

Abstract
Glioma is the most common primary malignant brain tumor and arises throughout the central nervous system (CNS). Recent focus on stem-like glioma cells has implicated neural stem cells (NSCs), a minor precursor population restricted to germinal zones, as a potential source of gliomas. In this review, we will focus on the relationship between oligodendrocyte progenitor cells (OPCs), the largest population of cycling glial progenitors in the postnatal brain, and gliomas. Recent studies suggest that OPCs can give rise to gliomas. Furthermore, signaling pathways often associated with NSCs also play key roles during OPC lineage development. Recent advances suggesting that gliomas can undergo a switch from progenitor- to stem-like phenotype after therapy, implicating that an OPC-origin is more likely than previously recognized. Future in-depth studies of OPC biology may shed light on the etiology of OPC-derived gliomas and reveal new therapeutic avenues.
Keywords: brain, cancer, glioma, glioblastoma, neural stem cell, oligodendrocyte progenitor cell
1. Introduction
Gliomas are the most common malignant primary brain tumor and associated with approximately 16,000 cancer-related deaths in United States per year (Louis et al., 2007). Recent advances in the molecular characterization of gliomas have defined subgroups of tumors that are genetically and epigenetically distinct (Noushmehr et al., 2010; H. S. Phillips et al., 2006; Sturm et al., 2012; Verhaak et al., 2010). The temporal and regional specificity of genetically distinct gliomas (Sturm et al., 2012), argue that either several discrete populations of precursor cells may be vulnerable to transformation, or that multiple glioma subgroups share a common cell of origin. Glial cells outnumber neurons by 10-fold in the human brain and are composed mainly of terminally differentiated cells and minor discrete precursor populations. Modeling of glioma in mice has demonstrated that cells at various differentiation stages throughout glial and neuronal lineages have the potential to generate gliomas. In this review, we present recent findings suggesting that the most wide-spread population of cycling cells in the pediatric and adult brain of mammalians, the oligodendrocyte progenitor cells (OPCs), represent a likely origin for large cohorts of gliomas. We propose that more in-depth studies of OPC biology will inform novel preventive measures and therapeutic interventions to reverse the fatal outcome of most glioma patients.
Gliomas can grossly be divided into astrocytic, oligodendrocytic, and ependymal phenotypes. Classification by the World Health Organization (WHO) distinguishes malignancy by grade (I-IV). Based on histological appearance, gliomas of most grades and types are found in children and adults. Recent molecular profiling of grade IV glioblastoma (GBM) exemplifies that subsets of tumors in children, young adults, and adolescents, that are indistinguishable by histology, can be segregated based on genetic alterations, broad-scale gene expression, and methylation patterns. Here, we will present recent experimental advances on the understanding of why humans are diagnosed with a certain type of glioma and where it came from.
Recent advances highlight the cellular heterogeneity in gliomas, the influence of the tumor microenvironment, and that treatment-resistant tumor cells display a high degree of stemness. Emerging research suggest that the failure to target glioma stem cells (GSCs) rather than the inability to debulk tumors through surgical resection, radiation and chemotherapy, explain the poor survival of glioma patients (Huse & Holland, 2010). In this review, we will discuss ways to identify GSCs, their interactions with tumor microenvironment, and therapeutic advances to target GSCs. In 2012, Yanoko Nishiyama and John Gurdon were awarded the Novel Prize in Medicine for identifying factors that can reprogram somatic cells into pluripotent stem cells. Since these factors are also expressed in stem-like cancer cells, it is possible that they arose from more differentiated cells. In fact, viral transduction of oncogenes into mature neurons and astrocytes generate gliomas in mice (Friedmann-Morvinski et al., 2012). Similarly, it is plausible that also OPCs can give rise to more stem-like gliomas.
2. Glial Cell Lineages
Recent advances describe a mosaic organization of neural stem cells (NSCs) and astrocyte precursors in the central nervous system (CNS) that generate neuron, astrocytes, and oligodendrocytes with a high degree of regional specificity of (Merkle, Mirzadeh, & Alvarez-Buylla, 2007; Tsai et al., 2012). The positional identity is an organizing principle underlying cellular subtype diversification in the brain and is controlled by a homeodomain transcriptional code (Hochstim, Deneen, Lukaszewicz, Zhou, & Anderson, 2008). During embryonic development, expansion and cell fate determination of neural precursors is controlled by gradients of secreted molecules along rostrocaudal and dorsoventral axes. Radial glia and embryonic NSCs generate neurons, glial cells, and ependymal cells in temporal waves during neural development (Rakic, 1990). As a remnant from fetal development, neurogenesis in mammalians is restricted after birth to the dentate gyrus of the hippocampus and the subventricular zone (SVZ) lining the lateral ventricles (Doetsch, 2003; Eriksson et al., 1998; Sanai et al., 2011). Recent studies suggest that NSCs are lining the third and fourth ventricles as well (S. Weiss et al., 1996; Xu et al., 2005). In the postnatal cerebellum, Bergmann glia express markers associated with NSCs (Koirala & Corfas, 2010; Sottile, Li, & Scotting, 2006). In contrast to rodents, SVZ neurogenesis in humans ceased after 18 months, indicating that few SVZ NSCs are present in the aging human brain (Sanai et al., 2011). Given the extensive self-renewal capacity of NSCs, these cells have been suggested as the cell of origin for gliomas (Figure 1). Considering the low abundance of NSCs and the wide distribution of gliomas throughout the human postnatal brain, it is more likely that an abundantly expressed cell type is susceptible to transform into glioma.
Figure 1.
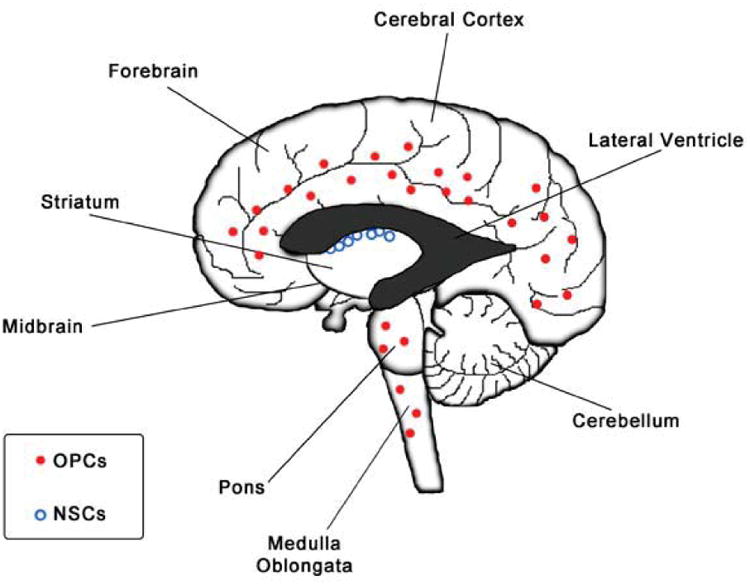
OPCs are the most widely distributed population of cycling cells in forebrain and hindbrain regions. In contrast, a discrete population of NSCs is found in the SVZ lining the lateral ventricles.
