensional gel. After protein blotting, immunodetection was carried out using antibodies against alpha subunit of ATP synthase, NDUFA9 (for complex I), MTCOI (for complex IV), SDHA (for complex II) from MitoSciences, Invitrogen, and against HSP60 (Abcam). Densitometric analysis was performed using Quantity One software (BioRad, Hercules, CA, USA).
Results
Muscle morphology, including histochemical reactions, was normal in all studied patients, with no RRF or COX-deficient fibres being present. Electron microscopy, performed only in P3 muscle, showed several swollen and irregularly shaped mitochondria with disruption of the central part and complete loss of the cristae. Some mitochondria were small with simplified cristae (Fig. 1a, b). MitoTracker-based visualization in P1 and P4 showed increased fragmentation of the mitochondrial network, suggesting altered fission/fusion dynamics (Fig. 1c–e)
Fig. 1.
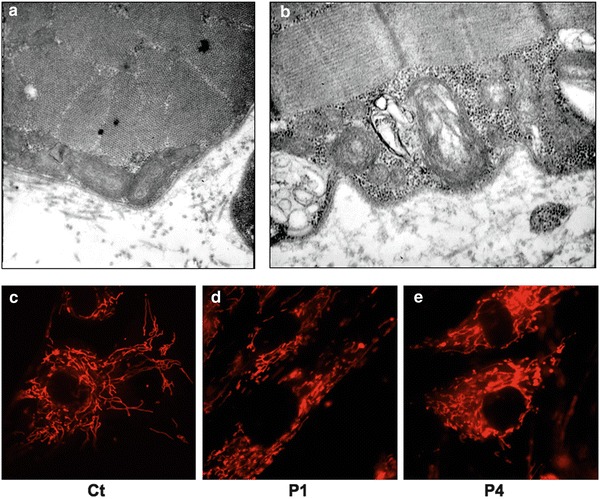
Morphological studies. (a, b) Electron microscopy in P3 muscle showing irregularly shaped mitochondria with disruption of the central part and loss of the cristae. (c–e) Representative images of mitochondrial morphology, showing the filamentous (c) or the more fragmented (d, e) mitochondrial network of fibroblasts from a control subject (c), P1 (d) and P4 (e)
In muscle homogenate we found isolated cI deficiency in P10 (15 %) and multiple defects in P1 (cI 48 %–cV 53 %) and P4 (cI 67 %–cII 48 %–cIII 55 %–cV 23 %), whereas in the other available samples, MRC activities were normal (Table 1). In autopsy skeletal and cardiac muscles from P8, all the RC activities were markedly reduced, probably because of post-mortem autolysis. In fibroblasts, we measured low cV activity in all but one analysed patients (P1 to P7), whereas all the other OXPHOS activities were in the control range. Normal cV activity was found in fibroblasts from P10, who displayed an isolated complex I deficiency in muscle.
In all patients, sequencing of mtDNA did not reveal any pathogenic variants. Because of the clinical and biochemical presentation, we sequenced TMEM70 (NM_017866.5) in patients 1 to 7. The DNA sample of P10, who presented an isolated cI defect in muscle, underwent whole-exome sequencing (Haack et al. 2012): the founder intronic mutation c.317-2A > G in TMEM70 was confirmed by Sanger sequence in both P10 and his affected siblings P8 and P9. The c.317-2A > G change was found in homozygosity in three additional patients (P2, P4, P6) and as a compound heterozygous mutation in P1 and P3, associated with a c.349_352del and c.783A > G mutations, respectively. The c.349_352del microdeletion causes a frameshift predicted to result in a truncated protein (p.Ile117Alafs*36); the c.783A > G change affects the termination codon and predicts the synthesis of a protein with an aberrant 17aa-long extra C-terminus (p.*261Trpext*17). We identified a novel homozygous missense mutation in P5 (c.701A > C, p.His234Pro) and a novel homozygous nonsense mutation in P7 (c.238C > T, p.Arg80*); these variants are not reported in public SNP databases, including dbSNP and the Exome Variant Server.
SDS-GE and immunodetection using an anti-TMEM70 in total lysates from P1, P2, P3, P4, P5 and P7 fibroblasts and from P8 cardiac muscle revealed virtual absence of TMEM70 protein in all tested patients’ samples, suggesting a deleterious effect of all identified TMEM70 mutations, including novel variants (Fig. 2a)
Fig. 2.
