

Abstract
Objective
Plasminogen activator inhibitor-1 (PAI-1) regulates angiogenesis via effects on extracellular matrix proteolysis and cell adhesion. However, no previous study has implicated PAI-1 in controlling vascular endothelial growth factor (VEGF) signaling. We tested the hypothesis that PAI-1 down-regulates VEGF receptor-2 (VEGFR-2) activation by inhibiting a vitronectin (VN)-dependent cooperative binding interaction between VEGFR-2 and αVβ3.
Approach and Results
We studied PAI-1's effects on VEGF signaling in human umbilical vein endothelial cells (HUVECs). PAI-1 inhibited VEGF-induced phosphorylation of VEGFR-2 in HUVECs grown on VN, but not on fibronectin or collagen. PAI-1 inhibited binding of VEGFR-2 to β3 integrin, VEGFR-2 endocytosis, and intracellular signaling pathways downstream of VEGFR-2. The anti-VEGF effect of PAI-1 was mediated by 2 distinct pathways, one requiring binding to VN and another requiring binding to very-low-density-lipoprotein receptor (VLDLR). PAI-1 inhibited VEGF-induced angiogenesis in vitro and in vivo, and pharmacological inhibition of PAI-1 promoted collateral arteriole development and recovery of hindlimb perfusion after femoral artery interruption.
Conclusions
PAI-1 inhibits activation of VEGFR-2 by VEGF by disrupting a VN-dependent, pro-angiogenic binding interaction involving αVβ3 and VEGFR-2. These results broaden our understanding of the roles of PAI-1, VN, and endocytic receptors in regulating VEGFR-2 activation and suggest novel therapeutic strategies for regulating VEGF signaling.
Keywords: Angiogenesis, vascular endothelial growth factor, plasminogen activator inhibitor-1, vitronectin, very-low-density-lipoprotein receptor
Introduction
Angiogenesis is an intensely regulated process that is integral to the evolution of many human diseases, including ischemic cardiovascular disease and cancer metastasis. Several studies have implicated plasminogen activator inhibitor-1 (PAI-1), the primary endogenous inhibitor of tissue- and urinary-type plasminogen activators (t-PA and u-PA, respectively), as playing an important role in regulating angiogenesis. PAI-1 exerts both pro- and anti-angiogenic properties, with the concentration of PAI-1 being an important determinant of which effect is observed.1,2 The pro-angiogenic effect of PAI-1 appears to be mediated by stabilizing the extracellular matrix (ECM) through inhibition of proteolysis,2,3 and by stimulation of fibronectin-dependent cell migration.4 PAI-1's anti-angiogenic effect appears to be mediated by binding of PAI-1 to vitronectin (VN),5 an ECM protein that promotes cell adhesion and migration by binding to cell surface receptors, including αVβ3 integrin and the receptor for urinary-type plasminogen activator (uPAR). The binding site for PAI-1 on VN overlaps with VN's binding sites for αVβ3 and uPAR. Therefore, binding of PAI-1 to VN blocks cell adhesion and migration, and hence inhibits angiogenesis.6
While PAI-1 regulates angiogenesis through downstream effects on ECM proteolysis and cell migration, no studies have implicated PAI-1 in controlling upstream signaling pathways that initiate angiogenesis. Critical among these is the activation of endothelial cells, mediated by binding of vascular endothelial growth factor (VEGF) to its main angiogenic receptor, VEGFR-2.7 A cooperative binding interaction between VEGFR-2 and αVβ3 integrin plays a key role in regulating VEGF signaling in endothelial cells.8 This receptor cross-talk depends on binding of αVβ3 to VN.9,10 Given that PAI-1 regulates binding of αVβ3 to VN, we hypothesized that PAI-1 regulates VEGF-mediated endothelial cell activation by disrupting VEGFR-2-αVβ3 cross-talk. In this study we performed a series of biochemical, cell culture, and in vivo experiments to test our hypothesis. Our results identify a previously unrecognized role of PAI-1 in regulating VEGF signaling which is VN-dependent and mediated by two distinct PAI-1 ligands.
Results
PAI-1 blocks VEGFR-2-β3 integrin complex formation
VN is a major ligand for αvβ3 and significantly enhances VEGF-mediated activation of endothelial cells via VEGFR-2.10,11 Given that PAI-1 binds VN and blocks its binding to αvβ3,6 we hypothesized that PAI-1 inhibits the VN-dependent binding interaction between αvβ3 and VEGFR-2. To test this hypothesis we cultured HUVECs in VN-coated wells and prepared cell extracts before and after treating cells with VEGF in the absence or presence of PAI-1-WT. VEGFR-2-αvβ3 complexes in extracts were captured with immobilized anti-VEGFR-2 antibody and detected by Western blotting with anti-β3 integrin antibody. Co-immunoprecipitation of β3 integrin and VEGFR-2 was significantly enhanced by VEGF and inhibited by PAI-1-WT (Fig. 1A), suggesting that VEGF induces VEGFR-2-αvβ3 complex formation and PAI-1 inhibits this process.
Figure 1.

PAI-1 inhibits VEGFR-2 signaling. (A) PAI-1 inhibits β3 integrin-VEGFR-2 complex formation. HUVECs were cultured on VN in presence of vehicle control, VEGF (50 ng/mL), or VEGF and PAI-1-WT (10 μg/mL), as indicated (“+” = added, “-” = omitted, replaced by vehicle). Cell lysates were prepared and incubated with resin-bound anti-VEGFR-2 antibody. Captured proteins were analyzed by Western blotting with anti-β3integrin antibodies. Representative images of 3 independent experiments are shown. *P<0.05 vs. negative control. (B) Inhibition of VEGF-induced VEGFR-2 phosphorylation by PAI-1 is VN-dependent. HUVECs were cultured on VN, collagen, or fibronectin in presence of vehicle control, VEGF (50 ng/mL), VEGF and PAI-1-WT (10 μg/mL), or PAI-1-WT, as indicated. Cell lysates were prepared and subjected to Western blotting to detect phosphorylated (p) and total VEGFR-2. (C) PAI-1 inhibits VEGFR-2 endocytosis. HUVEC cell surface proteins were biotinylated, after which cells were cultured in presence of vehicle control, VEGF, or VEGF and PAI-1-WT, as shown. Internalized VEGFR-2 was detected by Western blotting. (D) PAI-1 inhibits translocation of VEGFR-2 to perinuclear endosomes. HUVECs were cultured on VN in presence of vehicle control, VEGF (50 ng/mL), VEGF and PAI-1-WT (10 μg/mL), or PAI-1-WT, as indicated. VEGFR-2 and EEA1 were detected by immunofluorescence confocal microscopy. DAPI (nuclear) and merged VEGFR-2/EEA1/DAPI images are also shown. Images are representative of 3 independent experiments. Arrows indicate perinuclear location of VEGFR-2. (E) PAI-1 inhibits intracellular signaling pathways downstream of VEGFR-2. HUVECs were cultured on VN in presence of vehicle control, VEGF, or VEGF and PAI-1-WT, as indicated. Cell lysates were prepared and subjected to Western blotting to detect phosphorylated and total FAK and p44/p42. All graphs correspond to blots above them and represent densitometric analyses of 3 independent experiments. *P<0.05 vs. negative control group (1st bar).
