

Abstract
Background
Mounting evidence points to individual contributions of tumour necrosis factor-alpha (TNF) and the c-Jun N-terminal kinase (JNK) pathway to the induction and maintenance of various pain states. Here we explore the role of spinal TNF and JNK in carrageenan-induced hypersensitivity. As links between TNF and JNK have been demonstrated in vitro, we investigated if TNF regulates spinal JNK activity in vivo.
Methods
TNF levels in lumbar cerebrospinal fluid (CSF) were measured by enzyme-linked immunosorbent assay, spinal TNF gene expression by real-time polymerase chain reaction and TNF protein expression, JNK and c-Jun phosphorylation by western blotting. The role of spinal TNF and JNK in inflammation-induced mechanical and thermal hypersensitivity was assessed by injecting the TNF inhibitor etanercept and the JNK inhibitors SP600125 and JIP-1 intrathecally (i.t.). TNF-mediated regulation of JNK activity was examined by assessing the effect of i.t. etanercept on inflammation-induced spinal JNK activity.
Results
TNF levels were increased in CSF and spinal cord following carrageenan-induced inflammation. While JNK phosphorylation followed the same temporal pattern as TNF, c-jun was only activated at later time points. Intrathecal injection of TNF and JNK inhibitors attenuated carrageenan-induced mechanical and thermal hypersensitivity. TNF stimulation induced JNK phosphorylation in cultured spinal astrocytes and blocking the spinal actions of TNF in vivo by i.t. injection of etanercept reduced inflammation-induced spinal JNK activity.
Conclusions
Here we show that spinal JNK activity is dependent on TNF and that both TNF and the JNK signalling pathways modulate pain-like behaviour induced by peripheral inflammation.
What's already known about this topic?
Spinal tumour necrosis factor is involved in inflammation- (CFA) and nerve injury-induced pain.
The c-Jun N-terminal kinase (JNK) is activated in the spinal astrocytes in models of inflammatory (CFA) and neuropathic pain.
What does this study add?
Spinal TNF is important in carrageenan-induced hypersensitivity.
JNK is activated in the spinal cord with a similar temporal profile as the TNF release and JNK inhibition prevents the onset of carrageenan-induced hypersensitivity.
Spinal TNF regulates JNK activity in vivo.
1. Introduction
Tumour necrosis factor (TNF) is an important pro-inflammatory cytokine in the initiation and perpetuation of inflammatory responses. It is also a critical factor in pathological pain states (Wagner and Myers, 1996; Woolf et al., 1997; Vogel et al., 2006) and mounting evidence points to that TNF induces and maintains not only peripheral but also spinal sensitization (Kawasaki et al., 2008; Youn et al., 2008). For instance, intrathecal (i.t.) TNF induces both thermal and mechanical hypersensitivity (Kwon et al., 2005; Kawasaki et al., 2008; Gao et al., 2009). Moreover, increased gene and/or protein expression levels of spinal TNF have been reported in different pain models (Bao et al., 2001; Hao et al., 2007; Cao et al., 2010) and blockage of spinal TNF actions attenuates pain-like behaviour (Svensson et al., 2005; Choi et al., 2010; Christianson et al., 2011). It has been shown that spinal TNF can contribute to persistent pain states by activating mitogen activating protein kinases (MAPK) such as p38 and c-Jun N-terminal kinase (JNK; Boyle et al., 2006; Gao et al., 2010).
The JNK has three isoforms, JNK1, JNK2 and JNK3; all forms are expressed in the central nervous system. Spinal JNK1 and JNK2 are critical for the development and maintenance of chronic pain states (Zhuang et al., 2006; Gao and Ji, 2008; Wang et al., 2012). Activation of the JNK signalling pathway in experimental models of pain is further supported by the observation that c-Jun, one of the main JNK substrates, is activated in the spinal cord subsequent to peripheral inflammation and spinal nerve ligation (Gao and Ji, 2008; Gao et al., 2010). Intrathecal administration of the JNK inhibitors SP600125 and D-JNK-1 attenuates pain-like behaviour (Obata et al., 2004; Gao et al., 2010; Wang et al., 2012), indicating that JNK is an important regulator of pain transmission.
As we have previously found that i.t. injection of the TNF inhibitor etanercept attenuates mechanical hypersensitivity in the carrageenan model (Choi et al., 2010), the first aim of this study was to explore if peripheral inflammation drives spinal synthesis and release of TNF. Secondly, in order to examine the role of spinal JNK activation in inflammatory hypersensitivity, we tested if the inhibition of spinal JNK prevents carrageenan-induced pain-like behaviour and if inflammation-induced activation of spinal JNK is mediated by TNF. Although a link between TNF and JNK activation has been demonstrated in in vitro studies (Pollock et al., 2002; Gao et al., 2009, 2010; Guma and Firestein, 2012), this has not been hitherto characterized in an in vivo inflammatory pain model. Our studies show that spinal JNK activity is dependent on TNF and that both spinal TNF and JNK are involved in the hyperalgesia phenotype associated with peripheral inflammation.
2. Methods
2.1 Animals
Experiments were carried out according to protocols approved by the Institutional Animal Care Committee of the University of California, San Diego (UCSD) and the local Swedish ethical committee for animal experiments (Stockholms Norra Djurförsöksetiska Nämnd) and complied with ethical guidelines of the International Association for the Study of Pain (Zimmermann, 1983). Male Holzman Sprague-Dawley rats (300–350 g, Harlan Laboratories, Indianapolis, IN, USA and the Netherlands) were housed individually in micro isolator filter cages (UCSD) and open cage system (Karolinska Institutet) in a temperature-controlled room (∼23 °C) and maintained on a 12-h light/dark cycle with free access to food and water.
Each animal was only subjected to one condition.
2.2 Induction of inflammation and behavioural analysis
To induce a state of local inflammation, λ-carrageenan (Sigma Aldrich, St. Louis, MO, USA, 100 μL of 2% solution (w/v) in saline) was injected subcutaneously into the plantar surface of the left hind paw of a rat under brief isoflurane anaesthesia. Animals were habituated to the testing environment prior to baseline tests. To assess thermal hypersensitivity, a modified Hargreaves type device was employed (Dirig et al., 1997). Rats were placed individually in Plexiglas cubicles with glass surface and after habituation a radiant heat stimulus was applied to each paw and the latency defined as the time (seconds) required for the paw to show a brisk withdrawal. Etanercept was injected i.t. 60 min, SP600125 and JIP-1 15 min prior to carrageenan injection (time = 0). The data were also presented as hyperalgesic index, a calculation that defines the magnitude of carrageenan-induced sensitization. It represents the area between the extrapolated baseline and the time-response curve after carrageenan injection. Increasing values indicate increasing hypersensitivity.
For assessment of mechanical hypersensitivity, another set of animals was used. Rats were placed in individual Plexiglas compartments with wire mesh bottom and after acclimatization, von Frey filaments (0.41–15.2 g, Stoelting, Wood Dale, IL, USA) were applied perpendicularly to the mid hind paw and held for 4–6 s. A positive response was noted if the paw was sharply withdrawn. The 50% probability of withdrawal threshold was determined by Dixon's up-down method (Chaplan et al., 1994). Etanercept, JIP-1 and SP600125 were injected with the same pretreatment times as listed above. Data were also presented as hyperalgesic index.