A first wave of oligodendrocyte progenitors arise from the embryonic ventral forebrain, followed by a second wave originating from the lateral and caudal ganglionic eminences, and finally a third wave arises within the postnatal cortex (Kessaris et al., 2006). In the developing mouse brain and spinal cord, the first oligodendrocyte-lineage cells appear around embryonic day 12.5 (E12.5) (Zuo & Nishiyama, 2013). The cells are characterized by expression of the basic helix-loop-helix (bHLH) transcription factors OLIG1, OLIG2, NKX2.2, the Sry-related high mobility group box gene (SOX10), and platelet-derived growth factor receptor alpha (PDGFRA) (Zuo & Nishiyama, 2013). At E14.5 PDGFRA positive cells also express the chondroitin sulfate proteoglycan neuro-glial 2 (NG2) (in humans CSPG4) in the ventral mouse forebrain (Nishiyama, Lin, Giese, Heldin, & Stallcup, 1996). While OLIG2 is required for generation of oligodendrocyte specification, the bHLH factors ASCL1 promotes oligodendrogenesis by repressing DLX1/2, a transcriptional repressor of OLIG2 (Ligon et al., 2006; Petryniak, Potter, Rowitch, & Rubenstein, 2007). Other prerequisites for oligodendrogenesis include the SOXE proteins SOX8, SOX9, and SOX10 (C. C. Stolt et al., 2003). In contrast, the SOXD proteins SOX5 and SOX6 inhibit oligodendrocyte specification (C. Stolt et al., 2006). In addition, the developmentally expressed genes NOTCH-1, wingless (WNT), and sonic hedgehog (SHH), normally associated with NSCs, block differentiation and maintain an undifferentiated state in OPCs (Zuo & Nishiyama, 2013) (Figure 1B).
As the most widely distributed population of cycling cells in the postnatal brain, OPCs, also referred to as polydendrocytes, represent a fourth major type of glia in the CNS (Zuo & Nishiyama, 2013). In fact, approximately 70% of 5-bromo-2′-deoxyuridine (BrdU)-incorporating cells in the adult rat brain co-expressed NG2 (Dawson, Polito, Levine, & Reynolds, 2003; Lasiene, Matsui, Sawa, Wong, & Horner, 2009). An elegant study by Hughes et al. (2013) show that OPCs are under homeostatic control to ensure generation of appropriate numbers of myelin-producing oligodendrocytes (Hughes, Kang, Fukaya, & Bergles, 2013). As OPCs are recruited to focal injuries, a proliferative burst of OPCs surrounding the injury restore the cell density. It has been debated if all OPCs have the same proliferative capacity or respond to different environment cues. PDGFA acts as a potent mitogen of OPCs expressing PDGFRA (Hall, Giese, & Richardson, 1996). Hill et al., (2013) recently demonstrated that in white matter, but not grey matter, OPCs proliferate in response to PDGF by activating WNT and phosphatidylinositol 3-kinase (PI3K) (Hill, Patel, Medved, Reiss, & Nishiyama, 2013). Similar to NSCs, the mitogen epidermal growth factor (EGF) induces symmetrical cell division in adult OPCs (Sugiarto et al., 2011) (Figure 2). Treatment of human OPCs with histone deacetylase (HDAC) inhibitors prevented differentiation into oligodendrocytes, demonstrating the importance of post-translational modification of histones (Conway, O'Bara, Vedia, Pol, & Sim, 2012). In summary, several drivers (PDGFRA, EGFR, modulation of histone function) of gliomagenesis also play key roles in OPC development, arguing that proliferating OPCs, abundant throughout life, may transform in response to environmental pressure and play a key role in gliomagenesis.
Figure 2.
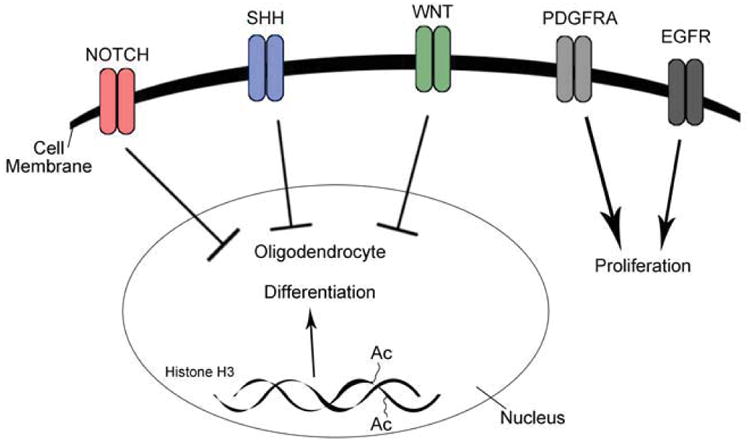
Activation of NOTCH, SHH, and WNT signaling inhibits OPC differentiation into oligodendrocytes. On the contrary, acetylation of histone H3 promotes differentiation of OPCs. Growth factor-mediated activation of PDGFRA or EGFR increases proliferation in OPCs.
