

Abstract
Cold inducible RNA-binding protein (CIRP) is a nuclear protein which has been recently identified as a novel inflammatory mediator in hemorrhagic shock and sepsis. We hypothesized that CIRP acts as a potent inflammatory mediator in hepatic ischemia-reperfusion (I/R), and thus blocking CIRP protects against I/R-induced liver injury. Male C57BL/6 mice were subjected to 70% hepatic ischemia by microvascular clamping of the hilum of the left and median liver lobes for 60 min, followed by reperfusion. Anti-CIRP antibody (1 mg/kg body weight) or vehicle (normal saline) in 0.2 mL was injected via the internal jugular vein at the beginning of the reperfusion. Blood and liver tissues were collected 24 h after I/R for various measurements and a 10-day survival study was performed. CIRP released into the circulation was significantly increased 24 h after hepatic I/R. Anti-CIRP antibody treatment markedly reduced hepatocellular damage markers and significantly improved the liver microarchitecture. Anti-CIRP also reduced the systemic and local inflammation demonstrated by attenuation in both serum and hepatic levels of interleukin 6. The expression of neutrophil-attracting chemokine as well as liver neutrophil infiltration was reduced by anti-CIRP treatment. Anti-CIRP also dramatically decreased the amount of apoptosis and nitrosative stress, evidenced by decrease in TUNEL staining and inducible nitric oxide synthase and cyclooxygenase-2 levels, respectively. Finally, the 10-day survival rate was increased from 37.5% in the vehicle group to 75% in the anti-CIRP treatment group. Thus, targeting CIRP offers potential therapeutic implications in the treatment of hepatic I/R injury.
Keywords: CIRP, hepatic ischemia/reperfusion, inflammation, neutrophil infiltration, apoptosis, nitrosative stress
Introduction
Hepatic ischemia-reperfusion (I/R) injury can occur in a variety of clinical settings that include but not limited to trauma, liver resections, orthotopic transplantation, or prolonged shock states (1, 2). In the setting of liver transplantation, hepatic I/R contributes up to 10% of early organ failure as well as creating a higher incidence of both acute and chronic rejection (3). There is a multifarious interaction between hepatocytes, sinusoidal endothelial cells, and Kupffer cells as well as infiltrating neutrophils, macrophages, and platelets. The proinflammatory signaling mediators on all of these cell types become activated as a consequence of tissue anoxia and are further exacerbated upon reperfusion (1). Many attempts have been made to apply pharmacologic therapies or modify surgical approaches to reduce or help protect against hepatic I/R injury with limited success (4, 5). Therefore, identification of novel targets for developing therapeutic interventions to treat liver I/R injury is still under intensive research.
In the clinic, it would be challenging to protect against the ischemic damage in advance. After its occurrence, one can explore only a few avenues to help alleviate the hepatic I/R injury. One way is to reduce the milieu of inflammation, which is a major contributing factor to the injury. The inflammatory response associated with hepatic I/R is considered as a sterile type, which is not triggered by bacterial infection (6, 7). Such an inflammatory reaction is in response to the free radicals generated from acute oxidative and nitrosative stress during I/R (6). Another initiation of sterile inflammation is caused by the release of endogenous damage-associated molecular patterns (DAMPs) from host cells during injury (6-8). DAMPs can interact with pattern-recognition receptors, such as toll-like receptor (TLRs) and receptors of advanced glycation end products (RAGEs), in immune cells to activate the NF-κB signaling pathway for proinflammatory cytokine release (7, 9)
Cold-inducible RNA-binding protein (CIRP) is a 172-amino acid nucleoprotein that belongs to the family of cold shock proteins (10, 11). It has one N-terminal consensus-sequence RNA-binding domain and one C-terminal glycine-rich domain (10). CIRP is originally identified to facilitate the translation by stabilizing mRNAs when cells are under certain stressful circumstances, including hypothermia, exposure to ultraviolet irradiation, or hypoxia (9, 11). Human CIRP mRNA is constitutively expressed in various cell lines from different organ origins, including blood, liver, kidney, cervix, and urinary bladder (10). However, the expression of CIRP in human tissues has not been reported yet, except our recent study shown in serum (11). Recently, our laboratory identified that extracellular CIRP acts as a DAMP to accentuate the inflammatory response through binding to TLR4 and myeloid differentiation factor 2 (MD2) complex (11-13). Also, neutralizing antiserum to CIRP attenuates the inflammatory response in the settings of hemorrhagic and septic shock (11).
In our recent study, we have observed a significant increased CIRP expression in the liver of rats which underwent hemorrhagic and septic shock (11). In this study, we hypothesized that CIRP plays an important role in the pathogenesis of I/R-induced hepatic injury. To investigate this, we used an established mouse model of hepatic I/R injury and first measure the levels and kinetics of CIRP release in the serum after I/R. We then further examined the effect of anti-CIRP antibody treatment on organ damage, inflammatory response, neutrophil infiltration, apoptosis, and oxidative stress. Finally, we evaluated the efficacy of anti-CIRP antibody treatment on the survival of mice after being subjected to hepatic I/R injury.
Materials and Methods
Experimental animals
Adult male C57BL/6 mice (20-25g) from Charles River Laboratories (Wilmington, Mass) were housed in a temperature controlled room on a 12-h light-dark cycle and fed a standard laboratory diet. Upon acquisition, mice were allowed to acclimate to the environment in our facility 5-7 days before the experiment. All mice were housed in cages of five members and animals were fasted overnight before undergoing surgery, but allowed water ad libitum. All experiments were performed in strict accordance with National Institute of Health guidelines for the use of experimental animals, and this study approved by the Institutional Animal Care and Use Committee of the Feinstein Institute for Medical Research.