2.3 Intrathecal injections and drugs
All drugs were delivered intrathecally in 10 μL vehicle followed by 10 μL saline flush. The JNK inhibitor SP600125 (3.3 and 33 μg, Sigma Aldrich) was dissolved in 5% Tween-80, 20% dimethyl sulfoxide (DMSO) in saline, while the JIP-1 amide fragment 53–63 (0.3–30 μg, Sigma Aldrich) and the neutralizing anti-TNF binding protein etanercept (30 and 100 μg, Pfizer, New York, NY, USA) were dissolved in saline. To allow delivery, chronic lumbar i.t. catheters, single-lumen polyethylene (OD 0.36 mm, 8.5 cm in length) from the cisterna were implanted under isoflurane anaesthesia as described previously (Yaksh and Rudy, 1976).
2.4 Cerebrospinal fluid (CSF) collection
CSF was collected 1, 2, 4 and 18 h after carrageenan injection by using micropipettes (0.8–1.1 × 100 mm, KIMAX-51, Kimble Chase, Vineland, NJ, USA). Isoflurane-anaesthetized rats were placed in sternal recumbency and a midline skin incision was made posterior to a length of 3 cm. The muscles were bluntly dissected and retracted to expose the interspinous space at L4/L5 and the interspinous ligament and L5 spinous process were carefully removed. The L4 spinous process was elevated and tissue from the dura carefully removed in order to eliminate blood pooling at the site of collection. The pulled microcapillary tube was introduced into the i.t. space and 40–50 μL of CSF was collected. The CSF was centrifuged to remove cells and the supernatant was immediately frozen. For soluble form of TNF analysis, 25 μL of CSF diluted with buffer (1:1) used per well and the enzyme-linked immunosorbent assay performed according to the manufacturer's instructions (R & D Systems, Minneapolis, MN, USA).
2.5 Western blot analysis
Under isoflurane anaesthesia ipsilateral lumbar spinal cords (L1-L6) were removed at 1, 2, 4, 8, 12 and 18 h after carrageenan injection. Flash frozen spinal cords were homogenized in lysis buffer. The proteins were separated using NuPAGE 4–12% Bis-Tris gel electrophoresis and 3-morpholin-4-ylpropane-1-sulfonic acid (MOPS) or 2-(N-Morpholino)ethanesulfonic acid hydrate (MES) running buffer (Invitrogen, Gran Island, NY, USA) and transferred to nitrocellulose membranes (Invitrogen). After blocking non-specific binding sites the blots were incubated with primary antibodies against TNF (rabbit, 1:1000, cat No ab66579, Abcam, Cambridge, UK), phosphorylated JNK (p-JNK, mouse, 1:1000, cat No 9255, clone G9, Cell Signaling, Boston, MA, USA), phosphorylated c-Jun (p-c-Jun, rabbit, 1:1000, cat No 9261 and 9164, Cell Signaling), total JNK (t-JNK, rabbit, 1:2000, cat No 9525, Cell Signaling), total c-Jun (t-c-Jun, rabbit, cat No 9162) and β-actin (mouse, 1:100000, cat No A2228, clone AC-74, Sigma Aldrich) and then with horseradish peroxidase-labeled secondary antibodies. Immunopositive bands were detected with chemiluminescent reagents and X-Ray film (Kodak, Rochester, NY, USA). Immunopositive bands were normalized against β-actin for the respective band and the data expressed as levels relative to the naïve controls (% of control) within the same membrane. Signal intensity was measured using Quantity One (BioRad, Hercules, CA, USA). A separated group was used to detect the effect of TNF inhibition on JNK phosphorylation. The ipsilateral lumbar spinal cord sections (L1-L6) were removed 1 h after etanercept or vehicle (saline) pretreatment. Relative levels of p-JNK and tot JNK were assessed as described above.
2.6 Quantitative real-time polymerase chain reaction (PCR)
Under isoflurane anaesthesia, ipsilateral lumbar spinal cords (L1-L6) were removed at 1, 2, 4, 8, 12 and 18 h after carrageenan injection. Flash frozen spinal cords were extracted with TRIzol (Invitrogen) according to the manufacturer's protocol. The RNA was reverse transcribed to complementary DNA (cDNA) and quantitative real-time PCR (GeneAmp 7000 Sequence Detection system, Applied Biosystems, Foster City, CA, USA) was performed with hydrolysis probes, both according to the manufacturer's instructions, to determine the relative mRNA levels. Pre-developed specific primers were used to detect Tnf (Rn99999017_m1) and the reference gene Hprt1 (Rn01527838_g1). Data were normalized to Hprt1 mRNA levels and presented as relative gene expression.
2.7 Statistical analysis
Western blot, qPCR and behavioural data were expressed as mean ± standard error of the mean (SEM). Western blot, qPCR and all behavioural hyperalgesic index data except data showing JIP-1 effect on mechanical hypersensitivity (Student's t-test was used) were analysed by one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test. For all of the behavioural time course analysis, two-way repeated measures ANOVA was used, followed by Bonferroni post hoc test for correction of multiple comparisons. The criterion for significance was set as p-values < 0.05.
3. Results
3.1 TNF levels are increased in the CSF and the spinal cord after carrageenan-induced peripheral inflammation
We examined if carrageenan-induced inflammation leads to elevated TNF protein levels in CSF. While TNF protein levels were not detected in naïve rats (n = 5), its levels reached detection limit 1 h post-injection of carrageenan (6.7 ± 4.7 pg/mL; n = 6), and in comparison to those levels, a significant increase of TNF was found in CSF sampled 2 (41.1 ± 16.9 pg/mL; n = 5) and 4 (32 ± 6.7 pg/mL; n = 6) h after induction of inflammation (p < 0.05; Fig. 1A). In order to determine if mRNA and protein levels in the spinal cord also changed in response to the peripheral inflammation, L1-L6 spinal cords were collected at different time points after the carrageenan injection. Tnf mRNA levels were increased 10-fold 8 h after carrageenan injection compared with naïve rats (p < 0.05; Fig. 1B). No difference in relative expression levels of the housekeeping gene (Hprt1) was observed between the different time points after injection of carrageenan (data not shown). TNF protein levels in the spinal cord were also increased 8 h after carrageenan injection compared with naïve rats (0 h: 100 ± 15.6%, n = 6; 8 h: 364.7 ± 71.5%, n = 6; p < 0.05), and remained significantly higher than in control tissues at the 12 and 18 h time points (12 h: 329.7 ± 41.1%, n = 6; 18 h: 493.1 ± 98.4%, n = 3; p < 0.05; Fig. 1C).
Figure 1.