PAI-1 inhibits VEGFR-2 phosphorylation, internalization, and signaling
To determine if PAI-1 inhibits VEGF-induced VEGFR-2 activation we cultured HUVECs in wells coated with collagen, fibronectin, or VN in the presence of PAI-1-WT or vehicle control. Four hrs later VEGF or vehicle control was added for 10 min, after which cell lysates were prepared, resolved by SDS-PAGE, and analyzed by immunoblotting. VEGF stimulated a significant increase in VEGFR-2 phosphorylation in HUVECs, regardless of whether they were cultured on collagen, fibronectin, or VN, though the magnitude of enhancement was greatest for cells grown on VN (Fig. 1B). The stimulatory effect of VEGF on VEGFR-2 phosphorylation was inhibited by PAI-1-WT in HUVECs cultured on VN. However, PAI-1-WT had no significant effect on VEGFR-2 phosphorylation in HUVECs grown on collagen or fibronectin. The inhibitory effect of PAI-1-WT on VEGFR-2 phosphorylation was dose-dependent (Supplementary Fig.1). The latent form of PAI-1, which has no protease inhibitory activity and binds VN with markedly reduced affinity, had no significant effect on VEGFR-2 phosphorylation, including in HUVECs cultured on VN (data not shown). PAI-1-WT did not bind to recombinant VEGF, nor did PAI-1-WT inhibit binding of VEGF to recombinant VEGFR-2 (Supplementary Fig. 2). We did not observe any significant detachment of HUVECs from wells in response to PAI-1 treatment (data not shown). As a whole, these results suggested that PAI-1 inhibits VEGF-induced VEGFR-2 activation by a VN-dependent mechanism.
After engagement by VEGF, VEGFR-2 undergoes endocytosis, which plays an important role in VEGFR-2 signaling.12,13 PAI-1-WT significantly inhibited the internalization of biotinylated VEGFR-2 by HUVECs (Fig. 1C). To better define the effect of PAI-1 on VEGFR-2 internalization, HUVECs were cultured in VN-coated wells and treated with PAI-1-WT or vehicle control, after which VEGF was added and cellular localization of VEGFR-2 was assessed by immunofluorescence. Consistent with a previous report,14 VEGF stimulated translocation of VEGFR-2 to perinuclear endosomes. PAI-1-WT significantly inhibited the perinuclear transfer of VEGFR-2 (Fig. 1D). We also examined the effect of PAI-1-WT on intracellular signaling pathways activated by binding of VEGF to VEGFR-2. PAI-1-WT inhibited VEGF-induced phosphorylation of FAK and p44/42 MAPK (Fig. 1E). PAI-1-WT also inhibited VEGF-induced co-immunoprecipitation of VEGFR-2 and β3 integrin and phosphorylation of both VEGFR-2 and p44/p42 in human microvascular endothelial cells (Supplementary Fig. 3), confirming that the anti-VEGF effect of PAI-1 was not restricted to HUVECs. As a whole, these experiments suggested that PAI-1 inhibits VEGFR-2 internalization and downstream signaling in vascular endothelial cells.
Inhibition of VEGF signaling by PAI-1 depends of VN and LDL receptor family member binding
To determine which of PAI-1's functional interactions mediate its effects on VEGFR-2 activation, we studied the capacity of recombinant PAI-1 proteins harboring specific loss-of-function mutations to inhibit VEGFR-2 phosphorylation. We performed these experiments with HUVECs whose endogenous PAI-1 expression was silenced with PAI-1 siRNA. This approach decreased PAI-1 expression by approximately 80% (Fig. 2A). Under these conditions PAI-1-WT efficiently inhibited VEGFR-2 phosphorylation, as did PAI-1-R, a mutant with preserved VN-binding, but lacking anti-protease activity (Fig. 2B). These results suggested that PAI-1's anti-protease activity was not absolutely required to inhibit VEGFR-2 activation and supported the hypothesis that PAI-1 inhibits VEGF signaling by a mechanism involving the PAI-1 VN-binding domain. Unexpectedly, PAI-1-AK, which retains full anti-protease activity, but does not bind VN, also inhibited VEGF-induced VEGFR-2 phosphorylation (Fig. 2B). This result suggested that the PAI-1-VN binding interaction was sufficient, but not required, for PAI-1 to inhibit VEGFR-2 signaling. It also suggested that while PAI-1-mediated down-regulation of VEGF signaling was VN-dependent, direct binding of PAI-1 to VN was not absolutely required to exert this effect. In addition to a VN binding site, PAI-1 contains a cryptic binding site for LRP1 and related endocytic receptors of the LDL receptor family, which becomes exposed after PAI-1 binds to u-PA.15 We hypothesized that binding of PAI-1 to an endocytic receptor inhibits VEGFR-2 activation by VEGF, as endocytic receptor binding capacity is retained in PAI-1-AK.16 Therefore, we studied the capacity of PAI-1-E, a mutant with markedly reduced binding capacity for LDL receptor family members,15 to inhibit VEGF-induced VEGFR-2 phosphorylation. PAI-E inhibited VEGF-induced VEGFR-2 phosphorylation (Fig. 2B). However, because the inhibitory effect of PAI-1-E could be mediated by its retained capacity to bind VN, we also constructed a compound mutant, PAI-1-AKE, which contained all of the mutations in PAI-1-AK and PAI-1-E. PAI-1-AKE retains full anti-protease activity, but does not bind VN or LDL receptor family members with high affinity. Unlike PAI-1-AK and PAI-1-E, PAI-1-AKE did not significantly inhibit VEGF-induced VEGFR-2 phosphorylation (Fig. 2B). Consistent with this result, RAP, which blocks binding of PAI-1 to LRP1 and other LDL receptor family members,17 completely inhibited the capacity of PAI-1-AK to down-regulate VEGFR-2 phosphorylation (Fig. 2C). As a whole, these findings suggested that PAI-1 binding to either VN or an endocytic receptor was sufficient to inhibit VEGFR-2 signaling. We also studied the capacity of PAI-AK to inhibit VEGFR-2 phosphorylation in the presence of a synthetic peptide, D2A-Ala, which blocks binding of uPAR to αVβ3.18 D2A-Ala potently inhibited the capacity of PAI-1-AK to disrupt VEGFR-2 activation, whereas a control (scrambled) peptide had no significant effect (Supplementary Fig. 4), suggesting that PAI-1 binding to an endocytic receptor family member inhibits VEGFR-2 signaling by a pathway involving a uPAR-integrin binding interaction.