Animal model of hepatic I/R
Mice were randomly assigned to three groups; sham, vehicle (normal saline), or anti-CIRP antibody treatment. Hepatic I/R was performed as described previously (2, 14). Briefly, animals underwent induction of anesthesia with 2.5% inhalational isoflurane, after which the ventral abdomen was shaved and cleansed with 10% povidone-iodine wash. A 3-4 cm midline incision was performed, and the hilum of the liver was exposed, allowing for identification of the hepatic artery and portal vein. A microvascular clip was placed across the hilum of the left lateral and median lobes to produce 70% ischemia for 60 min. At the beginning of the reperfusion, 0.2 mL normal saline, non-immunized control IgG (1 mg/kg body weight, [BW]), or anti-CIRP antibody (1 mg/kg BW) (11) was injected via internal jugular vein. We also administered anti-CIRP antibody at 10 mg/kg BW in our preliminary study and found its effects not significantly different from 1 mg/kg BW. The neutralizing polyclonal anti-CIRP antibody was generated as antisera against purified recombinant murine CIRP from rabbits and the IgG fraction was isolated from antisera by immobilized immunopure protein-A and -G chromatography as described previously by us (11). Anti-CIRP antibody inhibits the TNF-α release in macrophages stimulated with CIRP in a dose-dependent manner (11). After closure of the abdomen, 1 mL bolus of normal saline was given i.p. and the anesthesia was withdrawn. After operation, 1.0% lidocaine was applied topically to the surgical incision site and vein cannulation. Sham operated animals underwent midline laparotomy alone, without hepatic ischemia or administration of any treatments. Core body temperature was monitored throughout the entire operation and maintained between 35.5°C to 37.0°C by the use of an indwelling rectal thermometer and a heating pad placed below the animals. Blood and liver samples were collected 24 h after ischemia and stored at -80°C before analysis.
Survival study
Following the process of hepatic I/R as described in the animal model, the remaining 30% of the non-ischemic liver portion was resected at the onset of reperfusion. Then, normal saline or anti-CIRP antibody (1 mg/kg BW) was injected via internal jugular vein. Without resection, most of the animals which underwent hepatic I/R will live with 30% functionally non-ischemic liver for 10 days. The animals were then monitored daily to record the survival time.
Western blotting analysis
Liver tissues (100 mg) were lysed and homogenized in 300 μL lysis buffer (10 mM Tris-HCl pH 7.5, 120 mM NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate) containing a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, Ind) using a sonic dismembrator on ice. Samples were centrifuged at 14,000 revolutions/min for 15 min at 4°C, and the supernatant collected. Following measurement of sample protein concentration by BioRad DC protein assay kit (Bio-RAD, Hercules, Calif), 50 μg samples were separated on 4% to 12% Bis-Tris gels and transferred to nitrocellulose membranes. Membranes were incubated with primary antibody against CIRP or β-actin (SantaCruz Biotechnologies, Santa Cruz, Calif). All protein bands were detected by species-specific infrared fluorescence secondary antibodies (1:10,000) and analyzed by the LI-COR Odyssey Fc Imager (LI-COR, Lincoln, Neb) and band intensity measured using the NIH Image J densitometric software.
Measurement of injury markers and interleukin 6 (IL-6)
Whole-blood samples were centrifuged at 4,000 revolutions/min for 12 min to collect serum. The activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and lactate dehydrogenase (LDH) in the serum were determined by assay kits from Pointe Scientific (Lincoln Park, Mich). Serum and liver tissue IL-6 levels were determine by an enzyme-linked immunosorbent assay (ELISA) kit specific for mouse IL-6 (BD Biosciences, San Diego, Calif). These assays were carried out according to the manufacturer's instructions.
qPCR analysis
Total RNA was extracted from the liver by TRIzol reagent (Invitrogen, Carlsbad, Calif) and was reverse-transcribed into cDNA using murine leukemia virus reverse transcriptase (Applied Biosystems, Foster City, Calif). A PCR reaction was carried out in a 24 μL containing 0.08 μmol of each forward and reverse primer, 2 μL cDNA, 9.2 μL H2O, and 12 μL SYBR Green PCR Master Mix (Applied Biosystems). Amplification was conducted in an Applied Biosystems 7300 model under the thermal profile of 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. The level of mouse β-actin mRNA was used for normalization and each specific mRNA was conducted in duplicate. Relative expression of mRNA was calculated by the 2-ΔΔCt method, and results were expressed as fold change in comparison to the sham group. The sequence of primers for this study is listed as follows: IL-6, 5′-CCGGAGAGGAGACTTCACAG-3′ (forward) and 5′-CAGAATTGCCATTGCACAAC-3′ (reverse); MIP-2, 5′-CCCTGGTTCAGAAAATCATCCA-3′ (forward) and 5′-GCTCCTCCTTTCCAGGTCAGT-3′ (reverse); iNOS, 5′-GGCAAACCCAAGGTCTACGTT-3′ (forward) and 5′-GAGCACGCTGAGTACCTCATTG-3′ (reverse); COX-2, 5′-CTCAGCCAGGCAGCAAATC-3′ (forward) and 5′-ACATTCCCCACGGTTTTGAC-3′ (reverse); β-actin, 5′-CGTGAAAAGATGACCCAGATCA-3′ (forward) and 5′-TGGTACGACCAGAGGCATACAG-3′ (reverse).
Histological evaluation of liver injury and immunostaining for neutrophils
Liver tissues were taken from the median lobe following 24 h of reperfusion and stored in 10% formalin before being fixed in paraffin. Samples were then sectioned to 4 μm cuts and stained with hematoxylin–eosin (H&E). Liver parenchymal injury was then assessed in a blinded fashion using a semi-quantitative light microscopy evaluation. The histologic injury score for each sample was expressed as the sum of the individual scores given for five different parameters; cytoplasmic color fading, cellular vacuolization, sinusoidal congestion, necrosis, and erythrocyte stasis (15-17). Scores for each variable ranged from 0 (0%), to 1 (1%-10%), 2 (10%-30%), 3 (30%-60%), 4 (>60%), with a highest possible score of 20. Each sample score was then averaged over 10 microscopic fields. For neutrophil staining, paraffin-embedded liver tissues were dewaxed in xylene and rehydrated in a graded series of ethanol. The slides were heated in 0.92% citric acid buffer (Vector Laboratories, Burlingame, Calif) at 95°C for 30 min. After cooling, the slides were incubated with 2% H2O2/60% methanol and blocked in normal rabbit serum/Tris-buffered saline. The anti-Gr-1 antibody (BioLegend, San Diego, Calif) was applied and incubated overnight. The detection was carried out as per the instructions provided by an immunohistochemistry kit with NovaRED substrate from Vector Laboratories.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
Apoptosis in liver tissue was identified by TUNEL assay using an In Situ Cell Death Detection Kit (Roche). The assay was conducted according to the manufacturer's instructions. The nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI).