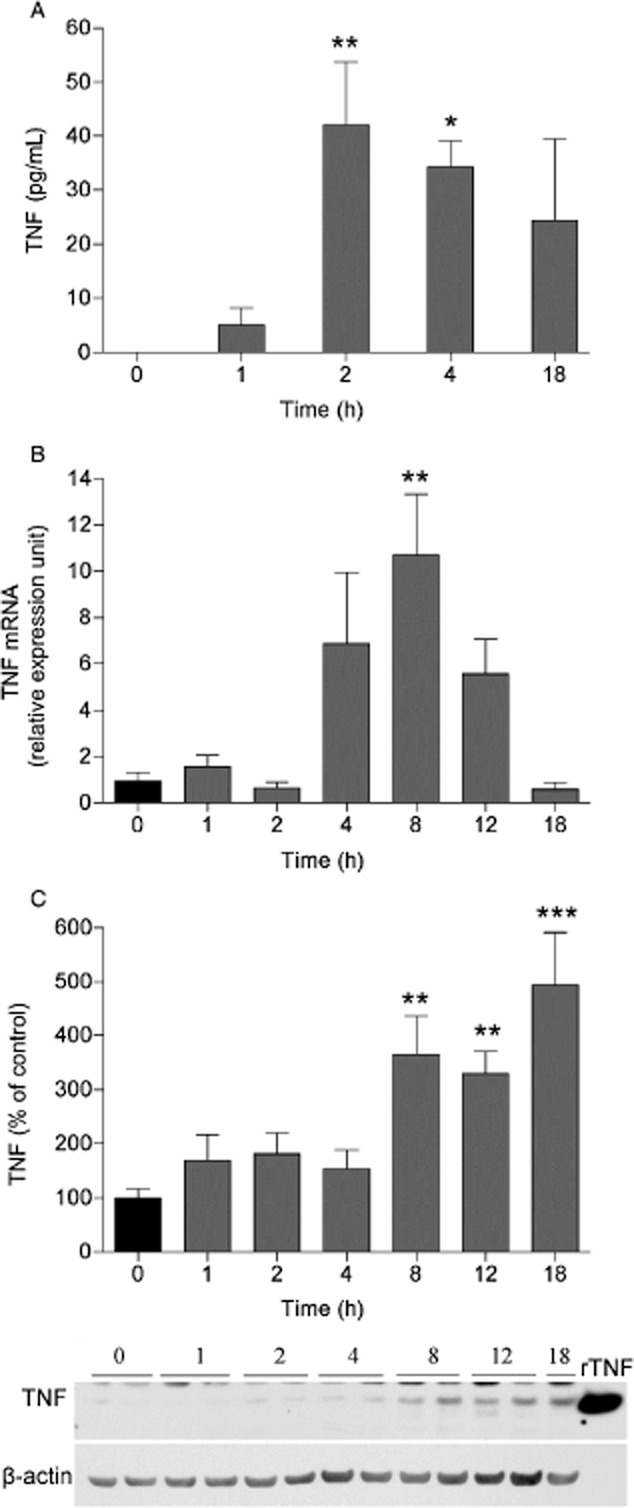
Carrageenan-induced peripheral inflammation increases TNF levels in the spinal cord. Bar graphs displaying (A) TNF levels in CSF (pg/mL) and (B) mRNA levels presented as relative expression units (REU) and (C) TNF protein expression as % of control in the lumbar ipsilateral spinal cord 1, 2, 4, 8, 12 and 18 h after carrageenan injection to the paw, compared with naïve (0 h time point) rats. Representative western blot images showing TNF and β-actin (loading control) immunopositive bands from spinal cord homogenates over time after carrageenan injection. Recombinant soluble TNF (rTNF) was used as positive control. All data are presented as mean ± SEM, *p < 0.05; **p < 0.01; ***p < 0.001, versus naive controls.
3.2 Inhibition of TNF reduces carrageenan-induced thermal and mechanical hypersensitivity
As we found that both mRNA and protein levels of TNF were increased in the spinal cord and TNF was detected in CSF after induction of peripheral inflammation the soluble TNF receptor etanercept was used to determine the role of TNF in carrageenan-induced thermal and mechanical hypersensitivity. Etanercept (30 and 100 μg) or the vehicle (saline, n = 5–6) was injected i.t. 1 h before carrageenan injection. While the 30 μg, i.t. dose did not change withdrawal latencies at any time points (n = 6; p > 0.05; Fig. 2A), it reduced mechanical thresholds at 60 and 120 min time points (n = 6; p < 0.05; Fig. 2C). The related hyperalgesic indices showed that there was no change in thermal hypersensitivity (p > 0.05; Fig. 2B), but significant decrease in mechanical hypersensitivity (p < 0.05; Fig. 2D). The withdrawal latencies were longer at 120 and 150 min time points (n = 6; p < 0.05; Fig. 2A) and the mechanical thresholds were higher at 60, 120 and 150 min time points in the 100 μg, i.t. etanercept group compared with the vehicle treated group (n = 7; p < 0.05; Fig. 2C). The related hyperalgesic indices showed that 100 μg, i.t. etanercept delayed the onset of both thermal and mechanical hypersensitivity (p < 0.05; Fig. 2B and D). No changes in withdrawal latencies or mechanical thresholds were observed in the control (without carrageenan injection) group (n = 6; p > 0.05) (Fig. 2) and contralateral sides (data not shown).
Figure 2.
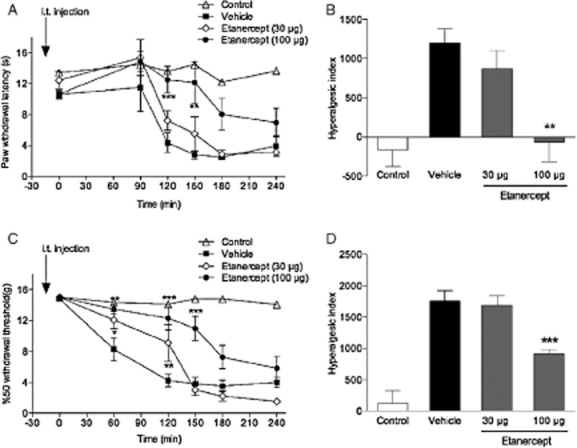
Intrathecal etanercept attenuates carrageenan-induced thermal and mechanical hypersensitivity. Graphs showing the effect of etanercept (30 and 100 μg) injected i.t. 1 h before carrageenan injection on (A) thermal withdrawal latencies and (C) mechanical withdrawal thresholds over time compared with the i.t. vehicle (saline)-injected group. (B and D) Bar graphs showing the related hyperalgesic indeces calculated for the 0–240 min time period after carrageenan injection. Control group represent naïve rats without carrageenan injection. All data are presented as mean ± SEM, *p < 0.05; **p < 0.01; ***p < 0.001, versus vehicle controls.