3. Glioma Subgroups and Cell of Origin
Neuropathologists classify gliomas based on grade (I-IV) that includes grade II diffuse gliomas and grade III-IV malignant gliomas. Ependymomas, astrocytomas, oligodendrogliomas, and oligoastrocytomas display fewer mitoses, necrosis, nuclear atypia, and vascular proliferation compared to grade IV GBM (Louis et al., 2007). To develop a more personalized medicine approach and improve survival in patients, research has focused on characterizing subsets of GBM patients. Microarray expression profiling was first able to separate low- versus high-grade gliomas and primary versus recurrent tumors (Godard et al., 2003; Rickman et al., 2001; van den Boom et al., 2003). New technological advances and the Cancer Genome Atlas network (TCGA) have defined subsets of GBMs based on epigenomic, genomic, and transcriptomal signatures (Noushmehr et al., 2010; H. S. Phillips et al., 2006; Verhaak et al., 2010). When Phillips et al. (2006) stratified primary GBMs based on survival, they identified a better prognosis for tumors displaying a proneural gene expression signature. Patients with worse prognosis could be separated into classical and mesenchymal subsets of tumors (H. S. Phillips et al., 2006). When primary GBMs were not classified based on survival, Verhaak et al. (2010) later identified a fourth neural GBM subgroup (Verhaak et al., 2010). A couple of years earlier, approximately 12% of GBM patients displayed isocitrate dehydrogenase (IDH) mutations in tumors and showed increased overall survival (Parsons et al., 2008). Verhaak et al. showed that IDH mutations were exclusively found in a subset of proneural GBMs (Verhaak et al., 2010). A recent study demonstrated 6 subgroups of GBMs (Sturm et al., 2012). Childhood GBMs showed a strong correlation to histone H3.3 (H3F3A) mutations at two critical residues; (K27(M) or G34(R/V), that added two additional GBM subgroups with unique epigenetic and gene expression signatures (Sturm et al., 2012). Interestingly, these H3F3A mutations give rise to GBMs in separate anatomic compartments, and in contrast to IDH1R132H mutant tumors, are diagnosed in adolescents (G34) or young children (K27). The authors refer to these 6 GBM subgroups as; IDH1, K27, G34, RTK I ‘PDGFRA’, mesenchymal, and RTK II ‘classic”, since they are associated with unique genetic alterations (Sturm et al., 2012) (Figure 2). Gene expression profiling of diffuse gliomas shows that approximately 75% and 50% of diffuse oligodendrogliomas and astrocytomas, respectively, belong to the proneural subgroup (L. a D. Cooper et al., 2010). Interestingly, the fraction of proneural tumors correlates well with the frequency of IDH mutations in diffuse gliomas (Yan et al., 2009). A recent study suggests that activity among signal transduction pathways at the protein level can define GBM subclasses (Brennan et al., 2009). However, future advances in proteomics are needed to effectively distinguish glioma patients at the protein level. Progress in subclassification of gliomas is used to develop biomarkers and gene expression signatures that can be used to stratify patients in clinical trials (Olar & Aldape, 2012). In parallel, magnetic resonance imaging (MRI) predictors that associate with GBM subgroups are in development (Gutman et al., 2013). MRI studies show that proneural GBMs had significantly lower levels of contrast enhancement and IDH1R132H mutant GBMs show accumulation of 2-hydroxyglutarate (2HG) (Chaumeil et al., 2013; Gutman et al., 2013). However, decision-making is still governed by genetic alterations as gliomas show regional heterogeneity for MRI parameters and association to GBM subgroups.
To develop subgroup-specific therapies in glioma, improved pre-clinical models are needed. Traditionally, researchers have studied therapeutics using cultures or xenografts of human GBM cell lines. Passaging of primary human GBM tumors in immunocompromised mice is an improved model system that better preserves tumor cell heterogeneity (Hodgson et al., 2009). However, the strong influence of the tumor microenvironment and the importance of blood-brain barrier permeability for drugs have led to generation of several genetically-engineered murine models (GEMM) of glioma (Table 1). These models are also useful for studies of premalignant events and the cell of origin for different types of gliomas. Although these models have been highly informative, a discrepancy of previously developed GEMM of glioma is the failure to recapitulate the genetic alterations observed in human counterparts. For example, whole-genome sequencing studies have identified B-RAFV600E, FGFR1, MYB1, MYBL1, H3F3A, and ATRX mutations in pediatric gliomas (J. Zhang et al., 2013). However, recent progress has employed BRAFV600E mutation and neurofibromin 1 (NF1) loss to generate pilocytic gliomas and high-grade GBMs (J. Chen et al., 2012; Robinson et al., 2011). Modeling of H3F3A and IDH1R132H mutations in murine GEMM will be useful tools to develop new therapeutics against larger cohorts of childhood and adult glioma patients.
Table 1.1. Murine glioma models.
| Cancer genes | Cell of Origin | Mechanism | Reference |
|---|---|---|---|
| Astrocytoma models (II—III) | |||
| c-myc | GFAP | Transgenic | Jensen et al. (2003) |
| K-ras | GFAP | Cre | Abel et al. (2009) |
| H-Ras | GFAP | Transgenic | Shannon et al. (2005) |
| H-Ras/Ptenloxp/loxp | GFAP | Cre | Wei et al. (2006) |
| EGFR/Ink4a/Arf−/− | Nestin | RCAS | Holland, Hively, DePinho, and Varmus (1998) |
| Oligodendroglioma models | |||
| H-ras/EGFRvlll | GFAP | Transgenic | Ding et al. (2003) |
| v-ErbB/Ink4a/Arf+/− | S100β | Transgenic | Weiss et al. (2003) |
| v-ErbB/Trp53−/− | S100β | Transgenic | Persson et al. (2010) |
| PDGFB/Ink4a/Arf−/− | Nestin | RCAS | Dai et al. (2001) |
| PDGFB/Akt | Nestin | RCAS | Dai et al. (2005) |
| PDGFB | CNP | RCAS | Lindberg, Kastemar, Olofsson, Smits, and Uhrbom (2009) |
| PDGFAL | GFAP | Transgenic | Nazarenko et al. (2011) |
| Glioblastoma (GBM) models | |||
| PDGFB | Nestin | RCAS | Shih et al. (2004) |
| PDGFB/Trp53−/− | GFAP | Transgenic | Hede et al. (2009) |
| K-Ras/Akt | Nestin | RCAS | Holland et al. (2000) |
| K-Ras/Ink4a/ArT−/− | GFAP, Nestin | RCAS | Uhrbom et al. (2002) |
| K-Ras/Akt/Ptenloxp/loxp | Nestin | RCAS/Cre | Hu et al. (2005) |
| Nflloxp/+/Trp53+/− | GFAP | Cre | Zhu et al. (2005) |
| Ptenloxp/+/Trp53loxp/loxp | GFAP | Cre | Zheng et al. (2008) |
| Ptenloxp/+/Nflloxp/+/Trp53loxp/− | GFAP | Cre | Kwon et al. (2008) |
| Ptenloxp/+/Nflloxp/+/Trp53loxp/loxp | SVZ, Nestin | CreER | Alcantara Llaguno et al. (2009) |
| Akt/H-Ras/Trp53+/− | SVZ, HC, GFAP | Lentiviral/Cre | Marumoto et al. (2009) |
| Ptenloxp/loxp/Trp53loxp/loxp/Rbloxp/loxp | SVZ, GFAP | Adenoviral/Cre | Jacques et al. (2010) |
| NfI+/−/Trp53loxp/loxp | GFAP | Transgenic/Cre | Wang et al. (2009) |
| PDGFB/Ptenloxp/loxp/Trp53loxp/loxp | Subcortical WM | Retroviral/Cre | Lei et al. (2011) |
| Ptcnloxp/loxp/Trp53loxp/loxpRblloxp/loxp | Astrocyte (GFAP) | CreER | Chow et al. (2011) |
| NfI−/−/Trp53−/− | OPC (NG2), NSCs (GFAP/Nestin) | MADM/Cre | Liu, Sage, et al. (2011) |
| EGFRvlll/Ink4a/Arf−/− | NSC, Astrocyte | Transplant | Bachoo et al. (2002) |
As a useful tool for pre-clinical studies and the delineation of cell of origins for different types of glioma, GEMMs of glioma generate tumors resembling grade II-IV human counterparts.