Statistical analysis
All data are expressed as a mean ± SE and compared by one-way analysis of variance (ANOVA) and the Student-Newman-Keuls (SNK) test. Survival rates were analyzed by the Kaplan-Meier estimator and compared using a log-rank test. Differences in values were considered significant if p < 0.05.
Results
Circulating levels of CIRP are increased after hepatic I/R
To explore the role of CIRP as a potential inflammatory mediator in hepatic I/R, we examined the release of CIRP in serum by Western blotting at 1, 4, and 24 h after the start of the 60-min hepatic ischemia followed by reperfusion in mice. Serum CIRP was detectable in sham, still comparable to sham at 1 h, increased by 2.6-fold at 4 h and significantly elevated by 5.9-fold at 24 h after hepatic I/R (Fig. 1). These findings are consistent with our prior studies of increased CIRP release in hemorrhagic shock and sepsis (11), suggesting that CIRP plays an important role in triggering inflammatory responses in hepatic I/R as well.
Fig. 1. Increased release of CIRP after hepatic I/R.
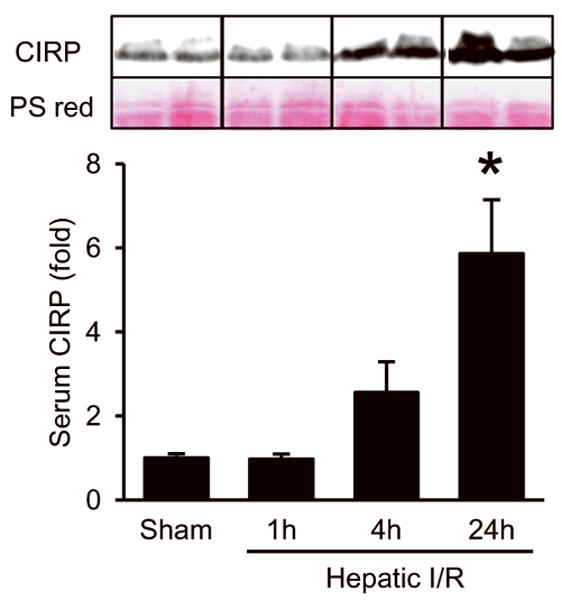
Western blot analysis of CIRP in the serum samples collected from sham and hepatic I/R groups at the indicated times after the start of hepatic I/R procedure. Representative blots against CIRP and Ponceau S red staining (PS red) are shown. Blots were scanned and quantified with densitometry. Band intensity of CIRP was normalized to the corresponding band intensity of PS red staining. The value in sham group is designated as 1 for comparison. Data presented as means ± SE (n=3-4/group) and compared by one-way ANOVA and SNK method; *p < 0.05 vs. sham.
Anti-CIRP antibody treatment attenuates liver injury after hepatic I/R
Serum levels of hepatic damage markers AST, ALT, and LDH were increased by 21-, 34- and 62-fold, respectively, 24 h after hepatic I/R (Fig. 2A-C). With the administration of neutralizing antibody against CIRP at the beginning of reperfusion, significant reduction was seen in organ injury markers by 74%, 83%, and 68%, respectively (Fig. 2A-C). However, the AST, ALT, and LDH levels in mice treated with non-immunized control IgG (1 mg/kg BW) were comparable to the vehicle group (AST, 1501 ± 567 vs. 1509 ± 205 IU/L; ALT, 543 ± 200 vs. 573 ± 236 IU/L; and LDH, 3502 ± 1256 vs. 2762 ± 656 IU/L - control IgG vs. vehicle). Further, these serum injury markers data correlated with the hepatic tissue architecture damaged observed by histological evaluation. Histological alterations seen in liver tissue samples 24 h after hepatic I/R were most evident for the degree of necrosis, micro-hemorrhage, and leukocyte infiltration in vehicle-treated animals in comparison to sham as well as anti-CIRP antibody-treated animals (Fig. 3A-C). Quantification of liver histologic injury score after hepatic I/R showed that vehicle-treated groups exhibited a 3.7-fold increase in severity compared to sham-operated animals, which was significantly reduced by 48% in anti-CIRP antibody-treated animals (Fig. 3D). Taken together, these results demonstrate that anti-CIRP antibody administration provides significant protection against liver injury from hepatic I/R.
Fig. 2. Effect of anti-CIRP antibody treatment on systemic organ injury markers after hepatic I/R.
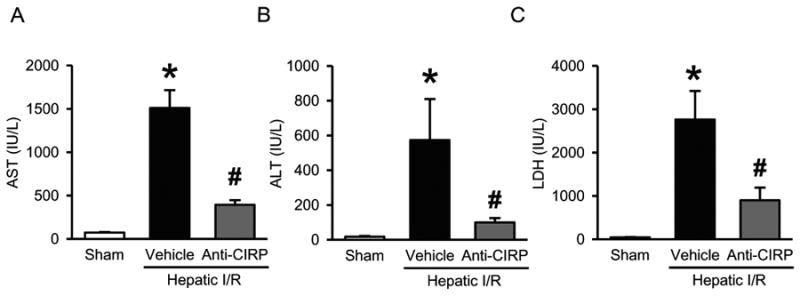
Serum samples were collected at 24 h post-hepatic I/R from sham, vehicle and anti-CIRP antibody-treated (Anti-CIRP) groups for measuring AST (A), ALT (B), and LDH (C). Data presented as means ± SE (n=6/group) and compared by one-way ANOVA and SNK method; *p < 0.05 vs. sham; #p < 0.05 vs. vehicle.
Fig. 3. Effect of anti-CIRP antibody treatment on tissue damage and cellular architecture in the liver after hepatic I/R.