3.3 Levels of phosphorylated JNK and c-Jun are increased in spinal cord after carrageenan-induced inflammation
To investigate if JNK1, JNK2 and the downstream substrate c-Jun are activated after carrageenan injection, L1-L6 spinal cords were harvested at different time points. Three bands were detected with the p-JNK antibody whereof the lowest was considered non-specific based on the molecular weight (approximately 40 kDa) and signal intensities for bands corresponding to 46 kDa (p-JNK1) and 54 kDa (p-JNK2) levels were quantified. The p-JNK1 signal was very strong compared with p-JNK2; therefore, the membranes were subjected to a shorter and a longer exposure for assessment of p-JNK1 and p-JNK2, respectively. p-JNK1 and p-JNK2 levels were significantly increased 1, 8, 12 h after carrageenan injection compared with basal phosphorylation levels of JNK1/2 (p-JNK1 0 h: 100 ± 9.4%, n = 11; 1 h: 340.4 ± 40%, n = 11; 8 h: 387.7 ± 99.3%, n = 10; 12 h: 572.5 ± 89.2%, n = 6; p < 0.05; p-JNK2 0 h: 100 ± 11.1%, n = 11; 1 h: 282.2 ± 22.3%, n = 11; 8 h: 226.4 ± 45.6%, n = 10; 12 h: 241.3 ± 33.4%, n = 6; p < 0.05) (Fig. 3A and B). The levels of total JNK1/2 were not changed in the spinal cord subsequent to the induction of peripheral inflammation (data not shown). The p-c-Jun levels were increased significantly only 12 h after injection compared with basal phosphorylation levels of c-Jun (0 h: 100 ± 5.2%, n = 6; 12 h: 300.2 ± 41.8%, n = 4; p < 0.05; Fig. 3C). The levels of total c-Jun were not changed after carrageenan-induced peripheral inflammation (data not shown).
Figure 3.
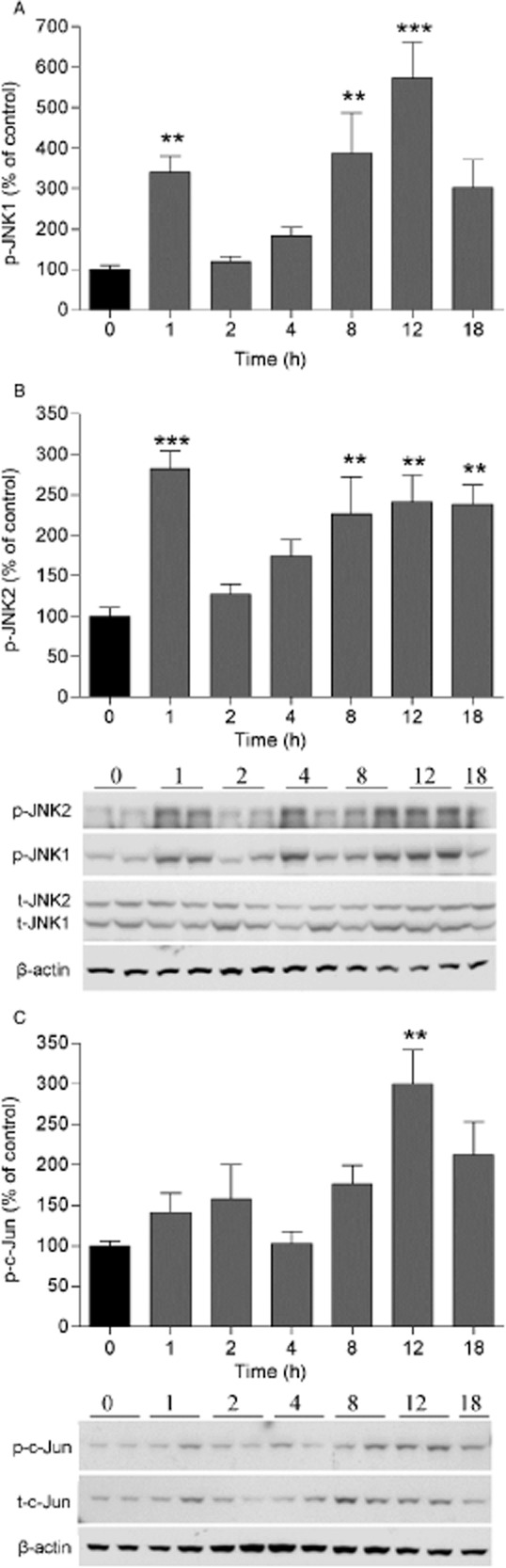
Carrageenan-induced peripheral inflammation increases JNK1/2 and c-Jun activity in the spinal cord. Bar graphs showing percent (%) change of (A) p-JNK1, (B) p-JNK2 and (C) p-c-Jun in the lumbar ipsilateral spinal cord 1, 2, 4, 8, 12 and 18 h after carrageenan injection compared with naïve (0 h time point) rats. Representative western blot images showing p-JNK1/2, p-c-Jun, total JNK1/2 (t-JNK1/2), total c-Jun (t-c-Jun) and β-actin (loading control) immunopositive bands from spinal cord homogenates over time after carrageenan injection. All data are presented as mean ± SEM, **p < 0.01; ***p < 0.001, versus naive controls.
3.4 JNK inhibitors attenuate carrageenan-induced thermal and mechanical hypersensitivity
To test the role of spinal JNK in carrageenan-induced thermal and mechanical hypersensitivity two different JNK inhibitors, SP600125 (3.3 and 33 μg, i.t.) and JNK-interacting protein-1 (JIP-1) (0.3–30 μg, i.t.) were used. Inhibitors and vehicles [5% Tween-80, 20% DMSO in saline (n = 8) and saline (n = 6–7), respectively] were injected i.t. 15 min before carrageenan injection. 3.3 μg, i.t. SP600125 at 90 and 120 min time points (n = 8; p < 0.05) and 33 μg, i.t. dose at 90, 120, 150 and 180 min time points (n = 9; p < 0.05) attenuated carrageenan-induced thermal hypersensitivity (Fig. 4A). The related hyperalgesic indeces showed that both doses attenuated carrageenan-induced thermal hypersensitivity (p < 0.05; Fig. 4A and B). However, we failed to show any antinociceptive effect on carrageenan-induced mechanical hypersensitivity with the same doses of SP600125 (data not shown). To examine if a higher dose of SP600125 (66 μg, i.t.) has effect on mechanical hypersensitivity it was injected in a volume of 20 μL (due to solubility issues at doses higher than 3.3 μg, i.t.). Unfortunately the 20 μL volume of vehicle had antinociceptive effects (data not shown) interfering with the assay and therefore, a second JNK inhibitor, JIP-1 was included in this study. While i.t. injection of 0.3 μg of JIP-1 attenuated carrageenan-induced thermal hypersensitivity only at 90 min time point (n = 4; p < 0.05), 3 μg, i.t. JIP-1 at 60, 90, 120, 150 min time points (n = 6; p < 0.05) and 30 μg, i.t. JIP-1 at 60, 90, 120 min time points (n = 6; p < 0.05) increased withdrawal latencies (Fig. 5A). The related hyperalgesic indeces showed that 3 and 30 μg (p < 0.05), but not the 0.3 μg (p > 0.05), i.t. JIP-1 reduced the thermal hypersensitivity (Fig. 5B). To test if JIP-1 also has effect on mechanical hypersensitivity, the highest dose (30 μg, i.t.) was injected. While we could not find any statistically difference at any time point in JIP-1-treated group compared with the vehicle-treated group (n = 8; p > 0.05; Fig. 5C), the related hyperalgesic indeces showed that there was a significant reversal of the carrageenan-induced mechanical hypersensitivity (p < 0.05; Fig. 5D). No changes in withdrawal latencies or mechanical thresholds were observed in the control group (without carrageenan injection) and contralateral sides (data not shown).