Figure 2.

Inhibition of VEGFR-2 phosphorylation by PAI-1 depends of VN and endocytic receptor binding. (A) HUVECs were transfected with human PAI-1 siRNA expression vector or control (Con-siRNA) plasmid. Western blot analysis of PAI-1 and β-actin expression in transfected HUVECs is shown. (B) PAI-1-silenced HUVECs were incubated with recombinant PAI-1 (WT, AKE, E, R, or AK mutant) or vehicle control (-) and stimulated with VEGF (+, 50 ng/mL) or vehicle control (-), as shown. Cell lysates were prepared and phosphorylated (p) and total VEGFR-2 were assessed by Western blotting. (C) Pre-incubation of HUVECs with RAP (0.2 μM) blocks PAI-1-AK's capacity to inhibit VEGF-induced VEGFR-2 phosphorylation. All graphs correspond to blots above them and represent densitometric analyses of 3 independent experiments. *P<0.05 vs. negative control (1st bar).
PAI-1 inhibits VEGF-induced endothelial cell adhesion, migration, and cord formation in a VN-dependent manner
We examined the effects of PAI-1 on the physiological responses of endothelial cells to VEGF stimulation. In the absence of added PAI-1, VEGF significantly stimulated adhesion and migration of HUVECs grown on VN, collagen, or fibronectin (Fig. 3A-B, white bars), with the stimulatory effect being maximal in cells grown on VN. In the absence of added VEGF, PAI-1-WT and PAI-1-R each significantly inhibited, but did not completely block, adhesion and migration of HUVECs cultured in VN-coated wells, whereas PAI-1-AK had no significant effect, consistent with its defect in VN binding. However, PAI-1-AK completely inhibited the up-regulation of adhesion and migration induced by VEGF in HUVECs cultured on VN, as did PAI-1-WT and PAI-1-R. PAI-1-WT, PAI-1-R, and PAI-1-AK had no significant effect on adhesion or migration of HUVECs cultured on fibronectin or collagen, either in the presence or absence of added VEGF.
Figure 3.

PAI-1 inhibits VEGF-induced HUVEC adhesion, migration, and tubule formation in a VN-dependent manner. (A,B) HUVECs were added to wells coated with VN, fibronectin, or collagen, as shown, incubated in the presence of recombinant PAI-1 (WT, R, or AK forms, 10 μg/mL) or vehicle control, as shown, and treated with VEGF (+, 50 ng/mL) or vehicle control (-), as indicated, after which cell adhesion (A) and migration (B) were measured. Data are mean of triplicate experiments. *P<0.05 vs. negative control (HUVECs not exposed to PAI-1 or VEGF). **P<0.05 vs. HUVECs incubated with VEGF only (no PAI-1). (C) Tubule formation. Representative images of HUVECs grown on Matrigel or Matrigel supplemented with VN (10 μg/mL) in the absence (-) of added VEGF or PAI-1-WT or in the presence (+) of VEGF (50 ng/mL), VEGF and PAI-1-WT (10 μg/mL), or PAI-1-WT, as shown. (D) Quantitative assessment of quadruplicate tubule formation experiments. *P>0.05 vs. HUVECs grown on Matrigel and exposed only to VEGF. **P<0.05 vs. HUVECs grown on Matrigel containing VN and exposed only to VEGF.
VEGF stimulates cultured endothelial cells to form cord-like structures that mimic forming blood vessels. To examine the effect of PAI-1 on this process, HUVECs were cultured on Matrigel formed in the presence or absence of supplemental VN. PAI-1-WT inhibited augmentation of tubule formation by VEGF in VN-supplemented Matrigel, but not in Matrigel lacking supplemental VN (Fig. 3C-D).
To further explore the functional significance of PAI-1's interactions with VN and endocytic receptor(s) in regulating VEGF signaling we studied the effects of recombinant PAI-1 mutants on VEGF-induced migration of VN-adherent HUVECs in the presence and absence of RAP. In the absence of RAP, PAI-1-WT, PAI-1-R, and PAI-1-AK each inhibited VEGF-induced migration (Supplementary Fig. 5A, bars 6-8). In the presence of RAP, PAI-1-WT and PAI-1-R each retained the capacity to inhibit VEGF-induced migration (bars 9-10), whereas the inhibitory effect of PAI-1 AK was lost (bar 11). Furthermore, in a separate set of experiments performed in the absence of RAP, PAI-1-AKE did not inhibit VEGF-induced HUVEC migration (Supplementary Fig. 5B, bar 4), while PAI-1-E, which is defective in endocytic receptor binding, but retains normal VN-binding affinity, inhibited VEGF-induced HUVEC migration (bar 5). Together, these results supported the functional significance of PAI-1 binding to either VN or an LDL receptor family member in regulating VEGF signaling. They also suggested that PAI-1's endocytic-receptor-dependent role in VEGF signaling, while not requiring binding of PAI-1 to VN, still exhibited VN-dependence, as the effect of PAI-1-WT, which binds LRP1 and other LDL receptor family members, was lost in the absence of VN.