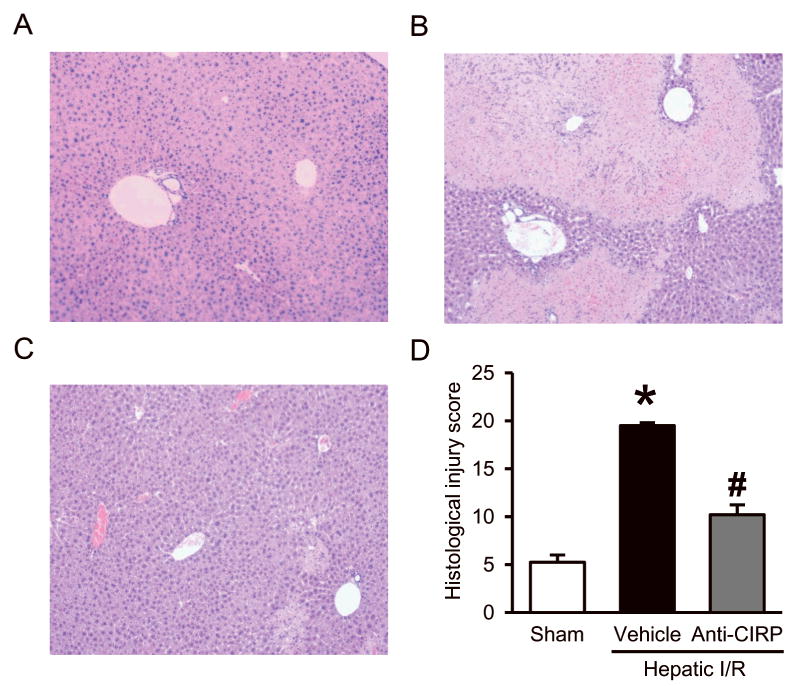
Histologic evaluation of the liver in the sham (A), vehicle (B), and anti-CIRP antibody-treated (C, Anti-CIRP) groups. Liver tissues were harvested 24 h after hepatic I/R, processed, and then stained with hematoxylin-eosin (H&E). Representative photomicrographs at 100× magnification. D, Semiquantitative histologic injury score measuring differences in cytoplasmic vacuolization, cytoplasmic fading, nuclear condensation, nuclear fading, nuclear fragmentation, and erythrocyte stasis examined on H&E staining as described in Materials and Methods. Data presented as means ± SE (n=4-5/group) and compared by one-way ANOVA and SNK method; *p < 0.05 vs. sham; #p < 0.05 vs. vehicle.
Anti-CIRP antibody treatment inhibits the inflammatory response after hepatic I/R
To ascertain the systemic and local inflammatory responses to hepatic I/R in our model, IL-6 levels were measured in both serum and liver tissue 24 h after hepatic I/R. Vehicle-treated animals showed an increase in serum IL-6 levels by 63-fold compared to sham group. When animals were administered anti-CIRP antibody, this increase in IL-6 levels was significantly reduced by 75% (Fig. 4A). In the liver tissue IL-6 protein levels increased by 2-fold in the vehicle-treated group compared to sham group, and anti-CIRP antibody treatment decreased it by 44% (Fig. 4B). Also, IL-6 mRNA increased by 18-fold in vehicle in comparison to sham, which was decreased by 94% in anti-CIRP treated group (Fig. 4C). Thus, anti-CIRP antibody administration resulted in downregulation of both systemic as well as local levels of proinflammatory marker IL-6.
Fig. 4. Effect of anti-CIRP antibody treatment on systemic and local proinflammatory cytokine IL-6 levels after hepatic I/R.
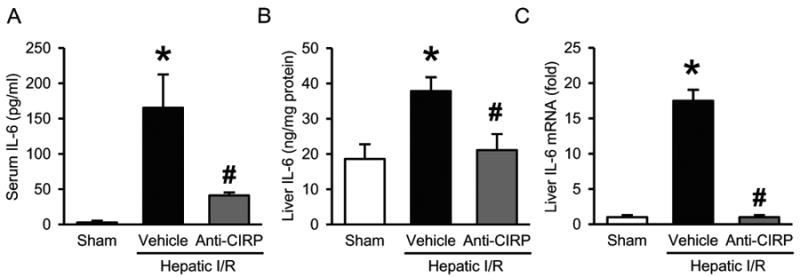
Serum samples and ischemic liver tissues were collected at 24 h post-hepatic I/R from sham, vehicle and anti-CIRP antibody-treated (Anti-CIRP) groups. IL-6 protein levels in serum (A) and liver tissue (B) were determined by enzyme-linked immunosorbent assay. (C) Liver IL-6 mRNA expression was determined by qPCR analysis and its levels were normalized to β-actin. The value in sham group is designated as 1 for comparison. Data presented as means ± SE (n=6-7/group) and compared by one-way ANOVA and SNK method; *p < 0.05 vs. sham; #p < 0.05 vs. vehicle.
Anti-CIRP antibody treatment reduces neutrophil infiltration into the liver after hepatic I/R
Hepatic I/R also results in overproduction of local proinflammatory chemokines, so we checked the expression of macrophage inflammatory protein 2 (MIP-2) in the liver tissue. The MIP-2 mRNA levels determined by qPCR dramatically increased by 179-fold in vehicle-treated mice compared to sham mice. However, MIP-2 levels were reduced by 80% in anti-CIRP antibody-treated group compared to vehicle group (Fig. 5A). Chemokines such as MIP-2 mediate tissue infiltration of neutrophils which release deleterious inflammatory mediators causing liver injury. To determine the degree of neutrophil infiltration associated with hepatic I/R, we performed immunohistochemistry staining of liver tissue for granulocyte receptor 1 (Gr-1), which is a marker for neutrophils. There is an abundance of neutrophils seen in vehicle group compared to sham, with a drastic reduction in anti-CIRP antibody-treated animals (Fig. 5B-D). Together, this data suggests that anti-CIRP antibody administration not only reduces proinflammatory chemokines, but also decreases associated neutrophil recruitment to the liver.
Fig. 5. Effect of anti-CIRP antibody treatment on neutrophil infiltration into the liver after hepatic I/R.