Figure 4.
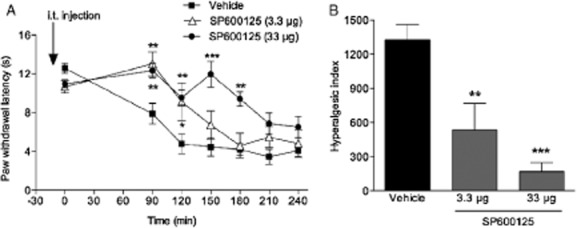
Intrathecal injection of the JNK inhibitor SP600125 attenuates carrageenan-induced thermal hypersensitivity. (A) Graphs depicting the effect of SP600125 (3.3 and 33 μg, i.t.), injected i.t. 15 min before carrageenan injection on thermal paw withdrawal latencies over time compared with i.t. vehicle (5% Tween-80, 20% DMSO in saline) injected group. (B) Bar graphs showing the hyperalgesic index calculated for the 0–240 min time period after carrageenan injection. All data are presented as mean ± SEM, *p < 0.05; **p < 0.01; ***p < 0.001, versus vehicle controls.
Figure 5.
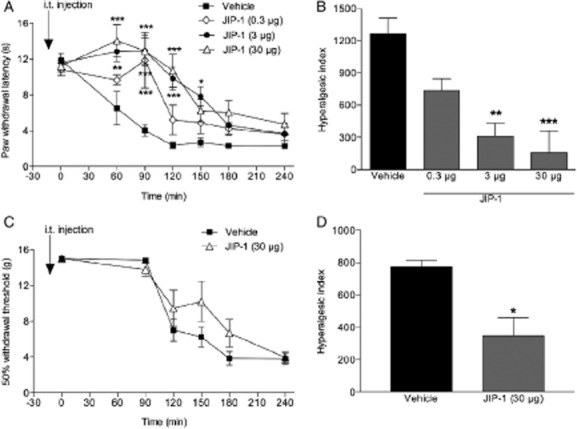
Intrathecal injection of the JNK inhibitor JIP-1 attenuates carrageenan-induced thermal and mechanical hypersensitivity. Graphs showing effects of JIP-1 (0.3–30 μg) or vehicle (saline)-injected i.t. 15 min before carrageenan injection on (A) thermal withdrawal latencies and (C) mechanical withdrawal thresholds. (B and D) Bar graphs showing the related hyperalgesic indeces for the 0–240 min time period after carrageenan injection. All data are presented as mean ± SEM, *p < 0.05; **p < 0.01; ***p < 0.001, versus vehicle controls.
3.5 Inhibition of TNF reduces increased phosphorylated levels of JNK after carrageenan injection
To determine if TNF drives spinal JNK activation after carrageenan injection, etanercept (100 μg, i.t.) was applied 15 min before the injection of carrageenan. L1-L6 spinal cords were removed 1 h (peak phosphorylation time of JNK) after etanercept pretreatment and JNK activation was evaluated. The increased levels of p-JNK1 and p-JNK2 were significantly inhibited by etanercept pretreatment (p-JNK1 control: 100 ± 10.3%, n = 8, carrageenan + vehicle: 294.6 ± 35%, n = 8, carrageenan + etanercept: 142.8 ± 16.5%, n = 8, p < 0.05; p-JNK2 control: 100 ± 16.1%, n = 8, carrageenan + vehicle: 271.5 ± 32%, n = 8, carrageenan + etanercept: 144.7 ± 29%, n = 8; p < 0.05; Fig. 6A and B). Total levels of JNK1 and JNK2 were not changed in any of the groups (data not shown).
Figure 6.
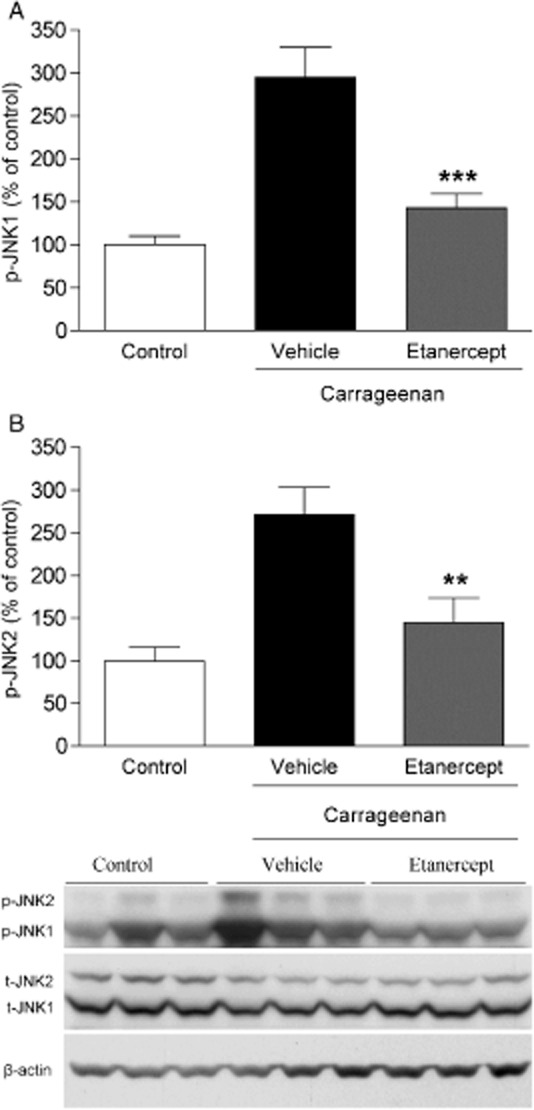
Intrathecal injection of TNF antagonist etanercept reduces carrageenan-induced spinal JNK1/2 activation. Bar graphs depicting percent (%) change of (A) p-JNK1 and (B) p-JNK2 in the lumbar ipsilateral spinal cord 1 h after carrageenan injection in the presence of i.t. etanercept (100 μg), compared with vehicle (saline) injected control rats. Representative western blot images showing immunopositive bands from spinal cord homogenates for p-JNK1/2, total JNK1/2 (t-JNK1/2) and β-actin (loading control). All data are presented as mean ± SEM, **p < 0.01; ***p < 0.001, versus vehicle controls.
4. Discussion and conclusions
In the current study, we show that induction of peripheral inflammation leads to an increased spinal gene and protein expression and release of TNF, as well as activation of JNK in the spinal cord. Inflammation-induced mechanical and thermal hypersensitivity were reduced by preventing the actions of spinal TNF and by inhibiting signalling through the JNK pathway. This is in line with accumulating evidence that JNK is activated in astrocytes in response to cytokine stimulation and that JNK activation is one important intracellular event in the development of neurological disorders following injury or disease (Fernandes et al., 2007; Gao et al., 2009; Zhang et al., 2013).