Binding of PAI-1 to VLDLR inhibits VEGF signaling
Since the VLDLR is thought to be the primary LDL receptor family member present on HUVECs, 19, 20 we examined if PAI-1-AK's ability to inhibit VEGF-induced cell migration could be reversed by an antibody that specifically blocks ligand binding to VLDLR. These studies demonstrated that the VLDLR antibody completely inhibited the anti-VEGF effect of PAI-1-AK (Supplementary Fig. 5C), suggesting that VLDLR is the LDL receptor family member that mediates PAI-1's anti-VEGF effect in HUVECs.
VEGF-induction of angiogenesis ex vivo and in vivo is VN-dependent and inhibited by PAI-1
To examine the role of VN in VEGF-induced angiogenesis under physiological conditions, we cultured segments of aorta from WT and Vn-/- mice ex vivo in Matrigel in the presence or absence of VEGF. The capacity of VEGF to stimulate microvessel sprouting was significantly greater in WT mice than in Vn-/- mice (Fig. 4A-B). Immunostaining confirmed the presence of VN in aorta of WT mice (Supplementary Fig. 6A). PAI-1-WT significantly inhibited VEGF-induced microvessel sprouting from WT aorta; however, PAI-1-AKE had no significant effect (Fig. 4C). To study the significance of our findings in vivo we injected VEGF-impregnated Matrigel into subcutaneous tissue of WT and Vn-/- mice. After 14 days solidified gels were retrieved and blood vessel invasion into them was measured. VEGF significantly increased endothelial cell invasion into Matrigels in WT mice, but not in Vn-/- mice (Fig. 4D-E). Immunostaining confirmed that host-derived VN diffused into Matrigel implants (Supplementary Fig. 6B). To determine if PAI-1 inhibits VEGF-induced angiogenesis in vivo, we injected Matrigel supplemented with PAI-1-WT (10 μg/mL) into WT- and Vn-/- mice. PAI-1-WT completely inhibited VEGF-induced angiogenesis in WT mice, but had no significant effect in Vn-/- mice (Fig. 4D-E). These results suggested that PAI-1 and VN play key roles in regulating VEGF-induced angiogenesis in vivo, and that PAI-1 could be used to pharmacologically inhibit VEGF-induced angiogenesis. However, down-regulation of fibrinolysis by active PAI-1 could promote thrombosis. Given that PAI-1-R does not inhibit PAs, we tested its capacity to inhibit VEGF-induced angiogenesis in vivo in WT mice. PAI-1-R significantly inhibited VEGF-induced angiogenesis (Fig. 4F-G).
Figure 4.

PAI-1 inhibits VEGF-induced angiogenesis. (A) Ex vivo angiogenesis is VN-dependent. Aortic rings from wild-type (WT) and Vn-/- mice were cultured ex vivo for 14 days in media supplemented with VEGF or vehicle control, as shown. Representative images of microvessel sprouts (arrows) from aortic rings are shown. Magnification 40×. (B) Quantitative analysis of microvessel sprouts from aortic rings (n=4/group). Results are expressed as % of control (WT aortic rings treated with vehicle; i.e. 1st bar). *P<0.05 vs. control. (C) PAI-1-WT inhibits VEGF-induced angiogenic sprouting from WT aortic rings, but PAI-1-AKE does not. *P<0.05 vs. control (1st bar; n=3/group). (D) In vivo angiogenesis is VN-dependent and inhibited by PAI-1. Matrigel supplemented with vehicle control, VEGF, VEGF and PAI-1-WT, or PAI-1-WT was injected into subcutaneous tissue of WT mice and Vn-/- mice, as shown. PECAM-1-positive microvessels (arrows) in Matrigel plugs excised 14 days later are shown. Magnification 400×. (E) Quantitative assessment of microvessel formation in Matrigel plugs. *P<0.05 vs. control (Matrigel supplemented with vehicle control injected into WT mice, 1st bar; n=6/group). (F) PAI-1-R inhibits VEGF-induced angiogenesis in WT mice. Representative images of microvessels in Matrigel implants supplemented with vehicle control, VEGF, VEGF and PAI-1-R, or PAI-1-R are shown. Magnification 400×. (G) Quantitative assessment of microvessel formation in Matrigel plugs retrieved from WT mice. *P<0.05 vs. vehicle control (1st bar; n=6/group).
Pharmacological inhibition of PAI-1 promotes reperfusion of ischemic hindlimb tissue
To examine the significance of our findings in a pathological model of PAI-1 over-expression relevant to human cardiovascular disease, we fed mice high-fat chow (HFC) for 14 weeks, which produced obesity, hyperglycemia, and a significant increase in plasma PAI-1 concentration (Supplementary Fig. 7). Hindlimb ischemia was induced in HFC-fed mice by ligation and excision of the femoral artery, after which mice received PAI-039, a specific PAI-1 inhibitor, or vehicle control. After 14 days of treatment, plasma PAI-1 activity was significantly lower in PAI-039-treated mice (0.61±0.09 ng/mL) than in controls (2.66±0.35 ng/mL; P<0.05; n=6/group). Laser Doppler imaging revealed that the recovery of perfusion of ischemic hindlimb tissue after femoral artery interruption was significantly increased in PAI-039-treated mice compared to vehicle controls (Fig. 5A-B). Consistent with these results, arteriole density in ischemic gastrocnemius muscle 14 days after induction of ischemia was significantly increased in PAI-039-treated mice vs. controls (Fig. C-D). Capillary density in ischemic gastrocnemius muscle did not differ significantly between experimental groups (Fig. 5E)
Figure 5.