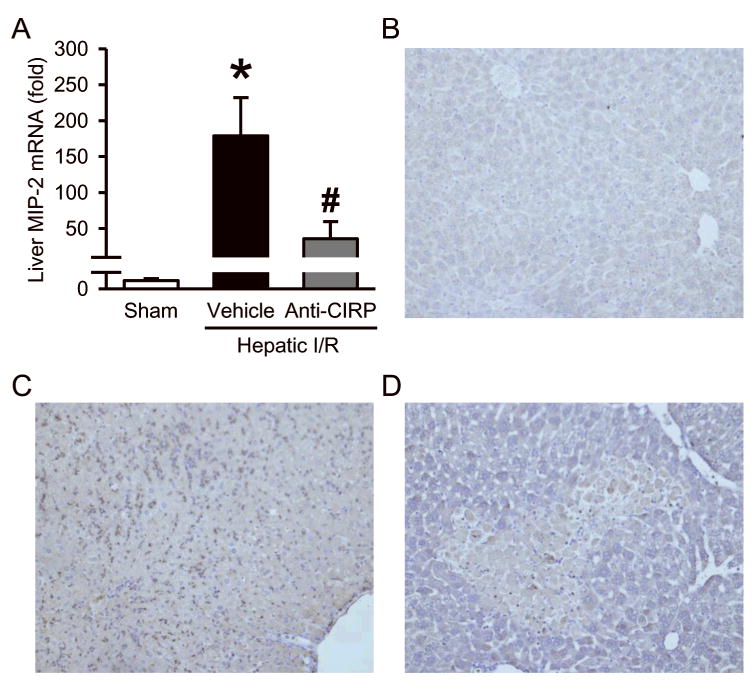
Ischemic liver tissues were collected at 24 h post-hepatic I/R from sham, vehicle and anti-CIRP antibody-treated (Anti-CIRP) groups. (A) MIP-2 mRNA expression was determined by qPCR analysis and its levels were normalized to β-actin. The value in sham group is designated as 1 for comparison. Data presented as means ± SE (n=6/group) and compared by one-way ANOVA and SNK method; *p < 0.05 vs. sham; #p < 0.05 vs. vehicle. Liver tissues were immunostained against Gr-1, a marker for neutrophil infiltration. Representative photomicrographs from sham (B), vehicle (C), and anti-CIRP antibody-treated (D, Anti-CIRP) groups at 100× magnification.
Anti-CIRP antibody treatment decreases apoptosis after hepatic I/R
To examine whether anti-CIRP antibody treatment has any effect on apoptosis after hepatic I/R, we performed TUNEL assay which detects DNA fragmentation resulting from apoptosis, in the liver tissues 24 h after hepatic I/R. There were no TUNEL-positive cells (green spots) in the sham livers (Fig. 6A), but vehicle treated animals showed very prominent TUNEL-positive areas (Fig. 6B). After anti-CIRP antibody treatment there is a significant decline in TUNEL-positive areas in comparison to vehicle treated animals (Fig 6C). Taking into account the H&E staining which showed necrosis, not apoptosis, in the majority of the hepatocellular damage, primary role of anti-CIRP antibody in hepatic I/R may not be protection from apoptosis, but reduction in apoptosis could be a secondary benefit.
Fig. 6. Effect of anti-CIRP antibody treatment on induction of apoptosis after hepatic I/R.
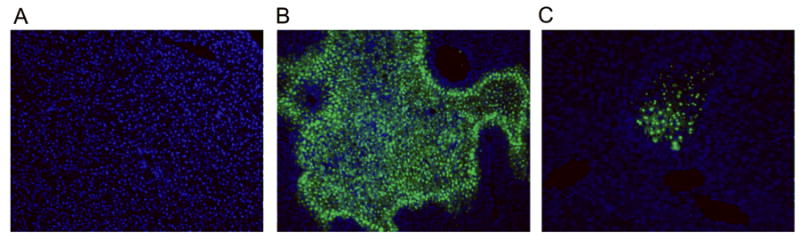
Liver tissues were harvested at 24 h after hepatic I/R and sections subjected to TUNEL assay to detect DNA fragmentation. Representative TUNEL staining (green fluorescent) and nuclear counterstaining (blue fluorescent) of liver sections from sham (A), vehicle (B), and anti-CIRP antibody-treated (C, Anti-CIRP) groups at 100× magnification.
Anti-CIRP antibody treatment reduces nitrosative stress after hepatic I/R
To determine the effect of anti-CIRP antibody treatment on the oxidative stress associated with hepatic I/R, we evaluated the liver tissue levels of iNOS and COX-2 at 24 h after hepatic I/R. We observed a 7.9-fold increase in iNOS mRNA expression in liver tissue of vehicle-treated I/R animals when compared to sham group, which was significantly reduced by 86% in anti-CIRP antibody-treated animals (Fig. 7A). COX-2 mRNA levels were also increased by 173-fold in vehicle treated group in comparison to sham, with a drastic reduction by 99% in anti-CIRP antibody-treated animals (Fig. 7B).
Fig. 7. Effect of anti-CIRP antibody treatment on iNOS and COX-2 expression after hepatic I/R.
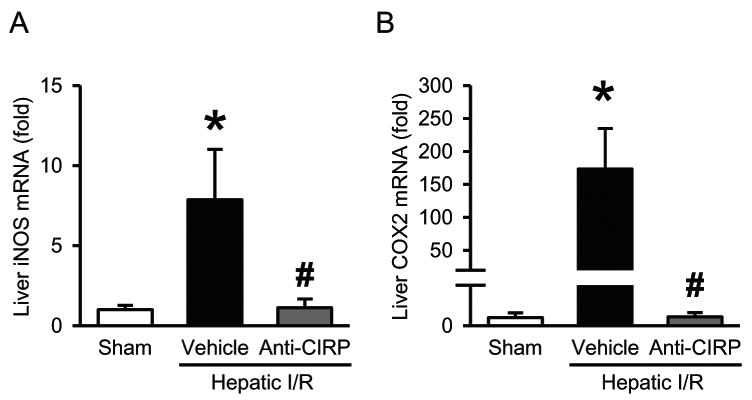
The ischemic portions of the livers were harvested at 24 h post-hepatic I/R for measuring iNOS (A) and COX-2 (B) mRNA levels by qPCR analysis. iNOS and COX-2 mRNA expression levels were normalized to β-actin. The value in sham group is designated as 1 for comparison. Data presented as means ± SE (n=6/group) and compared by one-way ANOVA and SNK method; *p < 0.05 vs. sham; #p < 0.05 vs. vehicle.