NSC, neural stem cell; OPC, oligodendrocyte progenitor cell; SVZ, subventricular zone; WM, white matter; HC, hippocampus.
4. H3F3A Mutations Drive Gliomagenesis in Separate Brain Regions
For NSCs and progenitor cells to achieve production of different types of neurons and glia at appropriate times and places during development, they must integrate cell-intrinsic programs and environmental cues. These developmental dynamics are reflected in changes in gene expression, which is regulated by transcription factors and at the epigenetic level. Methylation and acetylation of histones function as epigenetic modulators of differentiation in NSCs and progenitors (X.-L. Hu, Wang, & Shen, 2012). However, mutations of epigenetic sites on histones can block differentiation and transformation of cells into cancers. In this section of the review, we will discuss recent findings demonstrating mutations at two distinct residues (K27(M) or G34(R/V)) of H3F3A in childhood GBMs. Interestingly, K27 and G34 mutant pediatric GBMs are associated with distinct anatomical locations, have subgroup-specific gene expression signatures, and occur at different ages, suggesting that different precursor cells may represent the cell of origin for K27 and G34 mutant GBMs.
Whole-genome and whole-exome sequencing studies independently identified recurrent mutations in the gene H3F3A in pediatric glioma (Schwartzentruber et al., 2012; Wu et al., 2012). Interestingly, Sturm and colleagues found IDH mutations occurred in a distinct group of patient samples from H3F3A mutants, and the K27 and G34 mutations were mutually exclusive and clustered into their own subgroup when examining gene expression and methylation profile of tumors harboring these mutations (Sturm et al., 2012). Anatomically, the K27-mutant tumors arise in pontine and more rostral midline brain structures, whereas the G34-mutant tumors are usually hemispheric and found in the forebrain (Figure 3). K27 mutations occur in children while G34 mutations occur in adolescents and IDH mutations were found in young adults (Khuong-Quang et al., 2012; Sturm et al., 2012). This may be reflective of their prognosis as patients with K27 mutant tumors typically have lower overall survival rates compared to G34 mutant and H3F3A wildtype (WT) tumors, while patients with IDH mutant tumors had better prognosis compared to patients with tumors containing either H3F3A mutation (Sturm et al., 2012). Consistent with these findings, Wu et al identified 78% of patients with the lethal pediatric tumor, diffuse intrinsic pontine glioma (DIPG), contained the K27 mutation while no DIPG samples had a mutation at G34 (Wu et al., 2012). A separate study by Khuong-Quang et al. found a similar trend where K27 mutations were identified in 71% of DIPG samples, while none had G34 or IDH mutations (Khuong-Quang et al., 2012). Both K27 and G34 mutations frequently co-occur with other genetic mutations and associate with the expression of developmentally regulated genes. For example, G34 mutations correlated with hypermethylation and silencing of oligodendrocyte lineage genes such as OLIG1 and OLIG2, while expressing the forebrain marker, FOXG1. In contrast, K27 mutant tumors express OLIG1 and OLIG2, but not FOXG1 (Sturm et al., 2012). One of the initial studies that identified these recurrent H3F3A mutations also found mutations in the chromatin remodeling protein, ATRX, and the tumor suppressor TP53 (Schwartzentruber et al., 2012). ATRX mutations were more tightly associated with older patients and found in all tumors with G34 mutations in the Schwartzentruber et al study (Schwartzentruber et al., 2012; Sturm et al., 2012). TP53 mutations were also consistently found in tumors with G34 mutations, which may represent an alternate mechanism of p53 inactivation, as OLIG2, commonly lost in G34 tumors, has been shown to block p53 function (Mehta et al., 2011). Conversely, this may also explain why K27 mutant tumors (which are OLIG2+) do not need to correlate as strongly with TP53 mutations (although a high percentage (∼70%) of K27M mutant tumors do co-express mutant TP53). The frequent association of TP53 and H3F3A mutations in pediatric GBM is reminiscent with the strong link between TP53 and IDH1R132H mutations in adult GBM. To summarize, these studies suggest that H3F3A mutant GBMs may arise from regionally distinct OLIG2-expressing glial progenitors.
Figure 3.
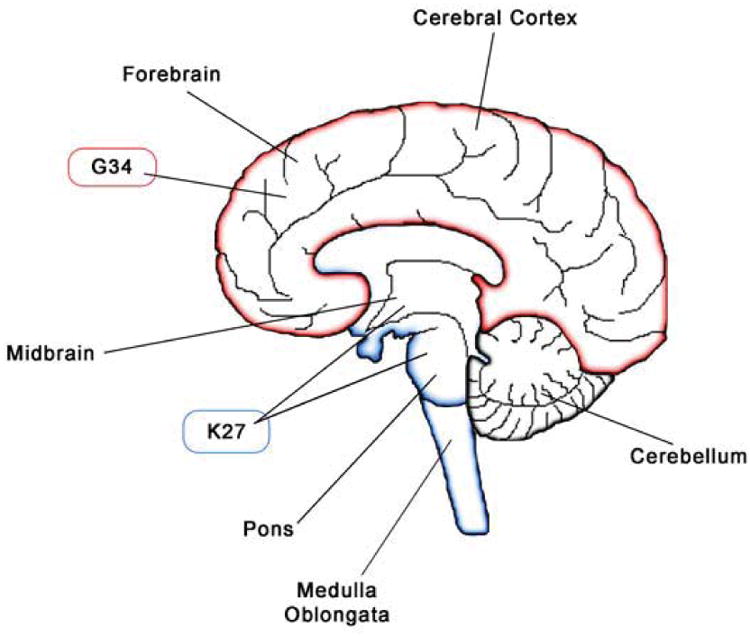
GBMs displaying G34 and K27 in the H3F3A mutations localize to forebrain and hindbrain regions, respectively.
