

Abstract
Background
The spontaneously hypertensive rat (SHR) strain exists in lines that contrast strongly in susceptibility to renal injury in hypertension. These inbred lines share common ancestry and only 13% of their genomes arise from different ancestors.
Methods and Results
We used next gen sequencing to detect natural allelic variation in 5 genes of the immunoreceptor signaling pathway (IgH, Dok3, Src, Syk and JunD) that arise from different ancestors in the injury-prone SHR-A3 and the resistant SHR-B2 lines. We created an intercross between these lines and in the F2 progeny we observed that the inheritance of haplotype blocks containing the SHR-A3 alleles of these 5 genes correlated with increased albuminuria and histological measures of renal injury. To test whether accumulated genetic variation in this pathway may create a therapeutic target in hypertensive renal injury, rats of both lines were treated with the immunosuppressant mycophenolate mofetil (MMF). MMF reduced proteinuria (albumin to creatinine ratio, uACR) from 6.6 to 1.2 mg/mg (p<0.001) in SHR-A3. Glomerular injury scores were reduced in MMF treated SHR-A3 from 1.6 to 1.4 (p<0.002). Tubulo-interstitial injury was reduced in MMF-treated SHR-A3 from 2.62 to 2.0 (p=0.001). MMF treatment also reduced renal fibrosis in SHR-A3, (3.9 vs. 2.0, p<0.001).
Conclusions
Polygenic susceptibility to renal injury in hypertension arises in association with genetic variation in genes that participate in immune responses and is dramatically improved by reduction of immune system activity.
Keywords: hypertension, kidney, renal disease, genetic hypertension, genome
Introduction
Chronic progressive kidney disease (CKD) risk is strongly predicted by CKD occurrence in family members. Among incident dialysis patients, 23% have a relative with ESRD 1. Essential hypertension (EH) is a major correlate of CKD, but does not predict renal disease risk as well as the occurrence of an affected relative. Rather hypertension appears to play a permissive role on which other factors determine disease risk. The heritable risk of CKD is well replicated in a common rat model of hypertension, the spontaneously hypertensive rat (SHR). Distinct inbred SHR lines sharing genetic susceptibility to hypertension have fixed different alleles across only 13% of their genome, yet strongly differ in susceptibility to CKD 2. Identification of the genes and biological pathways that participate in hypertension-associated CKD has been an important motivator of genome-wide genetic association studies in large human populations. However, these studies have uncovered genetic variation associated with only minor effects on renal function, leaving most of the heritable risk of CKD unexplained 3 and providing little insight into disease mechanism. One exception relates to allelic variation in African-Americans suggesting an important role of genetic evolution of the host immune response to pathogens in the origin of susceptibility to CKD in hypertension 4.
When systemic blood pressure is elevated, auto-regulation of renal capillary perfusion can break down 5. This may disturb the relationship between renal perfusion and local metabolic demand and initiate danger signals that in turn are activators of inflammation and immunity 6, 7. In SHR lines differing in CKD risk, injury susceptibility may arise within the genes participating in immune responses triggered by disturbed vascular auto-regulation. We have recently reported extensive functional natural allelic variation affecting the immunoglobulin heavy chain locus in this rat model of hypertensive renal disease 2, 8. This variation results in functional alteration to IgG sequence that is associated with severity of albuminuria. In addition to antigen recognition, IgG is a signaling ligand that drives downstream immune responses as a result of Fc receptor binding. Additional genetic variation in the Fc receptor signaling pathway may add to injury effects initiated by immunoglobulin.
In the present paper we identify additional genes in the immunoglobulin-signaling pathway that arise from different ancestors across injury-prone and resistant lines and show that they contain functional regulatory and structural genetic variation across the SHR lines. By examining the inheritance in SHR-A3 x SHR-B2 intercross F2 progeny of the haplotype blocks containing these immunoreceptor signaling alleles, substantial differences in renal injury susceptibility can be explained, supporting the conclusion that additive immune system genetic variation can be a key determinant of the renal injury response to hypertension. The hypothesis that heritable differences in immune signaling influence hypertensive renal injury in this genetic model was tested by comparing the response of renal injury-resistant and susceptible SHR lines to treatment with the immunosuppressant, mycophenolate mofetil (MMF).
Methods
Animals and treatments
Studies were performed on male rats of the stroke-prone spontaneously hypertensive-A3 (SHR-A3, SHRSP/Bbb) line and the injury resistant SHR-B2 line. The origin of these inbred lines has been previously reported 2, 9. Animals were housed in an AAALAC-approved animal facility and provided a standard rodent chow diet and drinking water ad libitum. No stroke-enhancing dietary manipulations were employed 10. The two lines are derived from a single pair of founders common to both lines 11. The ~87% of the genome shared identical by descent (IBD) across these lines is partitioned into discrete haplotype blocks 9. SHR-A3 (males) and SHR-B2 (females) were crossed to generate an F1 progeny. The F1 progeny was further crossed to generate a freely-segregating F2 progeny (n=210), as previously reported 9. All animal use was prospectively reviewed and approved by the University’s Animal Welfare Committee.