Anti-CIRP antibody treatment improves survival following a lethal model of hepatic I/R
To evaluate the potential survival benefit of neutralizing CIRP with anti-CIRP antibody, we used a total hepatic I/R model as previously described (2). We observed that only 37.5% of vehicle treated animals survived for 10 days after being subjected to hepatic I/R with subsequent partial hepatectomy of the non-ischemic portions of the liver. Whereas, the 10-day survival rate under these conditions increased significantly to 75% in the anti-CIRP antibody treated group (Fig. 8). Thus, anti-CIRP antibody treatment doubled the survival rate of mice after hepatic I/R compared to vehicle controls.
Fig. 8. Effect of anti-CIRP antibody treatment on survival in a lethal model of hepatic I/R.
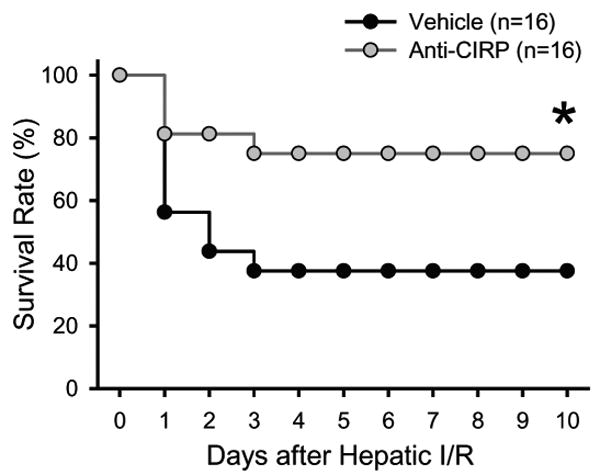
Survival rates over a 10-day period following 70% hepatic I/R with resection of the remaining non-ischemic 30% liver tissue are demonstrated for vehicle (black) and anti-CIRP antibody treatment (gray). Survival rates were analyzed by the Kaplan-Meier estimator using a log-rank test. *p < 0.05 vs. vehicle.
Discussion
CIRP is a member of the family of cold shock proteins that respond to cold stresses by functioning as RNA chaperone to promote translation giving a survival advantage (10). However, our previous study has established that CIRP can be released and acts as a DAMP to induce inflammatory responses (9, 11). We have also shown that neutralizing antisera to CIRP attenuates inflammatory responses and improves the survival of hemorrhagic and septic animals (11). The purpose of the current study is to test the hypothesis that CIRP can also act as a mediator of inflammation and injury in hepatic I/R. Indeed, we find that CIRP levels increase in the serum in a time dependent manner after hepatic I/R. In particular, we show that blockade of CIRP protects against hepatic I/R injury with a decrease in systemic and local hepatic proinflammatory cytokine expression. We also demonstrate that blockade of CIRP not only prevents the I/R associated neutrophil infiltration into the liver but also results in reduced hepatocyte apoptosis and oxidative stress. Finally, we reveal that administration of neutralizing antisera to CIRP significantly reduces the mortality rate to half in a lethal model of hepatic I/R.
Hepatic I/R injury has been shown to affect the function of multiple organ systems and cause systemic consequences (18, 19). Because hepatic I/R injury is seen in a variety of clinical settings and liver dysfunction creates such serious complications leading to possibly death, it makes this a topic for extensive research. The current understanding of the pathophysiology of hepatic I/R consists of complex interactions of multiple inflammatory pathways with the main contributors being from the production of reactive oxygen species, release of proinflammatory cytokines and chemokines, as well as the activation of immune cells that promote inflammation and tissue damage (20). The immune response generated during hepatic I/R lacks a microbial component, but instead is initiated by endogenous self proteins or DAMPs (6, 8). Although we did not examine the damage of other organs in this study, we expect there will be a systemic effect on other organs due to the elevation of CIRP in serum after hepatic I/R to induce inflammatory responses.
The extracellular delivery of DAMPs depends on their production by injured cells and the extent of the tissue injury (21). Activation of the immune system, specifically the innate immune cells by DAMPs has been under investigation for the past two decades. TLRs are pattern recognition receptors located both intra- and extra-cellularly, but differ from each other in ligand specificity, expression patterns, and in some instances signaling pathways (22-24). Recently, TLR4 has been revealed to play a pivotal role in multiple I/R models and its antagonism can attenuate the inflammatory response in I/R involving the heart, liver, and kidney (22, 25). Some DAMPs bind to the TLR4-MD2 complex on the surface of monocytes, macrophages, and neutrophils to generate a cytokine storm leading to damaging effect that is preceded by an ischemic insult (26).
CIRP has been found to be constitutively expressed in low amounts in various tissues and its expression is up regulated during cellular stress (11, 12, 27). The human CIRP gene has been mapped to chromosome locus 19p13.3 and is 95.3% identical to the amino-acid sequence to that of mouse CIRP (10). The redistribution of CIRP is not only attributed to necrosis, but it is also actively secreted from macrophages. The release of CIRP is not through the classical endoplasmic reticulum – Golgi apparatus, but through a lysosomal secretions process (11). This extracellular CIRP causes an induction of TNF-α, IL-6, and high-mobility group box 1(HMGB1) expression via engaging the TLR4-MD2 complex and activation of the NF-κB pathway (11, 27). Whether CIRP can suppress the production of anti-inflammatory cytokines needs further investigation. We believe that our study is first to report that CIRP plays a role in the generation of sterile inflammatory response in hepatic I/R. We found that serum CIRP levels increase in a time-dependent manner after hepatic I/R. Such increase is associated with the elevation of proinflammatory mediators IL-6 and MIP-2, and hepatocellular damage. These proinflammatory mediators drive neutrophils infiltrating the liver, which can exacerbate oxidative liver damages at the later stage of reperfusion (6). With administration of anti-CIRP antibody, the levels of local and systemic IL-6 as well as liver MIP-2 expression are decreased, which is associated with the reduction of neutrophil infiltration and hepatocellular damage.