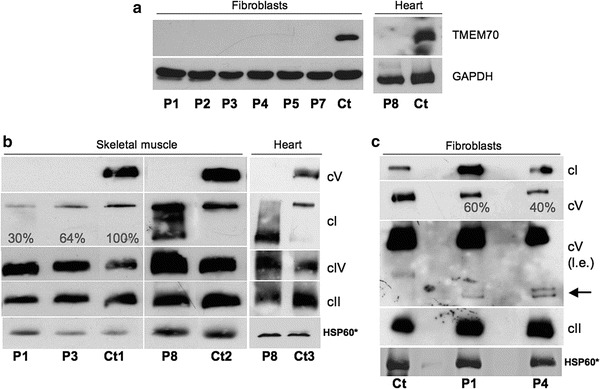
Protein characterization in muscle and fibroblasts from individuals with TMEM70 mutations. (a) SDS-gel electrophoresis of fibroblasts from patients 1,2,3,4,5,7 (P1, P2, P3, P4, P5, P7) and of autopsy cardiac muscle from patient 8 (P8), with corresponding controls (Ct). We used antibodies against TMEM70 and GAPDH (as loading control). (b) One-dimensional Blue Native Gel Electrophoresis (1D BNGE) of skeletal muscle homogenates from patients 1, 3 and 8 (P1, P3, P8) and controls (Ct1 and Ct2) and of cardiac muscle homogenates from patient 8 (P8) and a control (Ct3). Autopsy samples were used for P8, Ct2 and Ct3. We used an antibody against the subunit α of ATP synthase to detect complex V (cV), an antibody against SDH 70 kDa for complex II (cII), an antibody against NDUFA9 for complex I (cI), an antibody against subunit COX4 for complex IV (cIV) and an antibody against HSP60, used as loading control. The reported percentages correspond to the values of cI/cII signals obtained by densitometric analysis. (c) 1D BNGE of mitochondrial-enriched fibroblasts from patients 1 and 4 (P1, P4) and a control (Ct). We used the same antibodies reported above. For complex V a long exposure (l.e.) allowed the immunovisualization of subassembly species in samples from the patients (arrow). Asterisk for HSP60 immunodetection, the same samples used for 1D-BNGE were separated by SDS-GE and incubated with an anti-HSP60 antibody
1D BNGE showed undetectable cV in all tested skeletal muscle samples but displayed a moderate to severe reduction of cI in P4 (64 %) and P1 (30 %) compared to a control muscle (see Fig. 2b). In both P8 skeletal and cardiac autopsy muscles we confirmed the absence of cV, associated with accumulation of complex I sub-assembly species. However, since P8 samples were from an autopsy biopsy, we cannot exclude that these results are due to partial sample degradation (Fig. 2b). BNGE analysis in P1 and P4 fibroblasts showed a partial cV reduction, with accumulation of subassembly intermediates (F1 subcomplexes), as already reported in TMEM70 mutant cells (Cízková et al. 2008; Torraco et al. 2012) (Fig. 2c). Unfortunately no fibroblast samples were available for further investigation on P10, the only subject who showed normal cV activity in fibroblasts.
Discussion
Over 40 patients carrying mutations in TMEM70 have been thus far described (Cízková et al. 2008; Honzík et al. 2010; Spiegel et al. 2011; Honzik et al. 2012; Cameron et al. 2011), and though they originated from different European countries, most carried a homozygous splice site mutation and shared an identical Roma-gypsy origin that explains genetic homogeneity. Few additional mutations have been reported and often result in absent or prematurely truncated protein.