Pharmacological inhibition of PAI-1 promotes recovery of hindlimb perfusion after femoral artery ligation. (A) Representative laser-Doppler images of mouse hindlimbs at indicated time points after femoral artery ligation. “Pre” and “Post” are immediately before and after surgery, respectively. Arrows denote ischemic limb. Red color denotes normal perfusion. (B) Mean ratio of blood flow in ischemic and non-ischemic hindlimb foot pads for all animals at indicated time points (n=6/group; *P<0.05 vs. vehicle control). (C) Representative images of arterioles (arrows), assessed by anti-smooth muscle α-actin immunostaining, in ischemic gastrocnemius muscles recovered 14 days after femoral artery interruption. Distance bars = 100 μm. (D) Mean arteriole density in ischemic gastrocnemius muscle was significantly greater in PAI-039-treated mice (n=6/group; *P<0.05 vs. vehicle control). (E) Mean capillary density in ischemic gastrocnemius muscle, assessed by anti-PECAM-1 immunostaining, did not differ significantly between groups (P>0.5).
Discussion
We describe a previously unrecognized role of PAI-1 in regulating VEGF-induced activation of its main angiogenic receptor, VEGFR-2. Specifically, PAI-1 inhibits VEGFR-2 phosphorylation and translocation to perinuclear endosomes, as well as intracellular signaling downstream of VEGFR-2. The anti-protease function of PAI-1 is not absolutely required for it to inhibit VEGFR-2 activation, as PAI-1-R, a mutant devoid of anti-protease activity, inhibited VEGF signaling. This observation is consistent with the high-affinity binding interaction between PAI-1 and VN, which inhibits binding of VN to its cell surface receptors.6 Our data suggest that the anti-angiogenic property of PAI-1-R is mediated by its capacity to competitively inhibit binding of αVβ3 integrin to VN. However, an intriguing aspect of our study is the observation that inhibition of VEGFR-2 activation and angiogenic signaling by PAI-1, while requiring VN, is not mediated solely by binding to VN, as evidenced by the preserved anti-angiogenic effect of PAI-1-AK, which has no measurable VN binding affinity. Experiments examining how PAI-1 can inhibit VEGF signaling in a VN-dependent manner without binding VN led us to uncover another previously unrecognized regulator of VEGFR-2 activation, which appears to be the VLDLR. This conclusion is supported by three lines of evidence. First, the anti-angiogenic activity of PAI-1-AK is inhibited by RAP, an antagonist of LDL receptor family members; second, the anti-angiogenic activity of PAI-1-AK is lost upon introduction of an additional glutamate mutation (PAI-1-AKE) that ablates PAI-1's capacity to bind LDL receptor family members; and third, PAI-1-AK's ability to inhibit VEGF-induced HUVEC migration is blocked by an antibody directed against the ligand binding repeats of VLDLR. These results suggest that VLDLR, which is expressed by endothelial cells,19,20 is the LDL receptor family member that mediates PAI-1's anti-angiogenic effect and are consistent with a study showing that VLDLR signaling exerts an anti-angiogenic effect on retinal endothelial cells.21
Based on our data and published studies, we hypothesize that αvβ3 integrin and its ligand, VN, underpin the capacity of PAI-1 to inhibit VEGFR-2 activation, whether by binding to VN or VLDLR, as illustrated in Fig. 6. Previous studies showed that αvβ3 and VN potently enhance VEGFR-2 activation by VEGF,9,10 demonstrating a key role of αvβ3-VEGFR-2 cross-talk in VEGF-signaling and the VN-dependent nature of this interaction (Fig. 6A). By binding to the somatomedin B (SMB) domain of VN, PAI-1 competitively inhibits binding of αvβ3 to VN's immediately adjacent integrin binding site.6 Our experiments involving recombinant PAI-1 mutants suggest that blockade of αvβ3-VN binding interaction by binding of PAI-1 to VN directly uncouples αvβ3-VEGFR-2 cross-talk and down-regulates VEGFR-2 activation (Fig. 6B). Alternatively, binding of PAI-1 to VLDLR (most likely in the form of PAI-1-u-PA complex15,22) has the potential to indirectly uncouple αvβ3 from VEGFR-2, as it has been shown that PAI-1-uPA complex also binds to uPAR, which itself binds to integrins and regulates their adhesive and signaling properties.23 Specifically, formation of a macromolecular complex consisting of VLDLR, PAI-1, uPA, uPAR, and αVβ3 (or other integrins) can lead to integrin inactivation and disengagement from the ECM.24,25 Disengagement of αVβ3 from VN by this pathway would be anticipated to inhibit the VN-dependent cross-talk between αVβ3 and VEGFR-2, thereby down-regulating VEGF signaling (Fig 6C). Our data suggest that binding of PAI-1 to VLDLR is required for PAI-1 to functionally dissociate αvβ3 from VN via the pathway that does not require direct PAI-1 binding to VN, as the anti-angiogenic effect of PAI-1-AK (which does not bind VN, but binds uPA-uPAR complex) was lost upon introduction of an additional mutation into PAI-1-AK that ablated LDL receptor family member binding. The VLDLR-dependent, anti-angiogenic effect of PAI-1 could be mediated by endocytic-receptor-mediated internalization of αvβ3 (along with PAI-1, uPA, and uPAR, as has been described24,26) and/or by conformational changes in αVβ3 that are not accompanied by its cellular internalization.25 This issue was not resolved by our experiments. However, we showed that peptide-mediated inhibition of uPAR-integrin binding potently inhibited the anti-VEGF effect of PAI-1-AK, suggesting that PAI-1 disrupts VEGF signaling by a pathway involving uPAR-integrin complex formation. Consistent with our results, Alexander et al. showed that uPAR is an important determinant of VEGF signaling in endothelial cells.27 Further studies are necessary to more precisely define the molecular events underlying the VLDLR-dependent down-regulation of VEGFR-2 signaling by PAI-1.
Figure 6.