4.1 Regulation of DNA methylation by K27 and G34 H3F3A mutations
DNA is condensed in the nucleus by wrapping around histones and multiple histones assemble to form a nucleosome, which consists of nine histone subunits (two of each core histone [H2A, H2B, H3, H4] and the linker histone H1). In addition to compacting DNA, histones contribute to the epigenetic regulation of the genome via substitution of various histone isoforms and posttranslational modifications. These alterations that regulate gene expression are largely due to acetylation and methylation of lysine and arginine residues. Although methylation is generally associated with repression of gene expression, methylation can indicate active transcription depending on the residue that is methylated as well as the amount of methyl groups added at that site (mono-, di- and tri-methylation). This is illustrated with Histone H3 where tri-methylation of lysine 4 and 36 (K4, K36) associate with active transcription, while tri-methylation of K9 and K27 suggest gene silencing (Chi, Allis, & Wang, 2010). While di- and tri-methylation of K27 are linked to inhibition of gene expression, mono-methylated K27 is localized at highly expressed genes, as is acetylated K27 (Creyghton et al., 2010; Steiner, Schulz, Maksimova, Wong, & Gallagher, 2011).
Two DIPG K27 mutant cell lines demonstrated a reduction in tri-methylation (me3) and di-methylation (me2) status at K27 while not impacting the acetylation levels of K27 (H3K27ac) compared to normal NSCs (Chan et al., 2013). Similarly, another study found suppressed H3K27me3 levels in 6 GBMs containing K27 mutations (Venneti et al., 2013). Ectopic expression of H3F3A K27 in 293T cells, human astrocytes and mouse embryonic fibroblasts, decreased di-and tri-methylation of K27 on endogenous H3F3A proteins and the exogenously expressed mutant without affecting the methylation status of K36, a post-translationally modified site near G34 (Chan et al., 2013; Lewis et al., 2013). The mechanism for reduced methylation at K27 may be due to EZH2, the catalytic subunit of polycomb repressive complex 2 (PRC2), which is a methyltransferase that targets Histone 3.3 K27. Since the expression levels of EZH2 is not altered between patient samples expressing either WT H3F3A or the K27 mutation, the mutation may act in a dominant negative fashion to block EZH2 function (Venneti et al., 2013). This was supported by the finding that a K27 peptide demonstrated a higher affinity for EZH2 compared to wildtype H3F3A and inhibited PRC2 activity via interaction with EZH2 (Chan et al., 2013; Lewis et al., 2013). In K27 mutant DIPG cells, the sites which retained H3K27me3 had increased levels of trimethylation, as well as EZH2, and were genes associated with cancer pathways, such as the tumor suppressor CDKN2A (Chan et al., 2013). Although overexpression of mutant K27 lowered the methylation status of both the endogenous and exogenous K27, mis-expression of an H3F3A G34 mutant only decreased tri-methylation of K36 on the exogenous peptide, while not affecting the endogenous H3F3A K36 (Chan et al., 2013; Lewis et al., 2013). This would suggest that the G34 site would need to be mutated on both alleles to have a tumorigenic effect while the K27 mutation on only one allele may be sufficient for transformation. The requirement for an additional hit on G34 may explain why these tumors appear later in life than K27 mutant tumors. Sturm et al. found global hypomethlation of genomic DNA in G34 mutant tumors, which stands in contrast to global hypermethylation in IDH1R132H mutant tumors (Sturm et al., 2012). Hypomethylation in G34 mutant tumors is especially prominent at chromosome ends, which may provide a link with alternative lengthening of telomeres (ALT), a phenomenon commonly observed with ATRX mutations (Schwartzentruber et al., 2012; Sturm et al., 2012).
4.2 Chromosome and Myc aberrations in H3F3A mutant glioblastoma
Chromosomal aberrations have been investigated between H3F3A WT and K27 mutant DIPG tumors (Khuong-Quang et al., 2012). As expected due to the role histones play in chromatin remodeling, K27 mutant tumors exhibit different chromosomal aberrations compared to tumors with H3F3A WT. H3F3A WT samples exhibited gains of 2p and 7p as well as losses in 9p and 12q. K27 tumors harbored losses of 5q, 6q, 17p and 21q and gains in 19p. Focal recurrent gains were also observed in 4q12 (containing PDGFRA), and 8q24.21 (MYC/PVT1 locus). Intriguingly, while MYC is amplified in K27 tumors, chromosome 2p24.3 (which includes MYCN) is significantly gained in H3F3A wildtype tumors. Although amplification of MYCN has not been observed in G34 mutant samples, overexpression of the G34 mutation induces expression of MYCN in normal human astrocytes and fetal glial cells (Bjerke et al., 2013; Khuong-Quang et al., 2012). This implies that amplification and overexpression of MYC and MYCN appear to segregate based on the H3F3A mutations. MYC proteins are known to promote apoptosis via activation of TP53 (Chesler et al., 2008; Elson, Deng, Campos-Torres, Donehower, & Leder, 1995) and a number of cancer models utilize overexpression of MYC in combination with loss of TP53 function (Kawauchi et al., 2012; Pei et al., 2012; Schmitt et al., 2002; Yu & Thomas-Tikhonenko, 2002). Since MYC is also known to promote proliferation and block differentiation, the combination of amplified MYC, mutant TP53 and K27 may provide insight to the genetic etiology of DIPG.
4.3 Delineating the cell of origin for K27 and G34 H3F3A mutant glioblastoma
Neural stem cells, astrocyte precursors, and OPCs are all abundant at birth in humans and represent possible targets for transformation by K27 mutation. In mice, viral transduction of Nestin-expressing precursors with mutant TP53 and K27 failed to induce gliomagenesis in the newborn murine brainstem, but did induce proliferating ectopic cell clusters (Lewis et al., 2013). In humans, Nestin and OLIG2 are co-expressed in the ventral pons around 6 years of age, the peak for DIPG occurrence (Monje et al., 2011). In contrast to K27 mutant GBMs, G34 mutant tumors express the forebrain marker FOXG1 and other forebrain development-related transcription factors such as ARX, DLX5, FOXA1, NR2E1, POU3F2, and SP8 (Bjerke et al., 2013). Although NSC are not abundant in the forebrain in adolescents when most G34 mutant GBMs are diagnosed, these tumors express the NSC-associated genes Musashi-1 (MSI1), eyes absent homolog 4 (EYA4), and SOX2 under control of the active transcription marker, H3K36me3 (Bjerke et al., 2013). High expression of MYCN in G34 mutant GBMs may enable more restricted forebrain precursors to dedifferentiate into a NSC phenotype. Using a mosaic mouse model, Liu et al. (2011) elegantly demonstrated that even when combined loss of TP53 and NF1 were introduced in Nestin positive NSCs, transformation only occurred after NSCs had differentiated into OPCs (C. Liu et al., 2011). A similar approach can be used to delineate the cell of origin for K27 and G34 mutant GBMs.