Genome sequencing and bioinformatic analysis
Genomic DNA was extracted from samples of liver tissue obtained from male representatives of SHR-A3 and SHR-B2 (n = 1 per SHR line) and quantified by UV spectroscopy. DNA samples were submitted to a commercial genome sequencing company (Axeq Technologies, Rockville, MD, 20850, United States) for sequencing. Sequencing was performed using the Illumina HiSeq 2500 platform. An average of 1.37 billion reads of 100 bp were obtained from each sample providing an average genome coverage of approximately 45X. We mapped the paired-end reads to the rat assembly (RGSC3.4, Ensembl release 69) using Novoalign software (Novocraft.com, version V2.08.01 using default settings 12. After generating BAM files, the mapped reads were sorted by coordinate location using SamTools 13 and Picard tools 14. Finally, we used the Genome Analysis Tool Kit (GATK version v2.3-9-ge5ebf34) 15 to perform local realignment, recalibration and variant calling. We removed false positive calls by using a post-calling filter that enforces that each variant has a mapping quality >30, a base quality >20, and a coverage ≥10, with at least a 3:7 ratio of variant to reference and the presence of the variant in reads from both orientations. The resulting variant call format files (vcf version 4.1) were annotated using Annovar software 16 together with the rat genome annotations (RGSC3.4.69). Finally to identify location of haplotype blocks, we visually inspected the paired-end read data in the integrative genomics viewer (IGV) 17.
Blood pressure measurement
Blood pressure was measured by implanted telemetry (Data Sciences, St. Paul, MN) as previously reported 9. Animals were implanted at 16 weeks of age and allowed to recover for 1 week before blood pressure recordings began. Blood pressure was measured by continuous sampling for 30 seconds every 30 minutes for 24 hours. Blood pressure recording began 3–4 days after the initiation of drug/vehicle treatment and was repeated one day per week throughout the study.
Mycophenolate mofetil (MMF) study
After implantation of blood pressure telemetry devices and a 1 week recovery period, singly-housed male SHR-A3 and SHR-B2 were divided into two groups per line (n = 8–10 per group): one group received 25mg/kg/day MMF (CellCept, Genetech) as a single dose by gavage, while the other received a similar volume of water by gavage. Treatment commenced at 17 weeks of age, before emergence of histological renal injury 2, was administered 6 days per week and continued for 8 weeks. Animals were weighed once per week to allow adjustment of dosage as animals grew.
Histological assessment of renal injury
At the conclusion of the drug/vehicle treatment period, kidneys were harvested from isoflurane-anesthetized rats by ventral laparotomy. Kidneys were cut into radial segments by hand, fixed in 4% buffered formalin and embedded in paraffin using standard techniques. Five micron serial sections were stained with Periodic-Acid-Schiff’s stain (PAS) for assessment of renal injury, and Picro-sirius red to evaluate the extent of collagen accumulation as previously described 2.
Lymphocyte infiltration
We assessed kidney lymphocyte infiltration using immunohistochemistry of paraffin-embedded kidney blocks from control and MMF treated animals of both rat lines. An antibody against the lymphocyte cell surface protein sialophorin (anti-CD43, Clone: W3/13, ThermoFisher Scientific, Waltham, MA USA) was used to detect lymphocytes; this antibody detects T lymphocytes, granulocytes, monocytes and some B lymphocytes 18. Lymphocyte infiltration was assessed by counting CD43 positive cells present in 15 high power (x400) cortical fields per sample.
Albuminuria
We assessed albuminuria as urinary albumin:creatinine ratio (uACR). Urinary creatinine concentrations were determined by HPLC in urine samples collected by direct puncture of the urinary bladder. Urine was collected during implantation of the telemetry device and again at the conclusion of the study 19. Urine albumin was measured by an ELISA specific for rat serum albumin (Bethyl Labs, Montgomery, TX).
Statistical analysis
Normality testing of parametric data was performed using the Kolmogorov-Smirnov test as implemented in the package StatPlus (AnalystSoft, Inc., Vancouver, BC, Canada.) No data transformation was used. Comparison of data obtained from treated and untreated SHR lines was analyzed by ANOVA in the same software. Non-parametric data was analyzed by Kruskal-Wallis test after which significance testing of multiple groups was performed by Mann-Whitney U test with p values corrected for multiple testing using the Bonferroni correction. The effect of allelic variation in multiple immune signaling genes on renal injury was examined in F2 animals using the same package by linear regression of renal injury scores on the number of SHR-A3 alleles of these genes present per F2 individual. Trait effect size of parental alleles in the F2 progeny was estimated from the regression equation.
Results
We have reported extensive functional sequence variation in the Fc region of the immunoglobulin gamma heavy chains of SHR-A3 and its association with renal injury 8. Our analysis of the whole genome sequences for the two SHR lines reveals that the IgH locus contains extensive variation between the two SHR lines, for SHR-B2 this is the most divergent region of the entire genome 20. Variation at the IgH locus results in highly divergent serum levels of three of the four rat IgG subclasses and total serum IgG levels in SHR-A3. IgG can drive inflammation by binding to and activating Fc receptors. Others have reported that genetic variation in the locus containing the immunoglobulin receptor gene, FCGR3, strongly influences susceptibility to renal immune disease 21. These findings suggest that the Ig-Fc receptor signaling pathway may be relevant to hypertensive renal disease in the SHR model in which extensive renal infiltration with immune cells occurs 2.