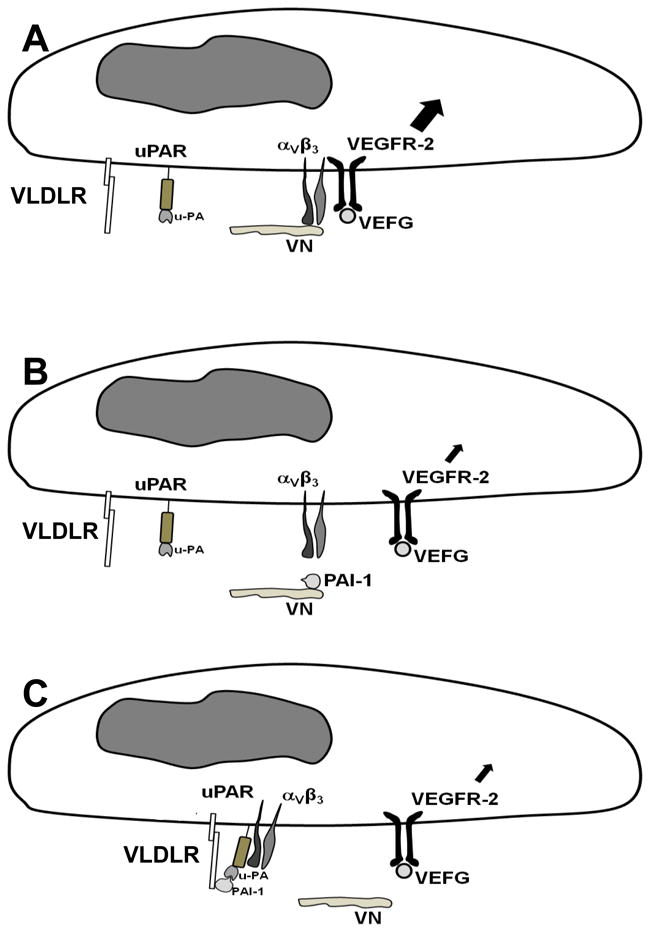
Proposed model by which PAI-1 inhibits VEGF signaling. (A) VEGF induces strong angiogenic signaling (large intracellular arrow) when αVβ3 is bound to VN and VEGFR-2. (B) PAI-1 binds VN and competitively blocks binding of αVβ3, which uncouples the pro-angiogenic binding interaction between αVβ3 and VEGFR-2 and down-regulates VEGF signaling (small intracellular arrow). (C) PAI-1-uPA complex binds to VLDLR and uPAR, which, through a uPAR-αVβ3 binding interaction, triggers dissociation of αVβ3 from VN, thereby uncoupling the pro-angiogenic binding interaction between αVβ3 and VEGFR-2, which down-regulates VEGF signaling.
Our experiments involving microvessel sprouting from aortic rings ex vivo and microvessel invasion into subcutaneous extracellular matrix implants in vivo suggest that PAI-1 is a physiologically relevant regulator of VEGFR-2 activation. Based on our data, we hypothesize that enhanced PAI-1 expression, which occurs under pathological conditions, such as diabetes mellitus,28 inhibits VEGFR-2 activation in vivo. Consistent with this hypothesis, we showed that PAI-039, a highly specific pharmacological inhibitor of PAI-1 with no discernable effect on angiogenesis in PAI-1-deficient mice,29,30 promoted recovery of tissue perfusion and development of collateral arterioles after induction of hindlimb ischemia in mice with diet-induced PAI-1 over-expression. Previous studies have shown that collateral arteriole development and recovery of tissue perfusion after femoral artery occlusion are dependent on VEGFR-2 activation by VEGF.31,32 Therefore, our findings with PAI-039, as well as another murine study involving pharmacological PAI-1 inhibition,33 support the in vivo relevance of the inhibition of VEGFR-2 activation by PAI-1. We did not observe an increase in capillary density in ischemic gastrocnemius muscle in response to PAI-039 treatment. However, this does not rule out an effect of PAI-1 on capillary remodeling. Furthermore, the dominant determinant of tissue reperfusion after femoral artery ligation is collateral arteriole development,34 and we saw a positive effect of PAI-039 on this angiogenic response. A limitation of our hindlimb ischemia model experiment is that we cannot definitively conclude that pharmacological inhibition of PAI-1 promoted tissue reperfusion solely by blocking the negative effect of PAI-1 on VEGFR-2 activation, as pharmacological inhibition of PAI-1 could potentially modulate the angiogenic response to ischemia by VEGFR-2-independent pathways. Nevertheless, our hindlimb ischemia model data support the significance of our proposed molecular mechanisms in a clinically-relevant, in vivo context, thereby complementing our cell culture, ex vivo aortic ring, and in vivo Matrigel plug data, which demonstrated that PAI-1 inhibits VEGFR-2 activation. Additional in vivo studies will be necessary to further dissect and better characterize the significance of our newly reported regulatory pathway on VEGF-dependent angiogenic signaling in normal vascular development and other disease models.
In summary, we have shown that PAI-1 inhibits VEGFR-2 activation by VEGF in a VN-dependent manner. Our data suggest that PAI-1 mediates its effect by inhibiting binding of αVβ3 to its ECM ligand, VN, thereby disrupting the pro-angiogenic binding interaction between αVβ3 and VEGFR-2. The anti-angiogenic effect of PAI-1 appears to be mediated, though not exclusively, by direct binding to VN, which competitively blocks binding of VN to αVβ3. We also have shown that binding of PAI-1 to an endocytic receptor of the LDL receptor family inhibits VEGFR-2 activation, and that this effect is inhibited by an antibody that blocks ligand binding to VLDLR. Our results suggest that VLDLR plays an important role in VEGF signaling, one which is sensitive to inhibition by PAI-1 and mediated by functional disengagement of αVβ3 from VN. We have shown that the anti-VEGF effect of PAI-1 is evident under physiological conditions in vitro and in vivo. Overall, our results broaden our understanding of the roles of PAI-1, VN, and the LDL receptor family in regulating angiogenic signaling. They also support proceeding to additional preclinical studies involving recombinant PAI-1 mutants and pharmacological PAI-1 inhibitors as therapeutic strategies to inhibit or promote angiogenesis in vivo.
Supplementary Material
Significance.
The plasminogen activation (PA) system regulates a diverse array of mammalian physiological and pathological processes. We report a previously unrecognized role of plasminogen activator inhibitor-1 (PAI-1), a central member of the PA system, in regulating the activation of vascular endothelial growth factor (VEGF) receptor-2 (VEGFR-2). We show that PAI-1 inhibits VEGFR-2 activation by blocking the pro-angiogenic binding interaction between VEGFR-2 and αVβ3 integrin, and that the anti-angiogenic effect of PAI-1 involves 2 pathways, one entailing binding to vitronectin; another requiring binding to very-low-density-lipoprotein (VLDL) receptor. Our findings significantly expand our understanding of the roles of PAI-1, vitronectin, and the LDL receptor family in regulating angiogenic signaling and suggest novel strategies for pharmacologically controlling angiogenesis.