5. Gliomagenesis and Mutations in Isocitrate Dehydrogenase Genes
In humans, five genes encode three isoforms of the metabolic enzyme IDH that are involved in the citric acid cycle. IDH1 is found both in the cytoplasm and peroxisomes while IDH2 and IDH3 are localized to the mitochondria. IDH1 and IDH2 are closely related and catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) while reducing nicotinamide adenine dinucleotide phosphate (NADP+) to NADPH (Figure 4). The unrelated multisubunit enzyme, IDH3, reduces NAD+ to NADH and is thought to play a central role in aerobic energy production in the tricarboxylic acid (TCA) cycle. A critical residue, involved in the binding of isocitrate, is arginine 132 (R132), found within the active site of human IDH1 and is evolutionary conserved in the functionally analogous R172 of IDH2. IDH1R132H results in an 80% reduction in enzyme activity. The R132 mutation abolishes normal catalytic activity by preventing the protein to bind to isocitrate. In normal cells, IDH genes mediate epigenetic changes that maintain cellular homeostasis. Yet, in cancer cells these epigenetic modifications represent a key hallmark of tumorigenesis. Interestingly, IDH mutations are generally mutually exclusive and occur uniquely on these arginine residues, which are conserved across species and malignancies.
Figure 4.
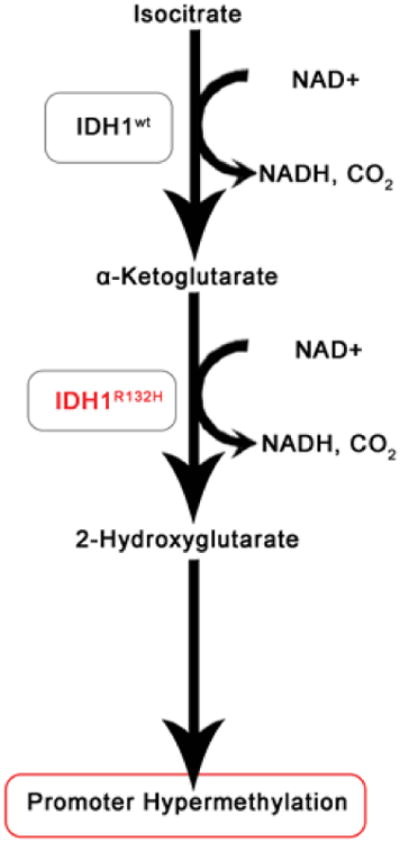
IDH1R132H mutant gliomas show reduced levels of α-ketoglutarate and overproduction of 2-hydroxyglutarate (2-HG) that leads to a hypermethylated phenotype.
The first observations of IDH1/2 mutations were identified in metastatic colon cancer. These mutations are also found in sarcomas, leukemias, and gliomas. Traditionally, oncogenic events are known to induce aberrant changes that lead to rapid cell cycle progression. However, pioneering studies in leukemia demonstrated that IDH mutation leads to a block in differentiation in hemapoietic precursor cells (Chaturvedi et al., 2013). The ability of IDH mutation to disrupt normal differentiation into defined cell lineages has also been demonstrated in murine forebrain NSCs (C. Lu et al., 2013). This block in differentiation was attributed to prevention in histone demethylations required for NSCs to terminally differentiate. Although the differentiation program was compromised in the NSCs, the authors found no indication of transformation, suggesting that IDH mutations may transform glial progenitor or dedifferentiated terminally differentiated glial cells. Parsons et al. (2008) sequenced over 20,000 protein coding genes in order to identify genetic alterations in GBMs and found that 12% of the samples analyzed harbored a recurrent point mutation in the active site of IDH1/2 (Parsons et al., 2008). Specifically, 5% of primary gliomas and 60-90% of secondary gliomas had IDH1/2 mutations. Further, they observed that IDH1R132H mutations occurred in a large fraction of young patients and that most patients with secondary GBMs were associated with increased overall survival. Mutations in IDH2 are much less common and mutually exclusive with IDH1R132H as mentioned before. Virtually all tumors with IDH mutations are of the proneural subtype. Other studies found IDH mutations in 80% grade II and grade III gliomas as well as in secondary GBMs. Importantly, Yan et al. (2009) reported that IDH1R132H is a favorable prognostic marker for glioma patients (Yan et al., 2009).
Hypoxia-inducible factor 1α (HIF-1α) is a transcription factor that facilitates tumor growth under conditions of low oxygen and its stability is regulated by α-ketoglutarate. Human gliomas with IDH1R132H mutation express higher levels of HIF-1α compared to IDH wildtype tumors. Additionally, expression of IDH1 in cultured cells reduces α-KG and increases HIF-1α levels (S. Zhao et al., 2009). These results indicate that IDH1R132H mutations may contribute to tumorigenesis in part through the HIF-1 pathway.
As previously mentioned, mutations occur at a single amino acid residue of the IDH1R132H active site. Interestingly, in tumors, only a single copy of the gene is mutated and the expression of a wildtype IDH1 allele is critical for the formation of a heterodimer with the mutated allele. Cancer-associated IDH1R132H mutations have been found to resulting in the production of 2-HG instead of α-KG. Remarkably, levels of 2-HG are elevated in human gliomas harboring IDH1R132H mutations. These findings indicate that the production of the oncometabolite 2-HG in IDH1R132H mutated tumors may contribute to glioma initiation and malignant progression.
The accumulation of 2-HG has been associated with increased histone methylation and decreased 5-hydroxylmehtylcytosine (5hmC), resulting in genome-wide histone and DNA methylation alterations (Figure 4). Furthermore, it has been demonstrated that a large number of loci in IDH1R132H mutant gliomas display hypermethylation (Noushmehr et al., 2010). Together, these findings suggest that IDH mutations may alter the expression of a large number of genes rather than a few specific genes. Reportedly, mutations of IDH1R132H occur at an early stage during gliomagenesis (Watanabe, Nobusawa, Kleihues, & Ohgaki, 2009) and may contribute to tumor initiation by globally changing the epigenetic landscape and control cellular state and fate. In fact, glioma samples with IDH mutations and increased histone methylation display a gene expression profile enriched for neural progenitor genes. In summary, 2-HG producing IDH mutations inhibit histone demethylation and can thereby block cell differentiation.