In addition, anti-CIRP antibody administration also decreases the cellular oxidative stress which plays an important role in the cellular damage during I/R injury (28). Upregulation of iNOS in I/R injury causes excessive production of nitric oxide which reacts with superoxide anion (O2-), creating peroxynitrite (ONOO-). Peroxynitrite then contributes to injury through lipid peroxidation, apoptosis, necrosis, and neutrophil recruitment by nitration of tyrosine residues on tissue proteins (29). Indeed, we observe that anti-CIRP antibody administration decreases the expression of iNOS and COX-2 as well as apoptosis in the liver after I/R. We believe this to be significant as previous studies have demonstrated that apoptosis is a key contributor to injury following hepatic I/R and that therapies aimed at reducing apoptosis have proven to be hepatoprotective (30). However, the initial I/R injury caused by reactive oxygen species, leading to activation of an inflammatory cascade and resulting in an increase of both apoptosis and necrosis (29, 31). There is a varying degree in the literature debating whether the primary mode of cell death in hepatic I/R is apoptosis or necrosis, and since apoptosis is an energy depended process necrosis should be the dominate pathway (28, 29, 31, 32). In our study, we show by H&E large areas of necrosis and by TUNEL assay large areas of apoptosis in the vehicle group, which are both drastically reduced in the anti-CIRP antibody-treated group. Therefore, regardless of which pathway is more prominent we show that blocking CIRP is beneficial in both ways.
Another DAMP protein HMGB1 has also been shown to be an early mediator of sterile inflammation (33, 34). Inhibition of HMGB1 activity with neutralizing antibody was also shown to significantly decrease liver damage after I/R, whereas administration of recombinant HMGB1 was shown to worsen I/R injury (35). We have previously shown that CIRP also drives HMGB1 release in macrophages and both bind to TLR4-MD2 complex and work additively in stimulating inflammatory response (11). It will be interesting to see if blocking both CIRP and HMGB1 together can provide an additive or synergistic protective benefit after hepatic I/R injury.
By attenuating the severe liver damage, anti-CIRP antibody provides a significant survival advantage to treat animals underwent hepatic I/R. To our knowledge, this represents the first study in which blocking of CIRP has improved a survival in animals when administered during the reperfusion phase of hepatic I/R. Antibody belongs to the biologics category in drug development and has less concern for toxicity when compared to chemical compounds. Whether the delayed administration of anti-CIRP antibody such as hours or even days after hepatic I/R still has a beneficial effect for clinical application in treatment needs to be investigated further. Although administration of anti-CIRP antibody improves the survival rate to 75%, it still does not provide a complete protection for animals from hepatic I/R injury. This suggests that factors other than CIRP may contribute to the lethality. Especially, when the liver undergoes such severe I/R damage, many intracellular components can be released and serve as DAMPs. It will be a challenge to identify each DAMP's contribution and its significance in deteriorating the organ; however, this study clearly demonstrates that CIRP is one of the major mediators to cause the mortality of animals after heptic I/R injury.
In summary, CIRP is released into the circulation after hepatic I/R and acts as an endogenous proinflammatory mediator. CIRP can be effectively blocked with anti-CIRP antibody, which results in attenuating liver injury induced by I/R. It does so by decreasing the inflammatory response, infiltration of neutrophils, and iNOS-mediated oxidative damage. In addition, it provides a distinct survival advantage in a lethal model of hepatic I/R. In the light of these observations, it indicates that extracellular CIRP contributes significantly to the deterioration of liver after I/R injury and targeting CIRP may provide another strategy to manage such clinical condition.
Acknowledgments
Source of Funding: This study was supported by the National Institutes of Health (NIH) grants HL076179 and GM053008 (PW).
Abbreviations
- DAMPs
damage-associated molecular patterns
- CIRP
cold-inducible RNA-binding protein
- AST
aspartate aminotransferase
- ALT
alanine aminotransferase
- LDH
lactate dehydrogenase
- IL-6
interleukin 6
- ELISA
enzyme-linked immunosorbent assay
- H&E
hematoxylin-eosin
- MIP-2
macrophage inflammatory protein-2
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- iNOS
inducible nitric oxide synthase
- and COX-2
cyclooxygenase-2
- TLR
Toll like receptors
Footnotes
Conflicts of Interest: All authors report no financial interests or potential conflicts of interest.
This work was presented for The Shock Society New Investigator Award at the 37th Annual Conference on Shock, Charlotte, North Carolina, June 7 to 10, 2014.