Acknowledgments
None.
Sources of Funding: This work was supported by NIH grants HL57346 and HL095951 (WPF), HL55374, HL57346, HL89407, and NS079639 (DAL), Department of Veterans Affairs Merit Review Award (CARA-007-12S; WPF), Missouri Life Sciences Research Board (WPF), American Heart Association Scientist Development Grant (JW), and National Natural Science Foundation of China (81172050, JW).
Non-standard Abbreviations and Acronyms
- HUVEC
Human umbilical vein endothelial cell
- PAI-1
Plasminogen activator inhibitor-1
- RAP
Receptor-associated protein
- VEGF
Vascular endothelial growth factor
- VEGFR-2
Vascular endothelial growth factor receptor-2
- VLDLR
Very-low-density-lipoprotein receptor
- VN
Vitronectin
- WT
Wild-type
Footnotes
Disclosures. The authors declare no competing financial interests.
References
- 1.McMahon GA, Petitclerc E, Stefansson S, Smith E, Wong MK, Westrick RJ, Ginsburg D, Brooks PC, Lawrence DA. Plasminogen activator inhibitor-1 regulates tumor growth and angiogenesis. J Biol Chem. 2001;276:33964–33968. doi: 10.1074/jbc.M105980200. [DOI] [PubMed] [Google Scholar]
- 2.Devy L, Blacher S, Grignet-Debrus C, Bajou K, Masson V, Gerard RD, Gils A, Carmeliet G, Carmeliet P, Declerck PJ, Nöel A, Foidart JM. The pro- or antiangiogenic effect of plasminogen activator inhibitor 1 is dose dependent. FASEB J. 2002;16:147–154. doi: 10.1096/fj.01-0552com. [DOI] [PubMed] [Google Scholar]
- 3.Bajou K, Masson V, Gerard RD, Schmitt PM, Albert V, Praus M, Lund LR, Frandsen TL, Brunner N, Dano K, Fusenig NE, Weidle U, Carmeliet G, Loskutoff D, Collen D, Carmeliet P, Foidart JM, Noël A. The plasminogen activator inhibitor PAI-1 controls in vivo tumor vascularization by interaction with proteases, not vitronectin. Implications for antiangiogenic strategies. J Cell Biol. 2001;152:777–784. doi: 10.1083/jcb.152.4.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Isogai C, Laug WE, Shimada H, Declerck PJ, Stins MF, Durden DL, Erdreich-Epstein A, DeClerck YA. Plasminogen activator inhibitor-1 promotes angiogenesis by stimulating endothelial cell migration toward fibronectin. Cancer Res. 2001;61:5587–5594. [PubMed] [Google Scholar]
- 5.Stefansson S, Petitclerc E, Wong MK, McMahon GA, Brooks PC, Lawrence DA. Inhibition of angiogenesis in vivo by plasminogen activator inhibitor-1. J Biol Chem. 2001;276:8135–8141. doi: 10.1074/jbc.M007609200. [DOI] [PubMed] [Google Scholar]
- 6.Stefansson S, Lawrence DA. The serpin PAI-1 inhibits cell migration by blocking integrin alpha V beta 3 binding to vitronectin. Nature. 1996;383(6599):441–443. doi: 10.1038/383441a0. [DOI] [PubMed] [Google Scholar]
- 7.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 8.Somanath PR, Malinin NL, Byzova TV. Cooperation between integrin alphaVbeta3 and VEGFR2 in angiogenesis. Angiogenesis. 2009;12:177–185. doi: 10.1007/s10456-009-9141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soldi R, Mitola S, Strasly M, Defilippi P, Tarone G, Bussolino F. Role of alphaVbeta3 integrin in the activation of vascular endothelial growth factor receptor-2. EMBO J. 1999;18:882–892. doi: 10.1093/emboj/18.4.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahabeleshwar GH, Feng W, Reddy K, Plow EF, Byzova TV. Mechanisms of integrin-vascular endothelial growth factor receptor cross-activation in angiogenesis. Circ Res. 2007;101:570–580. doi: 10.1161/CIRCRESAHA.107.155655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahabeleshwar GH, Chen J, Feng W, Somanath PR, Razorenova OV, Byzova TV. Integrin affinity modulation in angiogenesis. Cell Cycle. 2008;7:335–347. doi: 10.4161/cc.7.3.5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, Carmeliet P, Simons M. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev Cell. 2010;18:713–724. doi: 10.1016/j.devcel.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Constantino Rosa Santos S, Miguel C, Domingues I, Calado A, Zhu Z, Wu Y, Dias S. VEGF and VEGFR-2 (KDR) internalization is required for endothelial recovery during wound healing. Exp Cell Res. 2007;313:1561–1574. doi: 10.1016/j.yexcr.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 14.Nakayama M, Nakayama A, van Lessen M, et al. Spatial regulation of VEGF receptor endocytosis in angiogenesis. Nat Cell Biol. 2013;15:249–260. doi: 10.1038/ncb2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stefansson S, Muhammad S, Cheng XF, Battey FD, Strickland DK, Lawrence DA. Plasminogen activator inhibitor-1 contains a cryptic high affinity binding site for the low density lipoprotein receptor-related protein. J Biol Chem. 1998;273:6358–6366. doi: 10.1074/jbc.273.11.6358. [DOI] [PubMed] [Google Scholar]
- 16.Xu Z, Balsara RD, Gorlatova NV, Lawrence DA, Castellino FJ, Ploplis VA. Conservation of critical functional domains in murine plasminogen activator inhibitor-1. J Biol Chem. 2004;279:17914–17920. doi: 10.1074/jbc.M314197200. [DOI] [PubMed] [Google Scholar]
- 17.Hussain MM, Strickland DK, Bakillah A. The mammalian low-density lipoprotein receptor family. Annu Rev Nutr. 1999;19:141–172. doi: 10.1146/annurev.nutr.19.1.141. [DOI] [PubMed] [Google Scholar]