Our study presents a cohort of ten patients harbouring TMEM70 mutations. All subjects were Italians; however, P5 parents were from Macedonia (Table 1). Although most of our patients did not have a Roma origin, six patients were homozygous and 2 compound heterozygous for the common Roma mutation, suggesting spreading of an ancestral mutant allele. In the compound heterozygous subjects, the second mutation is predicted to result in a truncated (P1) or in an aberrant (P3) protein, with an extended C-terminus that can impair the function or triggers accelerated degradation. P7 is homozygous for a novel nonsense mutation, predicted to produce a truncated protein (p.Arg80*). On the other hand, P5 is the second patient carrying a homozygous missense mutation. Two missense changes have been described, both in compound heterozygosity with the “common” variant: the p.Gly165Asp mutation was described in a patient with a milder phenotype characterized by normal growth parameters and cognitive development, no dysmorphic features, improvement of symptoms with age and persistence of hypertrophic cardiomyopathy (Shchelochkov et al. 2010). The p.Thr210Pro was instead identified in two siblings presenting with hypertrophic cardiomyopathy that reversed spontaneously at around 1 year of age. Recently, a homozygous c.535C > T (p.Tyr179His) mutation has been described in a Turkish patient presenting with neonatal lactic acidaemia, severe hypotonia and hypertrophic cardiomyopathy (Atay et al. 2013). At the latest examination at age 1 year, the child showed stable cardiac hypertrophy and had not suffered of metabolic crises requiring hospital admissions. Our P5 was born at term, with a weight >3rd percentile, and he is still alive at the age of 4 years. Taken together, these observations may suggest that infants with missense, rather than nonsense variants in TMEM70, have a milder clinical course. However, the virtual absence of TMEM70 protein in P5 as observed in all the other patients carrying different loss-of-function mutations does not agree with this conclusion. Moreover, genotype to phenotype correlation is inconsistent, especially for the common splice mutation, which in our as well as in other studies (Honzik et al. 2012) is associated with variable degree of severity, ranging from fatal outcome at two days of age (P9 in this study) to survival at the age of 17 years (Cízková et al. 2008). The same variability has been reported for other TMEM70 mutations: the oldest patient reported to date (a 24-year-old man) harboured the same homozygous c.578_579delCA mutation detected in his brother, deceased at the age of 3.5 years (Spiegel et al. 2011).
The biochemical presentation was an interesting feature in our cohort of patients. Mutations in TMEM70 have been reported to cause isolated cV deficiency (Cízková et al. 2008). Often the spectrophotometric analysis for cV (ATP hydrolysis) is not performed (Spinazzi et al. 2012) because of its insufficient reliability in frozen muscle (Jonckheere et al. 2012) and the presence of oligomycin-insensitive ATPase activity in cultured cells (Barrientos et al. 2009). In our investigation, the measurement of cV activity in total homogenate from frozen muscles was unreliable, since normal values were obtained in samples with nearly absent cV holocomplex analysed by 1D BNGE Western blot. On the contrary, the ATPase activity in digitonin-treated fibroblasts was reliable, being reduced in all but one patient (P10).
In muscle, the spectrum of defective OXPHOS activities was broader than isolated cV deficiency, including low cI (in P1) or multiple OXPHOS defects (in P4). Likewise, a profound, apparently isolated reduction of cI was found in muscle from P10, which prompted us to carry out WES analysis and, unexpectedly, identify the common splicing mutation in TMEM70. Interestingly, a compound heterozygous patient harbouring the common splice site mutation and a missense variant has recently been reported to have no biochemical OXPHOS defects in muscle and fibroblasts (Shchelochkov et al. 2010). Therefore, the screening of TMEM70 should be performed in patients presenting a mitochondrial disorder characterized by early-onset cardiomyopathy, irrespective of the biochemical profile. In male patients, the analysis of TMEM70 should also be considered in the differential diagnosis with Barth syndrome (MIM#302060), an X-linked disease characterized by dilated or hypertrophic cardiomyopathy, skeletal myopathy, growth retardation and organic aciduria, including 3-MGA.
The exact molecular function of TMEM70 is still not completely clear and its specific role in ATP synthase assembly is still unknown, since no direct interaction has been demonstrated between this protein and the holocomplex V. In addition to synthesis of ATP from ADP and inorganic phosphate as a consequence of energy derived from proton translocation, cV has an important role for the correct structure of mitochondria. It constitutes 17–30 % of the inner membrane-bound proteins and is important for the formation and invaginations that form the cristae (Ozawa and Asai 1973). Dimeric and higher oligomeric forms of ATP synthase (Paumard et al. 2002) seem critical to maintain the shape of mitochondria by promoting the formation of the inner membrane cristae. Studies in yeast (Paumard et al. 2002; Lefebvre-Legendre et al. 2005) indicate that the cV structure rather than its enzymatic activity dictates cristae morphology. We found ultrastructural alterations in mitochondrial shape of the single TMEM70 patient where electron microscopy could be performed (P3), supporting the link between ATP synthase and mitochondrial morphology.
In conclusion, clinical features, including cardiomyopathy, respiratory distress at birth, lactic acidosis and muscular hypotonia often associated with dysmorphic features, sometimes supported by suggestive biochemical data (cV deficiency and 3-MGA), are still extremely important to point to the diagnosis.