It is established that acquired alterations in the methylation landscape cause dysregulation and can thereby be involved in onconenesis (Jones & Baylin, 2007). A distinct subclass of human glioblastomas has the CpG island methylator phenotype (CIMP) and is associated with the proneural subgroups of tumors and IDH mutation (Noushmehr et al., 2010). The fact that both IDH mutation and the CIMP phenotype occur in glioblastomas raises the question of cause and effect. Turcan et al. (2013) demonstrate elegantly that IDH1R132H mutation is the cause of CIMP, and establishes the CIMP phenotype by remodeling the epigenome (Turcan et al., 2013). Importantly, primary human astrocytes introduced with IDH1R132H mutation display epigenomic alterations mirroring low-grade gliomas positive for the CIMP phenotype. These observations further our mechanistic understanding of the role of IDH mutation and CIMP in gliomanenesis, providing targets for development of novel therapies.
5.1 Models of IDH-mutant gliomas
Studying IDH-mutant gliomas has been obstructed by the lack of models of IDH-mutant glioma-producing mice. With respect to the effects of IDH mutations on tumorigenesis Sasaki et al. (2012) generated brain-specific IDH1R132H knock-in (IDH1-KI) mice (Sasaki et al., 2012). These mice are embryonically lethal, display brain hemorrhage and elevated 2-HG levels but decreased reactive oxygen species (ROS). Interestingly, the increased levels of 2-HG stabilize HIF-1α proteins and upregulate HIF-target gene transcription. Moreover, an ER stress response is triggered which causes intrinsic cell death. These effects may increase vascular endothelial growth factor (VEGF) levels, leading to aberrant blood vessel formation and ultimately resulting in brain hemorrhage.
Cell lines with IDH1R132H mutation can only be maintained transiently in vitro, as the mutation does not persist in non-immortalized cells. Furthermore, primary IDH-mutant gliomas from patient tumors do not grow well in vitro (Piaskowski et al., 2011). In contrast to normal cells, introduction of IDH mutations into glioma cells decreases the proliferation rate, which may ultimately cause a selection pressure against cultured glioma cells harboring IDH mutations (Bralten et al., 2011). Despite these challenges, at least one group has reported the isolation of a glioma cell line with an endogenous R132H mutation in IDH1R132H (Luchman et al., 2012). The researchers established neurosphere cultures from an IDH1R132H mutant anaplastic oligoastrocytoma sample and confirmed retention of the mutation over passages. These cells were also used in orthotopic xenografts of non-obese diabetic/severe combined immune deficiency (NOD/SCID) mice. Mass spectroscopy experiments were performed to confirm production of 2-HG by glioma cells with endogenous IDH1R132H mutations both in vivo and in vitro. In summary, there is a great need for glioma models with IDH mutations to interrogate the roles of IDH mutations during gliomagenesis since efforts to model IDH mutant gliomas have failed and the ability to grow primary IDH mutant tumors has been challenging.
5.2 Glial progenitor-origin for IDH-mutant gliomas
Development of IDH-mutant sarcoma or leukemia-models, have allowed researcher to search for drugable targets up- or down-stream of IDH in a cell-type specific context (Sasaki et al., 2013). To date, most studies of IDH-mutations in glioma have relied on overexpression in different glioma cell lines (Rohle et al., 2013). However, genes important for terminal differentiation present in OPCs are lacking in these artificial cell lines and without those genes in place methylation inhibition cannot be studied. Since IDH mutations are most common in oligodendroglioma, development of IDH-mutant oligodendroglioma models and the ability to xenograft/culture IDH-mutant human oligodendrogliomas will generate excellent tools to better understand the earliest transformation events and to perform pre-clinical studies. As IDH mutations are found in 12% of all gliomas, much effort is currently focused on defining the molecular events following IDH mutations to find drugable targets. To improve the survival of patients diagnosed with glioma, defining the cell of origin and the first molecular events that initiate transformation will be fundamental for development of new therapies. IDH1R132H mutant oligodendrogliomas are thought to arise in white matter regions of the cerebral hemispheres. Oligodendrocyte progenitor cells represent the most widely spread population of cycling cells in the adult brain. OPCs in adults are scattered throughout the brain, while NSCs mainly reside in the subventricular zone and the hippocampus. The genetics and location of IDH1R132H gliomas suggest that OPCs may be susceptible to transformation. This raises the question if IDH1R132H mutation is sufficient to transform OPCs? Oligodendroglioma show distinct histological features including fried-egg morphology that distinguish them from other gliomas. Co-deletion of chromosomal arms 1p and 19q is a well-known prognostic marker found frequently (>60%) in grade II-III oligodendrogliomas. Recent exomic sequencing has identified tumor suppressor genes: homolog of Drosophila capicua (CIC) and far-upstream binding protein 1 (FUBP1) in 53% and 15% of oligodendrogliomas, respectively (Yip et al., 2011). Since CIC and FUBP1 are located on chromosomal arm 19q and 1p, respectively, loss of these regions inactivates the genes and could potentially contribute to oligodendroglioma development. A strong association between IDH1R132H mutation and 1p/19q co-deletion is found in grade II-III oligodendrogliomas, suggesting that these genetic alterations are important for gliomagenesis. While a subfraction of oligodendrogliomas display IDH1R132H and TP53 mutations, it is more commonly observed in astrocytic tumors.
The reason for the favorable prognosis of IDH1R132H mutant patients is still unknown. Several studies have suggested that IDH1R132H mutation and 2-HG block cell differentiation (Chaturvedi et al., 2013; Figueroa et al., 2010; C. Lu et al., 2013). The reduced aggressiveness in IDH-mutant tumors may be a result of inhibited differentiation, rather than regulation of cell cycle genes. Alternatively, the better prognosis for IDH-mutant patients may reflect an OPC origin. Mutant IDH1R132H accelerate onset of myeloproliferative disease (MPD)-like myeloid leukemia in mice in cooperation with Homeobox protein Hox-A9 (HOX9A) (Chaturvedi et al., 2013). Further, mutant IDH1R132H accelerated cell cycle transition through repression of cyclin-dependent-kinase inhibitors CDKN2A and CDKN2B, and reduced activation of MAPK signaling. Interestingly, a complex transcriptional regulatory network of HOX genes specify neural progenitor cell identity during early development (Gavalas, Trainor, Ariza-McNaughton, & Krumlauf, 2001; Lumsden & Krumlauf, 1996; Trainor & Krumlauf, 2001) Further, it has been reported that sustained activation of MAPK activation in oligodendrocytes stimulates oligodendrocyte progenitor expansion (Ishii, Furusho, & Bansal, 2013). Together, these data may support the idea that OPC are the cells of origin of IDH1R132H mutant cancers.