- 18.Degryse B, Resnati M, Czekay RP, Loskutoff DJ, Blasi F. Domain 2 of the urokinase receptor contains an integrin-interacting epitope with intrinsic signaling activity: generation of a new integrin inhibitor. J Biol Chem. 2005;280:24792–24803. doi: 10.1074/jbc.M413954200. [DOI] [PubMed] [Google Scholar]
- 19.Wyne KL, Pathak K, Seabra MC, Hobbs HH. Expression of the VLDL receptor in endothelial cells. Arterioscler Thromb Vasc Biol. 1996;16:407–415. doi: 10.1161/01.atv.16.3.407. [DOI] [PubMed] [Google Scholar]
- 20.Multhaupt HA, Gåfvels ME, Kariko K, Jin H, Arenas-Elliot C, Goldman BI, Strauss JF, 3rd, Angelin B, Warhol MJ, McCrae KR. Expression of very low density lipoprotein receptor in the vascular wall. Analysis of human tissues by in situ hybridization and immunohistochemistry. Am J Pathol. 1996;148:1985–1997. [PMC free article] [PubMed] [Google Scholar]
- 21.Xia CH, Lu E, Liu H, Du X, Beutler B, Gong X. The role of Vldlr in intraretinal angiogenesis in mice. Invest Ophthalmol Vis Sci. 2011;52:6572–6579. doi: 10.1167/iovs.10-7082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rettenberger PM, Oka K, Ellgaard L, Petersen HH, Christensen A, Martensen PM, Monard D, Etzerodt M, Chan L, Andreasen PA. Ligand binding properties of the very low density lipoprotein receptor. Absence of the third complement-type repeat encoded by exon 4 is associated with reduced binding of Mr 40,000 receptor-associated protein. J Biol Chem. 1999;274:8973–8980. doi: 10.1074/jbc.274.13.8973. [DOI] [PubMed] [Google Scholar]
- 23.Wei Y, Lukashev M, Simon DI, Bodary SC, Rosenberg S, Doyle MV, Chapman HA. Regulation of integrin function by the urokinase receptor. Science. 1996;273:1551–1555. doi: 10.1126/science.273.5281.1551. [DOI] [PubMed] [Google Scholar]
- 24.Czekay RP, Aertgeerts K, Curriden SA, Loskutoff DJ. Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J Cell Biol. 2003;160:781–791. [Google Scholar]
- 25.Czekay RP, Loskutoff DJ. Plasminogen activator inhibitors regulate cell adhesion through a uPAR-dependent mechanism. J Cell Physiol. 2009;220:655–663. doi: 10.1002/jcp.21806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Webb DJ, Nguyen DH, Sankovic M, Gonias SL. The very low density lipoprotein receptor regulates urokinase receptor catabolism and breast cancer cell motility in vitro. J Biol Chem. 1999;274:7412–7420. doi: 10.1074/jbc.274.11.7412. [DOI] [PubMed] [Google Scholar]
- 27.Alexander RA, Prager GW, Mihaly-Bison J, Uhrin P, Sunzenauer S, Binder BR, Schütz GJ, Freissmuth M, Breuss JM. VEGF-induced endothelial cell migration requires urokinase receptor (uPAR)-dependent integrin redistribution. Cardiovasc Res. 2012;94:125–135. doi: 10.1093/cvr/cvs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pandolfi A, Cetrullo D, Polishuck R, Alberta MM, Calafiore A, Pellegrini G, Vitacolonna E, Capani F, Consoli A. Plasminogen activator inhibitor type 1 is increased in the arterial wall of type II diabetic subjects. Arterioscler Thromb Vasc Biol. 2001;21:1378–1382. doi: 10.1161/hq0801.093667. [DOI] [PubMed] [Google Scholar]
- 29.Elokdah H, Abou-Gharbia M, Hennan JK, McFarlane G, Mugford CP, Krishnamurthy G, Crandall DL. Tiplaxtinin, a novel, orally efficacious inhibitor of plasminogen activator inhibitor-1: design, synthesis, and preclinical characterization. J Med Chem. 2004;47:3491–3494. doi: 10.1021/jm049766q. [DOI] [PubMed] [Google Scholar]
- 30.Leik CE, Su EJ, Nambi P, Crandall DL, Lawrence DA. Effect of pharmacologic plasminogen activator inhibitor-1 inhibition on cell motility and tumor angiogenesis. J Thromb Haemost. 2006;4:2710–2715. doi: 10.1111/j.1538-7836.2006.02244.x. [DOI] [PubMed] [Google Scholar]
- 31.Jacobi J, Tam BY, Wu G, Hoffman J, Cooke JP, Kuo CJ. Adenoviral gene transfer with soluble vascular endothelial growth factor receptors impairs angiogenesis and perfusion in a murine model of hindlimb ischemia. Circulation. 2004;110:2424–2429. doi: 10.1161/01.CIR.0000145142.85645.EA. [DOI] [PubMed] [Google Scholar]
- 32.Babiak A, Schumm AM, Wangler C, Loukas M, Wu J, Dombrowski S, Matuschek C, Kotzerke J, Dehio C, Waltenberger J. Coordinated activation of VEGFR-1 and VEGFR-2 is a potent arteriogenic stimulus leading to enhancement of regional perfusion. Cardiovasc Res. 2004;61:789–795. doi: 10.1016/j.cardiores.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 33.Tashiro Y, Nishida C, Sato-Kusubata K, Ohki-Koizumi M, Ishihara M, Sato A, Gritli I, Komiyama H, Sato Y, Dan T, Miyata T, Okumura K, Tomiki Y, Sakamoto K, Nakauchi H, Heissig B, Hattori K. Inhibition of PAI-1 induces neutrophil-driven neoangiogenesis and promotes tissue regeneration via production of angiocrine factors in mice. Blood. 2012;119:6382–6393. doi: 10.1182/blood-2011-12-399659. [DOI] [PubMed] [Google Scholar]
- 34.Scholz D, Ziegelhoeffer T, Helisch A, Wagner S, Friedrich C, Podzuweit T, Schaper W. Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J Mol Cell Cardiol. 2002;34:775–787. doi: 10.1006/jmcc.2002.2013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.