We sought to identify which genes in the Ig-Fc immunoreceptor signaling pathway might contain functional variation between SHR-A3 and SHR-B2, The FCGR3 gene lies in the 87% of the genome shared identical between SHR-A3 and SHR-B2. Targeted re-sequencing to detect copy number variation 21 led us to conclude that this locus does not differ across our SHR lines (data not shown). Pro-inflammatory Fc immunoreceptors are coupled to the protein tyrosine kinase, Syk. Coupling between Fc receptors and Syk occurs after phosphorylation of the ITAM (immunoreceptor tyrosine activation motif) domains of the Fc receptor by Src kinases 22. A scaffold protein, Dok3, is involved in spatial coordination of Src signaling 23. Important outputs of this signaling pathway include the proliferation of immune cells and increased reactive radical production 22. Both of these responses involve the transcription factor JunD 24–26. Each of these immunoreceptor signaling genes (Src kinase, Dok3, Syk, JunD) lies in the small part of the genome that has arisen from different ancestors in SHR-A3 and SHR-B2 9. Our analysis of the genome sequences of the two SHR lines in these regions is presented in Supplemental Table 1. We defined the haplotype blocks containing each of these genes and used analytical methods to determine which genes in these blocks contain amino acid variation across the two SHR lines. In aggregate, these 4 blocks containing the 5 immunoreceptor signaling genes comprise 28Mb of the rat genome, or approximately 0.1% of the entire genome 27. Among all genes located in these blocks that contain amino acid substitutions across the two SHR lines, there is no clear biological or functional relationship that suggests a role in progressive renal disease. We further analyzed the immunoreceptor genes in these blocks to catalog both non-synonymous and other potentially functional variation. Variation in the immunoglobulin heavy chain gene is very extensive and the complex, segmentally duplicated nature of this gene prevents a complete description of that variation here. In another manuscript we have completed an analysis of the variable, diversity, junctional and Fc gene segments of the IgH locus and the extensive divergence of the pre-immune immunoglobulin repertoire in these two rat lines 20. Our findings for the other loci are summarized below and detailed in the Data Supplement.
Src kinase (chr 3)
This gene is bi-allelic across the two SHR lines and contains 12 SNP variations. The SHR-A3 has a substitution in the conserved −3 position of the Kozak sequence. The substitution has been assessed in vitro and found to alter protein translation efficiency 28.
JunD (chr 16)
We re-sequenced the JunD promoter and its single coding exon in SHR-A3 and SHR-B2 and identified extensive variation in the promoter. Altered JunD expression has been previously reported in rats in association with a promoter SNP 29. SHR-B2 was found to share this promoter variant, while SHR-A3 possessed the genotype associated with reduced expression. We examined our previously reported Affymetrix gene expression array data from the kidneys of SHR-A3 and SHR-B2 30 and observed abundant expression of JunD in kidney RNA from SHR-B2. In contrast, JunD expression in SHR-A3 was not detected. In F2 animals in which free segregation of these two JunD alleles can occur, the level of JunD gene expression assessed by Affymetrix array correlated strongly with the inheritance of 2, 1, or 0 SHR-A3 JunD alleles (see Supplementary Data, Figure 1).
Dok3 (chr 17)
Two non-synonymous amino acid substitutions were detected in SHR-A3 (Ile71Val and Ala83Val). These substitutions are located in the pleckstrin homology domain of Dok3. Dok3 is an adapter molecule that recruits molecules to the immunoreceptor signaling complex 23. The Ile71Val substitution replaces an isoleucine that occurs uncommonly at this position in mammalian Dok3 with a valine that is the most common mammalian residue at this location. The alanine/valine substitution at position 83 replaces a highly conserved residue among higher vertebrates, suggesting potential deleterious consequences of substitution in SHR-A3.
Syk (chr 17)
Syk is bi-allelic across SHR-A3 and SHR-B2. Most genetic variation occurs in introns and no non-synonymous variation was detected. However, sequence variation was detected in the proximal promoter of the gene that may influence transcriptional control of expression.
Verification of allelic variation in other SHR lines
The selective breeding of distinct SHR lines has led to several injury-resistant SHR lines 31 that are not genetically identical. We examined genomic sequence variation (Rat Genome Database 32) from 2 additional injury-resistant SHR lines and one additional SHR-A3 line to determine whether sequence variation identified in the immunoreceptor signaling pathway was consistently observed in other lines that share the same end organ injury phenotype (see Data Supplement). We found that injury-resistant SHR lines share the same haplotype IgG, Dok3, Syk and JunD, while the two injury-prone SHR-A3 lines share the contrasting haplotype of these genes. For Src, the majority of variants observed also fell into dichotomous haplotypes distinguishing injury-prone from injury-resistant SHR lines, (see Data Supplement).