References
- 1.Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Pina E, Geller DA. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res. 2008;147(1):153–159. doi: 10.1016/j.jss.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuncewitch M, Yang WL, Molmenti E, Nicastro J, Coppa GF, Wang P. Wnt agonist attenuates liver injury and improves survival after hepatic ischemia/reperfusion. Shock. 2013;39(1):3–10. doi: 10.1097/SHK.0b013e3182764fe8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kupiec-Weglinski JW, Busuttil RW. Ischemia and reperfusion injury in liver transplantation. Transplant Proc. 2005;37(4):1653–1656. doi: 10.1016/j.transproceed.2005.03.134. [DOI] [PubMed] [Google Scholar]
- 4.Clavien PA, Selzner M, Rudiger HA, Graf R, Kadry Z, Rousson V, Jochum W. A prospective randomized study in 100 consecutive patients undergoing major liver resection with versus without ischemic preconditioning. Ann Surg. 2003;238(6):843–850. doi: 10.1097/01.sla.0000098620.27623.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petrowsky H, McCormack L, Trujillo M, Selzner M, Jochum W, Clavien PA. A prospective, randomized, controlled trial comparing intermittent portal triad clamping versus ischemic preconditioning with continuous clamping for major liver resection. Ann Surg. 2006;244(6):921–928. doi: 10.1097/01.sla.0000246834.07130.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Golen RF, Reiniers MJ, Olthof PB, van Gulik TM, Heger M. Sterile inflammation in hepatic ischemia/reperfusion injury: present concepts and potential therapeutics. J Gastroenterol Hepatol. 2013;28(3):394–400. doi: 10.1111/jgh.12072. [DOI] [PubMed] [Google Scholar]
- 7.Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med. 2011;17(11):1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143(5):1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 9.Ward PA. An endogenous factor mediates shock-induced injury. Nat Med. 2013;19(11):1368–1369. doi: 10.1038/nm.3387. [DOI] [PubMed] [Google Scholar]
- 10.Nishiyama H, Higashitsuji H, Yokoi H, Itoh K, Danno S, Matsuda T, Fujita J. Cloning and characterization of human CIRP (cold-inducible RNA-binding protein) cDNA and chromosomal assignment of the gene. Gene. 1997;204(1-2):115–120. doi: 10.1016/s0378-1119(97)00530-1. [DOI] [PubMed] [Google Scholar]
- 11.Qiang X, Yang WL, Wu R, Zhou M, Jacob A, Dong W, Kuncewitch M, Ji Y, Yang H, Wang H, Fujita J, Nicastro J, Coppa GF, Tracey KJ, Wang P. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med. 2013;19(11):1489–1495. doi: 10.1038/nm.3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou M, Yang WL, Ji Y, Qiang X, Wang P. Cold-inducible RNA-binding protein mediates neuroinflammation in cerebral ischemia. Biochim Biophys Acta. 2014;1840(7):2253–2261. doi: 10.1016/j.bbagen.2014.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajayer SR, Jacob A, Yang WL, Zhou M, Chaung W, Wang P. Cold-inducible RNA-binding protein is an important mediator of alcohol-induced brain inflammation. PLoS One. 2013;8(11):e79430. doi: 10.1371/journal.pone.0079430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuda A, Jacob A, Wu R, Zhou M, Aziz M, Wang P. Milk fat globule--EGF factor VIII ameliorates liver injury after hepatic ischemia-reperfusion. J Surg Res. 2013;180(1):e37–46. doi: 10.1016/j.jss.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heijnen BH, Straatsburg IH, Gouma DJ, van Gulik TM. Decrease in core liver temperature with 10 degrees C by in situ hypothermic perfusion under total hepatic vascular exclusion reduces liver ischemia and reperfusion injury during partial hepatectomy in pigs. Surgery. 2003;134(5):806–817. doi: 10.1016/s0039-6060(03)00125-9. [DOI] [PubMed] [Google Scholar]
- 16.Desmet VJ, Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, Kiernan TW, Wollman J. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. J Hepatol. 2003;38(4):382–386. doi: 10.1016/s0168-8278(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 17.Behrends M, Martinez-Palli G, Niemann CU, Cohen S, Ramachandran R, Hirose R. Acute hyperglycemia worsens hepatic ischemia/reperfusion injury in rats. J Gastrointest Surg. 2010;14(3):528–535. doi: 10.1007/s11605-009-1112-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nastos C, Kalimeris K, Papoutsidakis N, Tasoulis MK, Lykoudis PM, Theodoraki K, Nastou D, Smyrniotis V, Arkadopoulos N. Global Consequences of Liver Ischemia/Reperfusion Injury. Oxid Med Cell Longev. 2014;2014:906965. doi: 10.1155/2014/906965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. J Hepatol. 2012;57(3):692–694. doi: 10.1016/j.jhep.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 20.Pahlavan PS, Feldmann RE, Jr, Zavos C, Kountouras J. Prometheus' challenge: molecular, cellular and systemic aspects of liver regeneration. J Surg Res. 2006;134(2):238–251. doi: 10.1016/j.jss.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 21.van Golen RF, van Gulik TM, Heger M. The sterile immune response during hepatic ischemia/reperfusion. Cytokine Growth Factor Rev. 2012;23(3):69–84. doi: 10.1016/j.cytogfr.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR, Billiar TR. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204(12):2913–2923. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Neill LAJ, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. 2013;13(6):453–460. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- 24.Beutler BA. TLRs and innate immunity. Blood. 2009;113(7):1399–1407. doi: 10.1182/blood-2008-07-019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howell J, Gow P, Angus P, Visvanathan K. Role of toll-like receptors in liver transplantation. Liver Transpl. 2014;20(3):270–280. doi: 10.1002/lt.23793. [DOI] [PubMed] [Google Scholar]
- 26.Foell D, Wittkowski H, Roth J. Mechanisms of disease: a ‘DAMP’ view of inflammatory arthritis. Nat Clin Pract Rheumatol. 2007;3(7):382–390. doi: 10.1038/ncprheum0531. [DOI] [PubMed] [Google Scholar]
- 27.Brochu C, Cabrita MA, Melanson BD, Hamill JD, Lau R, Pratt MA, McKay BC. NF-kappaB-dependent role for cold-inducible RNA binding protein in regulating interleukin 1beta. PLoS One. 2013;8(2):e57426. doi: 10.1371/journal.pone.0057426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varadarajan R, Golden-Mason L, Young L, McLoughlin P, Nolan N, McEntee G, Traynor O, Geoghegan J, Hegarty JE, O'Farrelly C. Nitric oxide in early ischaemia reperfusion injury during human orthotopic liver transplantation. Transplantation. 2004;78(2):250–256. doi: 10.1097/01.tp.0000128188.45553.8c. [DOI] [PubMed] [Google Scholar]
- 29.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87(1):315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin FS, Shen SQ, Chen ZB, Yan RC. 17beta-estradiol attenuates reduced-size hepatic ischemia/reperfusion injury by inhibition apoptosis via mitochondrial pathway in rats. Shock. 2012;37(2):183–190. doi: 10.1097/SHK.0b013e31823f1918. [DOI] [PubMed] [Google Scholar]
- 31.Datta G, Fuller BJ, Davidson BR. Molecular mechanisms of liver ischemia reperfusion injury: insights from transgenic knockout models. World J Gastroenterol. 2013;19(11):1683–1698. doi: 10.3748/wjg.v19.i11.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klune JR, Tsung A. Molecular biology of liver ischemia/reperfusion injury: established mechanisms and recent advancements. Surg Clin North Am. 2010;90(4):665–677. doi: 10.1016/j.suc.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Lu B, Wang C, Wang M, Li W, Chen F, Tracey KJ, Wang H. Molecular mechanism and therapeutic modulation of high mobility group box 1 release and action: an updated review. Expert Rev Clin Immunol. 2014;10(6):713–727. doi: 10.1586/1744666X.2014.909730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201(7):1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]