TNF is a membrane-bound cytokine (mTNF) that is cleaved by TNF-alpha-converting enzyme to release the 17 kDa soluble form of TNF (sTNF). TNF is released by many immune cells, including macrophages and glial cells and both forms are biologically active and exert their actions by binding to TNF receptor 1 and 2. Our findings are in parallel with earlier reports showing TNF in CSF (Bianchi et al., 2007) and elevated gene and protein expressions of TNF and its role in mechanical and thermal hypersensitivity in inflammatory pain states (Cunha et al., 1992; Woolf et al., 1997; Raghavendra et al., 2004; Christianson et al., 2011). The fact that we could detect TNF in cell-free (centrifuged) CSF and observed a TNF immunoreactive band at approximately 17 kDa by western blotting of spinal cord homogenates suggests that TNF is cleaved from cellular membranes in response to a peripheral inflammation. This is in contrast to previous work showing that mTNF but not sTNF levels were significantly increased in the spinal cord after formalin injection (Zhou et al., 2008). Interestingly, in the same study, direct exposure of cultured microglia to a potent inflammatory stimulus resulted in increased mTNF levels followed by increase in released sTNF. Thus, it is possible that carrageenan-induced inflammation, which is more pronounced and lasts longer than the formalin-induced reaction, activates proteases that cleave TNF from cellular membranes in the spinal cord, releasing TNF that is detectable in CSF prior to increases of Tnf mRNA in the spinal cord tissue. One alternative explanation for the early increase of TNF protein levels in CSF is that production of TNF at the site of inflammation is redistributed to CSF. However, as recent findings have shown that carrageenan-induced paw inflammation does not lead to elevated plasma levels of TNF (Loram et al., 2007) or increased blood-spinal cord barrier permeability (Xanthos et al., 2012), it is not likely that the elevated spinal TNF levels observed in the current study is the result of transport of TNF from the periphery to the spinal cord.
In order to block the actions of spinal TNF, we injected etanercept intrathecally. Etanercept consists of two extracellular domains of the human 75 kD (p75) TNF receptor linked by the constant Fc portion of human immunoglobulin. This decoy receptor binds TNF and prevents it from acting on membrane-bound receptors (Hareoko and Bykerk, 2007). We found that etanercept prevented both carrageenan-induced mechanical and thermal hypersensitivity, which is in agreement with previous works (Inglis et al., 2005; Boettger et al., 2010; Choi et al., 2010; Christianson et al., 2010, 2011). Although the exact actions by which TNF is involved in nociception at the spinal level remains to be delineated, there are some possible mechanisms. In the spinal cord, TNF receptors are expressed on neurons, astrocytes and microglia (Gruber-Schoffnegger et al., 2013), thus TNF can act both in a paracrine and autocrine fashion. Once released, it is thought that TNF facilitates glutamatergic synaptic transmission between C fibres and lamina II neurons (Kawasaki et al., 2008; Zhang and Dougherty, 2011; Zhang et al., 2011) by binding to neuronally expressed TNF receptors (Ohtori et al., 2004; Xu et al., 2006; Zhang and Dougherty, 2011; Gruber-Schoffnegger et al., 2013). TNF drives neuronal excitability by decreasing inhibitory synaptic transmission in the spinal dorsal horn (Kawasaki et al., 2008; Zhang and Dougherty, 2011), up-regulating TRPV1 channels in spinal cord (Boettger et al., 2010) and modulating α-amino-3-hydroxy-5-methyl-4-isoxazole proprionic acid receptor trafficking (Choi et al., 2010). Noteworthy, recent work suggests that the induction of hyperalgesia by TNF is not a direct neuronal effect but is mediated indirectly via activation of spinal glial cells and subsequent release of other neuronally active factors (Gruber-Schoffnegger et al., 2013).
Despite similar levels of JNK1 and JNK2 proteins in the spinal cord, we found more pronounced phosphorylation of JNK1 than JNK2, both at baselines and after induction of inflammation. Consistently, the activation of both JNK1 and JNK2 with a stronger activation of JNK1 has been shown in the spinal cord after complete Freund's adjuvant (CFA) induced inflammation. Moreover, in the same study, JNK1 modulated spinal pain signalling to a greater extent than JNK2 (Gao et al., 2010). Of note, in our studies JNK1 and JNK2 were both activated in a biphasic fashion, following the same temporal pattern of the TNF increase in CSF. A link between TNF and JNK activation has been established in earlier work demonstrating that i.t. TNF leads to spinal activation of JNK (Gao et al., 2010). In our study, i.t. etanercept injected prior to induction of inflammation prevented activation of JNK at the early time point, providing further support for that TNF receptor activation drives signalling through the JNK pathway in the spinal cord. Although we did not assess the antinociceptive actions of etanercept or the effect of TNF blockage on JNK activity at the later time points, the matching time profile between the second wave of TNF release and JNK activation may suggest that these factors are linked and involved not only in induction of hypersensitivity but also at later time points. However, further studies are warranted to explore this causal relationship in detail. Interestingly, c-Jun, one of the major downstream substrates of JNK, was only activated at later time points suggesting that JNK couples to different intracellular signalling pathways at different phases of inflammatory pain.
By immunohistochemistry, we were unable to show the cellular localization of activated JNK in the spinal cord. However, earlier works have showed that JNK is activated in spinal astrocytes, but not in neurons and microglia in spinal nerve ligation and CFA models (Gao et al., 2009, 2010). After stimulation with TNF, rapid activation of both JNK1 and JNK2 was found demonstrating that phosphorylation of JNK in astrocytes is feasible, in support with other studies showing that astrocytes respond to TNF by MAPK activation and release of IL-1β, MCP-1, MMP (Guo et al., 2007; Gao et al., 2009; Guma and Firestein, 2012). It is important to note however that JNK activation was detected in spinal neurons in a bone cancer model (Wang et al., 2012). Even though localization of JNK activation may be model dependent, we cannot rule out that JNK was not activated in neurons in the current study.
Overexpression of JIP-1 deactivates the JNK pathway and JIP-1 has a higher JNK specificity than SP600125 (Heo et al., 2004), the second JNK inhibitor that was used in this study. We have previously found that i.t. JIP-1 attenuated antibody-induced hypersensitivity in mice (Bas et al., 2012) and here we show that it also attenuates carrageenan-induced mechanical and thermal hypersensitivity. Although the DMSO-containing vehicle of SP600125 interfered with assessment of mechanical hypersensitivity, it was effective on carrageenan-induced thermal hypersensitivity. Previously, it has been shown that i.t. SP600125 has predominant effect on thermal hypersensitivity (Doya et al., 2005; Daulhac et al., 2006; Cao et al., 2007; Ikeda et al., 2012), and given the modest effect of JIP-1 on mechanical hypersensitivity, it is possible that spinal JNK is to a greater extent involved in thermal than mechanical hypersensitivity.