Association of immunoreceptor gene variation with renal injury traits in the F2 progeny of an SHR-A3 x SHR-B2 intercross
We hypothesized that the accumulation of allelic variation in the immunoreceptor signaling pathway may cause differences in hypertensive renal injury observed in SHR-A3 compared with SHR-B2. To test this hypothesis we examined the relationship in the F2 progeny of an SHR-A3 x SHR-B2 intercross, between renal injury and inheritance of the 4 haplotype blocks containing SHR-A3 alleles of IgH (chr6), Src (chr3), Dok3 and Syk (chr17) and JunD (chr16). We used SNP genotypes at the following positions to represent inheritance of these blocks: chr3:146,943,779; chr6:143,652,058; chr16:18,825,153; and chr17: 14,963,716 (Rn4). Table 1 contrasts the phenotypic values observed for distinct renal injury phenotypes in SHR-A3 and SHR-B2 animals at 25 weeks of age. The table also shows what portion of the phenotypic differences between these lines was explained by accumulation of SHR-A3 alleles at these four blocks in the F2 progeny. For fibrosis and albuminuria two thirds of the trait difference could be attributed to SHR-A3 alleles at these four loci. While about half of the parental line trait difference for glomerular and tubulo-interstitial injury was explained. These findings suggest that accumulation of genetic variation in loci encoding genes of the immunoglobulin signaling pathway can influence susceptibility to hypertensive renal disease in this rat model.
Table 1.
Relationship in the F2 progeny of an SHR-A3 x SHR-B2 cross between inheritance of SHR-A3 marker alleles at 4 proposed renal injury loci and renal injury measures. Injury scores are presented for each of the parental lines, along with the difference between the lines (Delta A3 vs B2). We were interested to know, in F2 animals, whether a relationship existed between inheritance of SHR-A3 alleles at the 4 proposed renal injury loci and observed renal injury and how much of the parental injury Delta might be accounted for by such inheritance. The relationship between inheritance of SHR-A3 alleles and renal injury scores was observed by linear regression of scores for each injury measure and the number of SHR-A3 injury loci alleles inherited in each of 210 F2 animals. The injury measure in F2 animals that inherit from 0 of 8 or 8 of 8 SHR-A3 haplotype block marker alleles was estimated from the regression equation. This estimation did not require extrapolation beyond the actual data obtained (i.e. F2 animals included individuals inheriting as many as 8 SHR-A3 marker alleles and as few as 0). The combined effect size of these loci on injury is shown as the delta between the estimated injury score of F2 animals inheriting 0 or 8 SHR-A3 alleles. The percentage of parental strain renal injury difference that can be attributed inheritance of 0 or 8 SHR-A3 alleles in F2 animals is shown. The p values provided indicate the significance of the regressions obtained by ANOVA. uACR indicates urinary albumin to creatinine ratio, TI indicates tubulo-interstitial, Glom indicates glomerular.
| Parental lines | F2 animals | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Injury Trait | SHR-A3 | SHR-B2 | Delta (A3 vs B2) | A3 alleles 8/8 | A3 alleles 0/8 | Delta (8 vs 0 alleles) | % parental difference explained | p value |
| uACR (mg/mg) | 21.94 | 4.86 | 17.08 | 14.28 | 3.03 | 11.25 | 65.9 | 0.0124 |
| Fibrosis score | 3.65 | 2.5 | 1.15 | 3.14 | 2.36 | 0.78 | 67.8 | 0.0016 |
| TI injury score | 2.62 | 1.44 | 1.18 | 2.02 | 1.32 | 0.69 | 58.9 | 0.0076 |
| Glom injury score | 1.65 | 1.26 | 0.38 | 1.4 | 1.24 | 0.16 | 41.9 | 0.2608 |
Effect of immunosuppression on renal injury and lymphocyte infiltration in SHR-A3 and SHR-B2
Albuminuria
Treatment for 8 weeks with MMF of SHR-A3 animals resulted in strong suppression of albuminuria compared with untreated SHR-A3 animals (Figure 1). In contrast, MMF had no effect on urine albumin-creatinine ratios in injury-resistant SHR-B2 animals, which increased only slightly during the treatment interval in both the MMF-treated and control animals.
Figure 1.
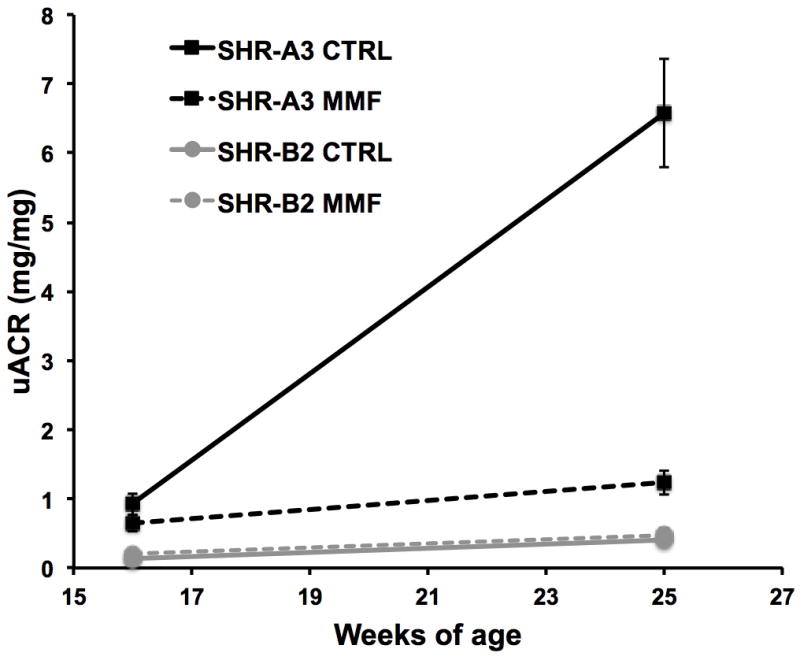
Urine samples were collected during implantation of blood pressure telemetry devices at 16 weeks of age and at the conclusion of the study. In untreated SHR-A3, albuminuria had become severe at 25 weeks of age. However, in SHR-A3, MMF treatment dramatically reduced albuminuria (broken black line) (p<0.001). In injury-resistant SHR-B2, the age-related increase in albuminuria is much less and is unaffected by MMF treatment (gray lines). Group sizes: SHR-A3 Control 16 and 25 weeks old, n = 10 and 8 respectively, SHR-B2 Control 16 and 25 weeks old, n = 8, SHR-A3 MMF treated 16 and 25 weeks old, n = 9, SHR-B2 MMF treated 16 and 25 weeks old, n = 9 and 8 respectively.