Acknowledgements
We thank Rosalba Carrozzo for the generous gift of anti-TMEM70 antibody. This work was supported by the Italian Ministry of Health (GR2010–2316392); Fondazione Telethon grants GGP11011 and GPP10005; CARIPLO grant 2011/0526; the Pierfranco and Luisa Mariani Foundation of Italy; and the Italian Association of Mitochondrial Disease Patients and Families (Mitocon). H.P. was supported by the Impulse and Networking Fund of the Helmholtz Association in the framework of the Helmholtz Alliance for Mental Health in an Ageing Society (HA-215) and the German Federal Ministry of Education and Research (BMBF) funded German Center for Diabetes Research (DZD e.V.) and Systems Biology of Metabotypes grant (SysMBo #0315494A), the grant RF-INN-2007-634163 of the Italian Ministry of Health, the BMBF funded German Network for Mitochondrial Disorders (mitoNET #01GM1113C/D) and the E-Rare project GENOMIT (01GM1207 and FWF I 920-B13). We acknowledge the “Cell lines and DNA Bank of Paediatric Movement Disorders and Neurodegenerative Diseases” of the Telethon Network of Genetic Biobanks (grant GTB12001J) and the EuroBioBank Network.
The authors declare that they have no conflict of interest.
Sentence Take-Home Message
TMEM70 patients present with a mitochondrial cardiomyopathy with early-onset hypotonia, respiratory distress and psychomotor delay, irrespective of the biochemical defect.
Compliance with Ethics Guidelines
Daria Diodato, Federica Invernizzi, Eleonora Lamantea, Gigliola Fagiolari, Rossella Parini, Francesca Menni, Giancarlo Parenti, Lina Bollani, Maria A.Donati, Denise Cassandrini, Elisabetta Pasquini, Filippo M. Santorelli, Tobias B. Haack, Holger Prokisch, Daniele Ghezzi, Costanza Lamperti and Massimo Zeviani declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Declaration of Helsinki (1975), as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Details of the Contributions of Individual Authors
DD, DG, CL and MZ designed the study; DD, FI, EL, GF, EP, FMS, TBH, HP, DG, CL and MZ performed experiments, collected and analysed data. RP, FM, GP, LB, MAD, DC, FMS and CL evaluated the patients and wrote case reports. DD, DG, CL and MZ wrote the manuscript. FI, EL, GF, RP, FM, GP, LB, MAD, DC, EP, FMS, TBH and HP critically revised the manuscript for important intellectual content.
Footnotes
Competing interests: None declared
Contributor Information
Costanza Lamperti, Email: Costanza.Lamperti@istituto-besta.it.
Massimo Zeviani, Email: mdz21@mrc-mbu.cam.ac.uk.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Atay Z, Bereket A, Turan S, et al. A novel homozygous TMEM70 mutation results in congenital cataract and neonatal mitochondrial encephalo-cardiomyopathy. Gene. 2013;15:197–199. doi: 10.1016/j.gene.2012.11.044. [DOI] [PubMed] [Google Scholar]
- Barrientos A, Fontanesi F, Diaz F. Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr Protoc Hum Genet. 2009;63:19.3.1–1. doi: 10.1002/0471142905.hg1903s63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugiani M, Invernizzi F, Alberio S, et al. Clinical and molecular findings in children with complex I deficiency. Biochim Biophys Acta. 2004;1659:136–147. doi: 10.1016/j.bbabio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Cameron JM, Levandovskiy V, Mackay N, et al. Complex V TMEM70 deficiency results in mitochondrial nucleoid disorganization. Mitochondrion. 2011;11:191–199. doi: 10.1016/j.mito.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Cízková A, Stránecký V, Mayr JA, et al. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat Genet. 2008;40:1288–1290. doi: 10.1038/ng.246. [DOI] [PubMed] [Google Scholar]
- Dubowitz V. Muscle biopsy, a practical approach. 2. London: Bailliere Tindal; 1985. [Google Scholar]
- Greaves LC, Preston SL, Tadrous PJ, et al. Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc Natl Acad Sci U S A. 2006;103:714–719. doi: 10.1073/pnas.0505903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, Haberberger B, Frisch EM, et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J Med Genet. 2012;49:277–283. doi: 10.1136/jmedgenet-2012-100846. [DOI] [PubMed] [Google Scholar]
- Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet. 1990;46:428–433. [PMC free article] [PubMed] [Google Scholar]
- Honzík T, Tesarová M, Mayr JA, et al. Mitochondrial encephalo-cardiomyopathy with early neonatal onset due to TMEM70 mutation. Arch Dis Child. 2010;95:296–301. doi: 10.1136/adc.2009.168096. [DOI] [PubMed] [Google Scholar]
- Honzik T, Tesarova M, Magner M, et al. Neonatal onset of mitochondrial disorders in 129 patients: clinical and laboratory characteristics and a new approach to diagnosis. J Inherit Metab Dis. 2012;35:749–759. doi: 10.1007/s10545-011-9440-3. [DOI] [PubMed] [Google Scholar]
- Jonckheere AI, Huigsloot M, Lammens M, et al. Restoration of complex V deficiency caused by a novel deletion in the human TMEM70 gene normalizes mitochondrial morphology. Mitochondrion. 2011;11:954–963. doi: 10.1016/j.mito.2011.08.012. [DOI] [PubMed] [Google Scholar]
- Jonckheere AI, Smeitink JA, Rodenburg RJ. Mitochondrial ATP synthase: architecture, function and pathology. J Inherit Metab Dis. 2012;35:211–225. doi: 10.1007/s10545-011-9382-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonckheere AI, Renkema GH, Bras M, et al. A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain. 2013;136:1544–1554. doi: 10.1093/brain/awt086. [DOI] [PubMed] [Google Scholar]
- Lefebvre-Legendre L, Salin B, Schaeffer J, et al. Failure to assemble the alpha 3 beta 3 subcomplex of the ATP synthase leads to accumulation of the alpha and beta subunits within inclusion bodies and the loss of mitochondrial cristae in Saccharomyces cerevisiae. J Biol Chem. 2005;6:18386–18392. doi: 10.1074/jbc.M410789200. [DOI] [PubMed] [Google Scholar]
- Mayr JA, Havlícková V, Zimmermann F, et al. Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 epsilon subunit. Hum Mol Genet. 2010;19:3430–3439. doi: 10.1093/hmg/ddq254. [DOI] [PubMed] [Google Scholar]
- Nijtmans LG, Henderson NS, Holt IJ. Blue native electrophoresis to study mitochondrial and other protein complexes. Methods. 2002;26:327–334. doi: 10.1016/S1046-2023(02)00038-5. [DOI] [PubMed] [Google Scholar]
- Ozawa T, Asai J. On the relationship between mitochondrial ATPase and the inner membrane. J Bioenerg. 1973;4:507–519. doi: 10.1007/BF01515942. [DOI] [PubMed] [Google Scholar]
- Paumard P, Vailllier J, Coulary B, et al. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002;21:221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S, Blok RB, Dahl HH, et al. Leigh syndrome: clinical features and biochemical DNA abnormalities. Ann Neurol. 1996;39:343–351. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- Shchelochkov OA, Li FY, Wang J, et al. Milder clinical course of Type IV 3-methylglutaconic aciduria due to a novel mutation in TMEM70. Mol Genet Metab. 2010;101:282–285. doi: 10.1016/j.ymgme.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Spiegel R, Khayat M, Shalev SA, et al. TMEM70 mutations are a common cause of nuclear encoded ATP synthase assembly defect: further delineation of a new syndrome. J Med Genet. 2011;48:177–182. doi: 10.1136/jmg.2010.084608. [DOI] [PubMed] [Google Scholar]
- Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc. 2012;31:1235–1246. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- Tiranti V, Galimberti C, Nijtmans L, Bovolenta S, Perini MP, Zeviani M. Characterization of SURF-1 expression and Surf-1p function in normal and disease conditions. Hum Mol Genet. 1999;8:2533–2540. doi: 10.1093/hmg/8.13.2533. [DOI] [PubMed] [Google Scholar]
- Torraco A, Verrigni D, Rizza T, et al. TMEM70: a mutational hot spot in nuclear ATP synthase deficiency with a pivotal role in complex V biogenesis. Neurogenetics. 2012;13:375–386. doi: 10.1007/s10048-012-0343-8. [DOI] [PubMed] [Google Scholar]
- Wortmann SB, Rodenburg RJ, Jonckheere A, et al. Biochemical and genetic analysis of 3-methylglutaconic aciduria type IV: a diagnostic strategy. Brain. 2009;132:136–146. doi: 10.1093/brain/awn296. [DOI] [PubMed] [Google Scholar]
- Wortmann SB, Duran M, Anikster Y et al (2013) Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J Inherit Metab Dis(Jan 8) [DOI] [PubMed]