In conclusion, our results emphasize that blocking spinal TNF inhibits both pain-like behaviour and the increased levels of phosphorylated JNK1 and JNK2 subsequent to carrageenan injection. Previous studies demonstrating a link between TNF and JNK pathway have been performed in vitro in astrocytes (Gao et al., 2009, 2010), synovial fibroblasts and chondrocytes (Guma and Firestein, 2012), as well as on sensory neurons (Pollock et al., 2002). To our knowledge, our work is the first to show TNF-dependent activation of spinal JNK after peripheral inflammation in vivo. TNF and signalling pathways downstream of TNF receptor such as JNK represent potential targets for pain relief.
Author contributions
Experimental design, conduction of experiments, data analysis, preparation of manuscript, careful editing of manuscript: D.B.B., S.A. Data analysis, preparation of manuscript, careful editing of manuscript: K.S. Experimental design, conduction of experiments, data analysis: S.C., B.F., J.S. Development of concept, experimental design, editing: X.Y.H. Development of concept, experimental design, preparation of manuscript, editing: T.L.Y. Development of concept, experimental design, conduction of experiments, preparation of manuscript, careful editing of manuscript: C.I.S. All authors discussed the results and commented on the manuscript.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
TNF induces JNK activation in spinal primary astrocytes.
Primary culture of astrocytes. Western blotting (Codeluppi et al., 2011).
TNF induces phosphorylation of JNK in cultured spinal astrocytes.
References
- Bao L, Zhu Y, Elhassan AM, Wu Q, Xiao B, Zhu J, Lindgren JU. Adjuvant-induced arthritis: IL-1beta, IL-6 and TNF-alpha are up-regulated in the spinal cord. Neuroreport. 2001;12:3905–3908. doi: 10.1097/00001756-200112210-00010. [DOI] [PubMed] [Google Scholar]
- Bas DB, Su J, Sandor K, Agalave NM, Lundberg J, Codeluppi S, Baharpoor A, Nandakumar KS, Holmdahl R, Svensson CI. Collagen antibody-induced arthritis evokes persistent pain with spinal glial involvement and transient prostaglandin dependency. Arthritis Rheum. 2012;64:3886–3896. doi: 10.1002/art.37686. [DOI] [PubMed] [Google Scholar]
- Bianchi M, Martucci C, Ferrario P, Franchi S, Sacerdote P. Increased tumor necrosis factor-alpha and prostaglandin E2 concentrations in the cerebrospinal fluid of rats with inflammatory hyperalgesia: The effects of analgesic drugs. Anesth Analg. 2007;104:949–954. doi: 10.1213/01.ane.0000258060.89380.27. [DOI] [PubMed] [Google Scholar]
- Boettger MK, Weber K, Grossmann D, Gajda M, Bauer R, Bar KJ, Schulz S, Voss A, Geis C, Brauer R, Schaible HG. Spinal tumor necrosis factor alpha neutralization reduces peripheral inflammation and hyperalgesia and suppresses autonomic responses in experimental arthritis: A role for spinal tumor necrosis factor alpha during induction and maintenance of peripheral inflammation. Arthritis Rheum. 2010;62:1308–1318. doi: 10.1002/art.27380. [DOI] [PubMed] [Google Scholar]
- Boyle DL, Jones TL, Hammaker D, Svensson CI, Rosenberg S, Albani S, Sorkin LS, Firestein GS. Regulation of peripheral inflammation by spinal p38 MAP kinase in rats. PLoS Med. 2006;3:338. doi: 10.1371/journal.pmed.0030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao F, Gao F, Xu AJ, Chen ZJ, Chen SS, Yang H, Yu HH, Mei W, Liu XJ, Xiao XP, Yang SB, Tian XB, Wang XR, Tian YK. Regulation of spinal neuroimmune responses by prolonged morphine treatmentin a rat model of cancer induced bone pain. Brain Res. 2010;1326:162–173. doi: 10.1016/j.brainres.2010.02.039. [DOI] [PubMed] [Google Scholar]
- Cao FL, Liu MG, Hao J, Li ZM, Chen J. Different roles of spinal p38 and c-Jun N-terminal kinase pathways in bee venom-induced multiple pain-related behaviors. Neurosci Lett. 2007;427:50–54. doi: 10.1016/j.neulet.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Choi JI, Svensson CI, Koehrn FJ, Bhuskute A, Sorkin LS. Peripheral inflammation induces tumor necrosis factor dependent AMPA receptor trafficking and Akt phosphorylation in spinal cord in addition to pain behavior. Pain. 2010;149:243–253. doi: 10.1016/j.pain.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson CA, Corr M, Firestein GS, Mobargha A, Yaksh TL, Svensson CI. Characterization of the acute and persistent pain state present in K/BxN serum transfer arthritis. Pain. 2010;151:394–403. doi: 10.1016/j.pain.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson CA, Dumlao DS, Stokes JA, Dennis EA, Svensson CI, Corr M, Yaksh TL. Spinal TLR4 mediates the transition to a persistent mechanical hypesensitivity after the resolution of inflammation in serum-transferred arthritis. Pain. 2011;152:2881–2891. doi: 10.1016/j.pain.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codeluppi S, Norsted Gregory E, Kjell J, Wigerblad G, Olson L, Svensson CI. Influence of rat substrain and growth conditions on the characteristics of primary cultures of adult rat spinal cord astrocytes. J Neurosci Methods. 2011;197:118–127. doi: 10.1016/j.jneumeth.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Cunha FQ, Poole S, Lorenzetti BB, Ferreira SH. The pivotal role of tumour necrosis factor alpha in the development of inflammatory hyperalgesia. Br J Pharmacol. 1992;107:660–664. doi: 10.1111/j.1476-5381.1992.tb14503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daulhac L, Mallet C, Courteix C, Etienne M, Duroux E, Privat AM, Eschalier A, Fialip J. Diabetes-induced mechanical hyperalgesia involves spinal mitogen-activated protein kinase activation in neurons and microglia via N-methyl-D-aspartate-dependent mechanisms. Mol Pharmacol. 2006;70:1246–1254. doi: 10.1124/mol.106.025478. [DOI] [PubMed] [Google Scholar]
- Dirig DM, Salami A, Rathbun ML, Ozaki GT, Yaksh TL. Characterization of variables defining hindpaw withdrawal latency evoked by radiant thermal stimuli. J Neurosci Methods. 1997;76:183–191. doi: 10.1016/s0165-0270(97)00097-6. [DOI] [PubMed] [Google Scholar]
- Doya H, Ohtori S, Fujitani M, Saito T, Hata K, Ino H, Takahashi K, Moriya H, Yamashita T. c-Jun N-terminal kinase activation in dorsal root ganglion contributes to pain hypersensitivity. Biochem Biophys Res Commun. 2005;335:132–138. doi: 10.1016/j.bbrc.2005.07.055. [DOI] [PubMed] [Google Scholar]