Histologically assessed injury
Similar effects of MMF to reduce renal injury in SHR-A3 were observed when kidney tissue harvested at the completion of the study was examined histologically. Tubulo-interstitial renal injury is prominent in the control SHR-A3 animals at 25 weeks of age. However, injury scores in MMF-treated SHR-A3 animals were not different than in SHR-B2 animals (Figure 2). MMF treatment had no discernable effect on tubulo-interstitial renal injury in SHR-B2 animals. Compared with tubulo-interstitial injury, glomerular damage is less extensive in SHR-A3 2. MMF treatment was associated with a significant reduction in glomerular damage in the SHR-A3 line (Figure 3). No effect on glomerular injury scores was observed as a result of MMF treatment in the SHR-B2 line. Examination of renal fibrosis using Picro-sirius red staining indicated that MMF treatment significantly reduced fibrosis in SHR-A3, but was without effect on the already low levels of fibrosis observed in SHR-B2 (Figure 4). In order to demonstrate that MMF altered immune cell infiltration into the kidney we quantitated CD43-immunreactive cells in paraffin-embedded sections of renal cortex. MMF treatment was associated with strongly reduced lymphocyte infiltration in SHR-A3. Lymphocyte infiltration in SHR-B2 was sparse and was not affected by MMF (Figure 5 and Supplemental Figure 2).
Figure 2.

Tubulo-interstitial injury was assessed on a 1–5 scale as described 2 in periodic acid-Schiff’s stained sections of kidney at the conclusion of the treatment period. Injury was more severe in untreated SHR-A3 than in SHR-B2. Treatment with MMF was associated with a marked reduction in tubulo-interstitial injury in SHR-A3 (p<0.001), but had no effect in SHR-B2. Group sizes: SHR-A3 control n = 10, SHR-A3 MMF treated n = 9, SHR-B2 control n = 9, SHR-B2 MMF treated n = 8.
Figure 3.

Glomerular injury was assessed on a 1–5 scale as described 2 in periodic acid-Schiff’s stained sections of kidney at the conclusion of the MMF treatment period. Injury was more severe in untreated SHR-A3 than in SHR-B2. Treatment with MMF reduced glomerular injury in SHR-A3 (p<0.002), but had no effect in SHR-B2. Group sizes as in Fig. 2.
Figure 4.

Renal fibrosis was assessed on a 1–5 scale as described 2 in Picro-sirius red stained sections of kidney at the conclusion of the treatment period. Fibrosis was more severe in untreated SHR-A3 than in SHR-B2. Treatment with MMF was associated with a marked reduction in tubulo-interstitial injury in SHR-A3 (p<0.001), but had no effect in SHR-B2. Group sizes: SHR-A3 control n = 10, SHR-A3 MMF treated n = 10, SHR-B2 control n = 8, SHR-B2 MMF treated n = 11.
Figure 5.

Renal lymphocyte infiltration was assessed by immunohistochemistry using a rat anti-CD43 antibody. Lymphocyte infiltration was extensive in untreated 25 week old SHR-A3, and was minimal in untreated SHR-B2. Treatment of SHR-A3 rats with MMF was associated with significant reductions in lymphocyte infiltration to levels indistinguishable from that in untreated SHR-B2 rats (p<0.001). Group sizes: SHR-A3 control n = 10, SHR-A3 MMF treated n = 6, SHR-B2 control n = 8, SHR-B2 MMF treated n = 8.
Blood pressure effects
We began our treatment with MMF at a time when renal injury is beginning to emerge in SHR-A3 2. Progression of renal injury during the following weeks may result in additional increase in blood pressure as renal inflammation increases and function declines. We hypothesized that, if immune system genetic variation contributes to increasing blood pressure during the emergence of renal injury, then treatment with MMF would reduce the increase in blood pressure during the period when renal injury progresses in SHR-A3. We observed that in vehicle-treated SHR-A3 animals, blood pressure continued to rise over the 8 week study interval (Figure 6). Treatment of SHR-A3 with MMF prevented elevation of blood pressure in SHR-A3 during the period from 18–25 weeks when renal injury develops in untreated animals 2. Blood pressure in 17–18 week old SHR-B2 animals is lower than that in SHR-A3 9. In SHR-B2, treatment with MMF produced no consistent significant effect on blood pressure in SHR-B2. Thus, immunosuppression in SHR-A3, during the period when renal injury is developing was associated with both reduced injury and a suppression of the progressive elevation in blood pressure seen in untreated SHR-A3 animals during this period.
Figure 6.

Blood pressure was recorded throughout the treatment period, starting 2–4 days after gavage with MMF or water. BP increased progressively in untreated SHR-A3, while MMF-treated SHR-A3 maintained BP. Significant differences in BP in treated versus untreated SHR-A3 animals emerged after 2 weeks of treatment and were sustained for the rest of the study. BP in MMF-treated SHR-A3 animals was lower than in untreated SHR-A3, but remained higher than in SHR-B2 animals. In MMF-treated SHR-B2 animals, only small changes in blood pressure were noted that were significant only at 4 weeks of treatment (NS = not significant, * = p<0.05. ** = p<0.01. *** = p< 0001). Group sizes: SHR-A3 control n = 10, SHR-A3 MMF treated n = 10, SHR-B2 control n = 8, SHR-B2 MMF treated n = 9. Polynomial regression (degree 2) analysis of the blood pressure trends over time was performed using R. Coefficient values and their significance are included as supplemental data.
Discussion
Susceptibility to hypertensive renal disease in both human populations and animal models of disease indicates the role of risk factors that are separate from those producing hypertension 2, 33, 34. The heritability of risk implies that studies to identify genetic variation associated with susceptibility may shed light on disease mechanism. However, with the exception of a locus that has major effects on disease risk in African Americans 35, 36, little progress has been made in defining the elements of genetic risk. This presumably reflects a complex genetic etiology including a polygenic inheritance so that studies of even large human populations have yielded little substantial insight. The missing heritability in genome-wide association studies of human populations may also reflect uneven sampling of genetic variation. This is a substantial problem at the highly divergent immunoglobulin heavy chain locus 37.
Hypertensive renal injury susceptibility in SHR-A3 is also a polygenic trait and the power of quantitative trait mapping to define loci involved is constrained by a genetic architecture that may involve multiple variant genes and functional interactions among these genes 2. Our hypothesis that hypertensive renal injury may reflect genetic variation in immune mechanisms arises from our prior work showing that injury-prone SHR-A3 have extensive immune cell infiltration in the kidney and foam cells in the glomeruli 2. It is further substantiated by the presence of extensive functional genetic variation, compared to SHR-B2, in the immunoglobulin heavy chain 8. This variation in the IgH locus alters the pre-immune immunoglobulin repertoire (the germ-line encoded combinations of VDJ gene segments that provide initial interaction with antigen in the B cell lineage) 20. IgH locus variation in SHR-A3 compared to SHR-B2 also determines large differences in serum immunoglobulin levels (and therefore Fc receptor ligand concentrations) and disrupts some IgG-Fc receptor interactions 8. The immunoreceptor signaling pathway is enriched with genetic variation that is much greater than expected by chance alone. Overall only ~1 in 8 genes arise from different ancestors across the two SHR lines we studied. The finding that 5 of the genes in this pathway arise from different ancestors is unexpected and may reflect the effect of trait selection during the fixation of hypertensive end-organ injury susceptibility in SHR-A3 31.
In the present study we have proposed that genetic variation in additional genes of the immunoglobulin signaling pathway links the injury initiated by hypertension to severity of renal damage. Fc receptor variation in other rat strains involving the presence or absence genomic sequence encoding a decoy Fc gamma receptor illustrates how genetic variation can modulate immunoreceptor signaling to affect renal disease risk 38. These observations prompted our investigation to uncover further genetic variation in this pathway as a mechanism influencing renal injury in hypertension. Sequence analysis of these genes has uncovered functional genetic variation in them that may contribute to differences in renal disease susceptibility in SHR lines. We have used both targeted re-sequencing of genes in the immunoglobulin signaling pathway as well as whole genome sequencing of both SHR lines. The former was performed in multiple animals of each SHR line, while the latter used DNA from one representative animal of each line. Such whole genome sequencing is not error free, but we have been able to confirm variation in SHR sequenced by other laboratories. The accumulation of functional genetic variation in the pathway suggested to us that immune signaling may be a specific therapeutic target by which hypertensive renal disease could be modulated. The capacity of MMF to markedly suppress distinct elements of renal disease in the injury-prone SHR line supports the involvement of natural allelic variants involved in immune signaling on loss of renal function and progression of tissue damage in hypertension.
MMF prevented renal injury-associated elevation of blood pressure in injury-prone SHR-A3, but was without consistent effects on blood pressure in injury-resistant SHR-B2. MMF is not a blood pressure lowering drug: it does not lower blood pressure in hypertensive SHR-B2 animals and clinical reports linking this drug to lower blood pressure occur only when immune-mediated renal disease co-exists. The capacity of MMF to prevent the progressive elevation of blood pressure as renal injury emerges in SHR-A3 can be reasonably attributed to its immunosuppressive effect and the reduction in progression of renal injury that occurs when MMF is administered.
Early evidence of the role of immunity in hypertensive kidney injury 39 has been extended by the introduction of knockout models, modern immunosuppressant drugs and growing evidence of interactions between angiotensin II, hypertension and immune function 40–47. The observations we report in the present study focus attention specifically on the role of B cell mediated immunity. However, MMF-mediated immunosuppression may also suppress immune mechanisms involving T cells, macrophages and possibly other cellular components of the immune system. Several of the genes we have investigated are known to also function in other components of the cellular immune system and genetic variation has been associated with altered immune cell function 29. Finally, our F2 intercross studies suggest that a large fraction, but not all, of the heritable risk of renal injury is attributable to loci containing the genes we have identified, but disease risk unaccounted in our studies may lie outside these genomic regions.