- Fernandes A, Falcao AS, Silva RF, Brito MA, Brites D. MAPKs are key players in mediating cytokine release and cell death induced by unconjugated bilirubin in cultured rat cortical astrocytes. Eur J Neurosci. 2007;25:1058–1068. doi: 10.1111/j.1460-9568.2007.05340.x. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Activation of JNK pathway in persistent pain. Neurosci Lett. 2008;437:180–183. doi: 10.1016/j.neulet.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Xu ZZ, Liu YC, Wen YR, Decosterd I, Ji RR. The c-Jun N-terminal kinase 1 (JNK1) in spinal astrocytes is required for the maintenance of bilateral mechanical allodynia under a persistent inflammatory pain condition. Pain. 2010;148:309–319. doi: 10.1016/j.pain.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29:4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber-Schoffnegger D, Drdla-Schutting R, Hönigsperger C, Wunderbaldinger G, Gassner M, Sandkühler J. Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-α and IL-1β is mediated by glial cells. J Neurosci. 2013;33:6540–6551. doi: 10.1523/JNEUROSCI.5087-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guma M, Firestein GS. c-Jun N-terminal kinase in inflammation and rheumatic diseases. Open Rheumatol. 2012;6:220–231. doi: 10.2174/1874312901206010220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, Wei F, Dubner R, Ren K. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Mata M, Glorioso JC, Fink DJ. Gene transfer to interfere with TNFalpha signaling in neuropathic pain. Gene Ther. 2007;14:1010–1016. doi: 10.1038/sj.gt.3302950. [DOI] [PubMed] [Google Scholar]
- Hareoko B, Bykerk V. Etanercept in the treatment of rheumatoid arthritis. Ther Clin Risk. 2007;3:99–105. doi: 10.2147/tcrm.2007.3.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo YS, Kim SK, Seo CI, Kim YK, Sung BJ, Lee HS, Lee JI, Park SY, Kim JH, Hwang KY, Hyun YL, Jeon YH, Ro S, Cho JM, Lee TG, Yang CH. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J. 2004;23:2185–2195. doi: 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Kiritoshi T, Murase K. Contribution of microglia and astrocytes to the central sensitization, inflammatory and neuropathic pain in the juvenile rat. Mol Pain. 2012;8:43. doi: 10.1186/1744-8069-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis JJ, Nissim A, Lees DM, Hunt SP, Chernajovsky Y, Kidd BL. The differential contribution of tumour necrosis factor to thermal and mechanical hyperalgesia during chronic inflammation. Arthritis Res Ther. 2005;7:807–816. doi: 10.1186/ar1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: Distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon MS, Shim EJ, Seo YJ, Choi SS, Lee JY, Lee HK, Suh HW. Differential modulatory effects of cholera toxin and pertussis toxin on pain behavior induced by TNF-alpha, interleukin-1beta and interferon-gamma injected intrathecally. Arch Pharm Res. 2005;28:582–586. doi: 10.1007/BF02977762. [DOI] [PubMed] [Google Scholar]
- Loram LC, Fuller A, Fick LG, Cartmell T, Poole S, Mitchell D. Cytokine profiles during carrageenan-induced inflammatory hyperalgesia in rat muscle and hind paw. J Pain. 2007;8:127–136. doi: 10.1016/j.jpain.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtori S, Takahashi K, Moriya H, Myers RR. TNF-α and TNF-α receptor type 1 upregulation in glia and neurons after peripheral nerve injury: Studies in murine DRG and spinal cord. Spine. 2004;29:1082–1088. doi: 10.1097/00007632-200405150-00006. [DOI] [PubMed] [Google Scholar]
- Pollock J, McFarlane SM, Connell MC, Zehavi U, Vandenabeele P, MacEwan DJ, Scott RH. TNF activated p38 mitogen-activated protein kinase (p38MAPK) and c-Jun N-terminal kinase (JNK) but not p42/p44 MAPK. Neuropharmacology. 2002;42:93–106. doi: 10.1016/s0028-3908(01)00163-0. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Schäfers M, Jones TL, Powell H, Sorkin LS. Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett. 2005;379:209–213. doi: 10.1016/j.neulet.2004.12.064. [DOI] [PubMed] [Google Scholar]
- Vogel C, Stallforth S, Sommer C. Altered pain behavior and regeneration after nerve injury in TNF receptor deficient mice. J Peripher Nerv Syst. 2006;11:294–303. doi: 10.1111/j.1529-8027.2006.00101.x. [DOI] [PubMed] [Google Scholar]
- Wagner R, Myers RR. Schwann cells produce tumor necrosis factor alpha: Expression in injured and non-injured nerves. Neuroscience. 1996;73:625–629. doi: 10.1016/0306-4522(96)00127-3. [DOI] [PubMed] [Google Scholar]
- Wang XW, Hu S, Mao-Ying QL, Li Q, Yang CJ, Zhang H, Mi WL, Wu GC, Wang YQ. Activation of c-jun N-terminal kinase in spinal cord contributes to breast cancer induced bone pain in rats. Mol Brain. 2012;5:21. doi: 10.1186/1756-6606-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Allchorne A, Safieh-Garabedian B, Poole S. Cytokines, nerve growth factor and inflammatory hyperalgesia: The contribution of tumour necrosis factor alpha. Br J Pharmacol. 1997;121:417–424. doi: 10.1038/sj.bjp.0701148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthos DN, Pungel I, Wunderbaldinger G, Sandkuhler J. Effects of peripheral inflammation on the blood-spinal cord barrier. Mol Pain. 2012;18:8. doi: 10.1186/1744-8069-8-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JT, Xin WJ, Zang Y, Wu CY, Liu XG. The role of tumor necrosis factor-alpha in the neuropathic pain induced by Lumbar 5 ventral root transection in rat. Pain. 2006;123:306–321. doi: 10.1016/j.pain.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- Youn DH, Wang H, Jeong SJ. Exogenous tumor necrosis factor-alpha rapidly alters synaptic and sensory transmission in the adult rat spinal cord dorsal horn. J Neurosci Res. 2008;86:2867–2875. doi: 10.1002/jnr.21726. [DOI] [PubMed] [Google Scholar]
- Zhang FF, Morioka N, Nakashima-Hisaoka K, Nakata Y. Spinal astrocytes stimulated by tumor necrosis factor-α and/or interferon-γ attenuate connexin 43-gap junction via c-jun terminal kinase activity. J Neurosci Res. 2013;91:745–756. doi: 10.1002/jnr.23213. [DOI] [PubMed] [Google Scholar]
- Zhang H, Dougherty PM. Acute inhibition of signaling phenotype of spinal GABAergic neurons by tumour necrosis factor. J Physiol. 2011;589:4511–4526. doi: 10.1113/jphysiol.2011.215301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Berta T, Xu ZZ, Liu T, Park JY, Ji RR. TNF-α contributes to spinal cord synaptic plasticity and inflammatory pain: Distinct role of TNF receptor subtypes 1 and 2. Pain. 2011;152:419–427. doi: 10.1016/j.pain.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Peng X, Hao S, Fink DJ, Mata M. HSV-mediated transfer of interleukin-10 reduces inflammatory pain through modulation of membrane tumor necrosis factor alpha in spinal cord microglia. Gene Ther. 2008;15:183–190. doi: 10.1038/sj.gt.3303054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: Respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TNF induces JNK activation in spinal primary astrocytes.
Primary culture of astrocytes. Western blotting (Codeluppi et al., 2011).
TNF induces phosphorylation of JNK in cultured spinal astrocytes.