In conclusion, we report here that functional genetic variation is enriched in the immunoreceptor signaling pathway of an injury-prone SHR line beyond that expected by chance. Examination of the inheritance of this variation, by tracking the transmission of the loci containing the variation in an F2 intercross, provides support that this variation is associated with susceptibility to renal injury. Finally, the potential causative role of these genes is supported by the effect of immunosuppression with MMF to reduce renal injury.
Supplementary Material
Acknowledgments
Funding Sources: The work reported has been supported by grants from NIH (R01DK069632 and R01DK081866) to PAD. PAD is grateful for additional endowed support from the Cullen Chair in Molecular Medicine at the University of Texas HSC Houston, which provided support for rat genome sequencing.
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Freedman BI, Volkova NV, Satko SG, Krisher J, Jurkovitz C, Soucie JM, et al. Population-based screening for family history of end-stage renal disease among incident dialysis patients. Am J Nephrol. 2005;25:529–535. doi: 10.1159/000088491. [DOI] [PubMed] [Google Scholar]
- 2.Braun MC, Herring SM, Gokul N, Monita M, Bell R, Hicks MJ, et al. Hypertensive renal disease: Susceptibility and resistance in inbred hypertensive rat lines. J Hypertension. 2013;31:2050–2059. doi: 10.1097/HJH.0b013e328362f9a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kottgen A, Pattaro C, Boger CA, Fuchsberger C, Olden M, Glazer NL, et al. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010;42:376–384. doi: 10.1038/ng.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freedman BI, Kopp JB, Langefeld CD, Genovese G, Friedman DJ, Nelson GW, et al. The apolipoprotein l1 (apol1) gene and nondiabetic nephropathy in african americans. J Am Soc Nephrol. 2010;21:1422–1426. doi: 10.1681/ASN.2010070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bidani AK, Polichnowski AJ, Loutzenhiser R, Griffin KA. Renal microvascular dysfunction, hypertension and ckd progression. Curr Opin Nephrol Hypertens. 2013;22:1–9. doi: 10.1097/MNH.0b013e32835b36c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, et al. Renal-associated tlr2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. 2005;115:2894–2903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, et al. Tlr4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herring SM, Gokul N, Monita M, Bell R, Boerwinkle E, Wenderfer SE, Braun MC, Doris PA. Immunoglobulin locus associates with serum igg levels and albuminuria. J Am Soc Nephrol. 2011;22:881–889. doi: 10.1681/ASN.2010111148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bell R, Herring SM, Gokul N, Monita M, Grove ML, Boerwinkle E, et al. High-resolution identity by descent mapping uncovers the genetic basis for blood pressure differences between spontaneously hypertensive rat lines. Circ Cardiovasc Genet. 2011;4:223–231. doi: 10.1161/CIRCGENETICS.110.958934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hazama F, Ooshima A, Tanaka T, Tomimoto K, Okamoto K. Vascular lesions in the various substrains of spontaneously hypertensive rats and the effects of chronic salt ingestion. Jpn Circ J. 1975;39:7–22. doi: 10.1253/jcj.39.7. [DOI] [PubMed] [Google Scholar]
- 11.Okamoto K, Aoki K. Development of a strain of spontaneously hypertensive rats. Jpn Circ J. 1963;27:282–293. doi: 10.1253/jcj.27.282. [DOI] [PubMed] [Google Scholar]
- 12.Novoalign software.
- 13.Samtools software.
- 14.Picard software.
- 15.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K, Li M, Hakonarson H. Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative genomics viewer (igv): High-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlsson SR, Fukuda M. Isolation and characterization of leukosialin, a major sialoglycoprotein on human leukocytes. J Biol Chem. 1986;261:12779–12786. [PubMed] [Google Scholar]
- 19.Yuen PS, Dunn SR, Miyaji T, Yasuda H, Sharma K, Star RA. A simplified method for hplc determination of creatinine in mouse serum. Am J Physiol Renal Physiol. 2004;286:F1116–1119. doi: 10.1152/ajprenal.00366.2003. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez-Garay ML, Cranford SM, Braun MC, Doris PA. Diversity in the pre-immune immunoglobulin repertoire of shr lines susceptible and resistant to end organ injury. Genes Immun. 2014 Jul 24; doi: 10.1038/gene.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, et al. Copy number polymorphism in fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 22.Mocsai A, Ruland J, Tybulewicz VL. The syk tyrosine kinase: A crucial player in diverse biological functions. Nat Rev Immunol. 2010;10:387–402. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemay S, Davidson D, Latour S, Veillette A. Dok-3, a novel adapter molecule involved in the negative regulation of immunoreceptor signaling. Mol Cell Biol. 2000;20:2743–2754. doi: 10.1128/mcb.20.8.2743-2754.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerald D, Berra E, Frapart YM, Chan DA, Giaccia AJ, Mansuy D, et al. Jund reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 25.Meixner A, Karreth F, Kenner L, Wagner EF. Jund regulates lymphocyte proliferation and t helper cell cytokine expression. Embo J. 2004;23:1325–1335. doi: 10.1038/sj.emboj.7600133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pillebout E, Weitzman JB, Burtin M, Martino C, Federici P, Yaniv M, et al. Jund protects against chronic kidney disease by regulating paracrine mitogens. J Clin Invest. 2003;112:843–852. doi: 10.1172/JCI17647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Worley KC, Gunaratne P. Encyclopedia of molecular cell biology and molecular medicine. Wiley-VCH Verlag GmbH & Co; KGaA: 2006. Rat genome (rattus norvegicus) [Google Scholar]
- 28.Kozak M. Point mutations define a sequence flanking the aug initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 29.Behmoaras J, Bhangal G, Smith J, McDonald K, Mutch B, Lai PC, et al. Jund is a determinant of macrophage activation and is associated with glomerulonephritis susceptibility. Nat Genet. 2008;40:553–559. doi: 10.1038/ng.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dmitrieva RI, Hinojos CA, Grove ML, Bell RJ, Boerwinkle E, Fornage M, et al. Genome-wide identification of allelic expression in hypertensive rats. Circ Cardiovasc Genet. 2009;2:106–115. doi: 10.1161/CIRCGENETICS.108.809509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okamoto K, Yamori Y, Nagaoka A. Establishment of the stroke-prone spontaneously hypertensive rat (shr) Circ Res. 1974;14:I143–I153. [Google Scholar]
- 32.Atanur SS, Diaz AG, Maratou K, Sarkis A, Rotival M, Game L, et al. Genome sequencing reveals loci under artificial selection that underlie disease phenotypes in the laboratory rat. Cell. 2013;154:691–703. doi: 10.1016/j.cell.2013.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Dijk SJ, Specht PA, Lazar J, Jacob HJ, Provoost AP. Synergistic qtl interactions between rf-1 and rf-3 increase renal damage susceptibility in double congenic rats. Kidney Int. 2006;69:1369–1376. doi: 10.1038/sj.ki.5000301. [DOI] [PubMed] [Google Scholar]
- 34.Satko SG, Sedor JR, Iyengar SK, Freedman BI. Familial clustering of chronic kidney disease. Semin Dial. 2007;20:229–236. doi: 10.1111/j.1525-139X.2007.00282.x. [DOI] [PubMed] [Google Scholar]
- 35.Kao WH, Klag MJ, Meoni LA, Reich D, Berthier-Schaad Y, Li M, et al. Myh9 is associated with nondiabetic end-stage renal disease in african americans. Nat Genet. 2008;40:1185–1192. doi: 10.1038/ng.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kopp JB, Smith MW, Nelson GW, Johnson RC, Freedman BI, Bowden DW, et al. Myh9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet. 2008;40:1175–1184. doi: 10.1038/ng.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watson CT, Breden F. The immunoglobulin heavy chain locus: Genetic variation, missing data, and implications for human disease. Genes Immun. 2012;13:363–373. doi: 10.1038/gene.2012.12. [DOI] [PubMed] [Google Scholar]
- 38.Page TH, D’Souza Z, Nakanishi S, Serikawa T, Pusey CD, Aitman TJ, et al. Role of a novel rat-specific fc receptor in macrophage activation associated with crescentic glomerulonephritis. J Biol Chem. 2012;287:5710–5719. doi: 10.1074/jbc.M111.260695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White FN, Grollman A. Autoimmune factors associated with infarction of the kidney. Nephron. 1964;1:93–102. doi: 10.1159/000179322. [DOI] [PubMed] [Google Scholar]
- 40.Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, et al. Stimulation of lymphocyte responses by angiotensin ii promotes kidney injury in hypertension. Am J Physiol Renal Physiol. 2008;295:F515–524. doi: 10.1152/ajprenal.00527.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the dahl salt-sensitive rat. Hypertension. 2006;48:149–156. doi: 10.1161/01.HYP.0000228320.23697.29. [DOI] [PubMed] [Google Scholar]
- 43.Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, et al. Immunosuppressive treatment protects against angiotensin ii-induced renal damage. Am J Pathol. 2002;161:1679–1693. doi: 10.1016/S0002-9440(10)64445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodriguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M, et al. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin ii exposure. Kidney Int. 2001;59:2222–2232. doi: 10.1046/j.1523-1755.2001.00737.x. [DOI] [PubMed] [Google Scholar]
- 45.Romero F, Rodriguez-Iturbe B, Parra G, Gonzalez L, Herrera-Acosta J, Tapia E. Mycophenolate mofetil prevents the progressive renal failure induced by 5/6 renal ablation in rats. Kidney Int. 1999;55:945–955. doi: 10.1046/j.1523-1755.1999.055003945.x. [DOI] [PubMed] [Google Scholar]
- 46.Shagdarsuren E, Wellner M, Braesen JH, Park JK, Fiebeler A, Henke N, et al. Complement activation in angiotensin ii-induced organ damage. Circ Res. 2005;97:716–724. doi: 10.1161/01.RES.0000182677.89816.38. [DOI] [PubMed] [Google Scholar]
- 47.Tian N, Gu JW, Jordan S, Rose RA, Hughson MD, Manning RD., Jr Immune suppression prevents renal damage and dysfunction and reduces arterial pressure in salt-sensitive hypertension. Am J Physiol Heart Circ Physiol. 2007;292:H1018–1025. doi: 10.1152/ajpheart.00487.2006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.