In contrast to classical and mesenchymal GBMs that express gene expression profiles reminiscent of NSCs, IDH-mutant gliomas display a proneural phenotype (Verhaak et al., 2010). Interestingly, Verhaak et al. (2010) found that the transcriptomal signature of proneural GBMs were closely associated with oligodendrocytes rather than astrocytes, neurons, or NSCs (Verhaak et al., 2010). Due to low activity of NADPH-producing dehydrogenases in rodent versus human brain, it was suggested that rodents are unsuitable to model gliomas (Atai et al., 2011). However, no studies have demonstrated expression of IDH isoforms at high resolution and in defined cell populations, why the cell-specific role of this gene family is still unclear. In the Verhaak et al. study, many genes that are normally found in OPCs, such as NKX2.2, PDGFRA, and OLIG2, were overexpressed in IDH-mutant proneural GBMs (Verhaak et al., 2010). Support from GEMMs show that proneural gliomas can derive from OPCs (Lindberg, Kastemar, Olofsson, Smits, & Uhrbom, 2009; Persson et al., 2010). The regional association of IDH1-mutant GBMs to frontal cortex and white matter regions, argue that OPCs, rather than NSCs, represent a likely origin for IDH-mutant gliomas.
6. Proneural-to-Mesenchymal Transition (PMT) in Glioma
Epithelial-to-mesenchymal transition (EMT) occurs during critical stages during development, such as gastrulation and neural crest formation (Kalluri & Weinberg, 2009; Thiery, 2002). As epithelial cancer cells undergo EMT, they become invasive, metastasize, and become drug-resistant (Craene & Berx, 2013; A. Singh & Settleman, 2010). In many cancers, including breast cancer, the mesenchymal phenotype is associated with increased activation of transforming growth factor beta (TGFβ) signaling, leading to increased expression of the transcription factor families of SNAIL, SLUG and TWIST, and down-regulation of E-cadherin (Craene & Berx, 2013; Mallini, Lennard, Kirby, & Meeson, 2013).
Transcriptomal profiling of gliomas, displaying a neuroepithelial origin, show that the mesenchymal phenotype is associated with stemness, invasiveness, and poor survival (H. S. Phillips et al., 2006; Sturm et al., 2012; Verhaak et al., 2010) (Figure 5). Although in a small cohort of patients, recurrent tumors shifted from a proneural to mesenchymal phenotype, reminiscent of EMT (Lai et al., 2011; H. S. Phillips et al., 2006). In this section, we will review new exciting data suggesting that expression of a single gene or changes in the tumor microenvironment can shift gliomas between proneural and mesenchymal phenotypes, which we will refer to as a proneural-to-mesenchymal transition (PMT). Since recurrent gliomas tend to have a mesenchymal phenotype (Lai et al., 2011), these findings have implications for therapy and suggest that even more stem-like gliomas can arise from OPCs.
Figure 5.
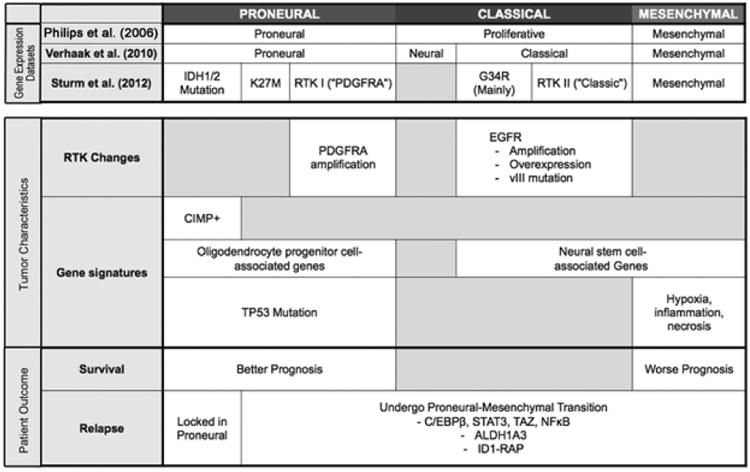
Transcriptomal profiling of human GBMs identified a proneural subgroup of patients with better overall survival compared to proliferative or mesenchymal subgroups. With no stratification based on survival, Verhaak et al. identified four subgroups and found that approximately 35% of proneural GBMs displayed IDH1/2 mutations along with a hypermethylated (CIMP+) phenotype. Sturm et al. extended these observations to also include childhood GBMs and found that hotspot mutations in H3F3A and IDH1 defined distinct epigenetic and biological subgroups. While proneural GBMs are associated with OPC-like gene expression signature, more aggressive GBMs express genes known to drive mesenchymal transcriptional networks.
6.1 Mesenchymal phenotype as a function of glioma subgroup
Mesenchymal GBMs are associated with genes expressed in immune cells, vasculature, invasive cells, but also markers expressed in mesenchymal stem cells and NSCs (H. S. Phillips et al., 2006; Sturm et al., 2012; Verhaak et al., 2010). In contrast, IDH mutant GBMs are strictly proneural (Lai et al., 2011; Sturm et al., 2012). IDH mutations are frequent in secondary (60-90%) GBMs and grade II-III gliomas compared to primary (5%) GBMs (Balss et al., 2008; Bleeker et al., 2009; Hartmann et al., 2009; Kang et al., 2009; Sanson et al., 2009; Watanabe et al., 2009; Yan et al., 2009). In Phillips et al., (2006), IDH wildtype, but not mutant, underwent PMT as they underwent recurrence (Lai et al., 2011; H. S. Phillips et al., 2006). This raises the question if methylation of target genes is required for PMT in gliomas. In epithelial cancers, EMT is associated with increased invasion, resistance to therapy, and acquisition of a ‘cancer stem cell’-like phenotype (Mani et al., 2008; Polyak & Weinberg, 2009). It becomes increasingly clear, that anti-angiogenic treatments and radiotherapy enrich for GSCs and generate highly invasive mesenchymal tumors (Bao et al., 2006; Diehn et al., 2009; Kraus et al., 2002; K. V Lu et al., 2012). Therefore, as we learn more about the mechanisms that drive PMT in glioma, a future goal should be to design therapies that block PMT and improve outcome in patients.
6.2 Transcriptional master regulators of PMT in glioma
In epithelial cancers, SNAIL, TWIST, ZEB1 and TGFB1 are known master regulators of EMT (Kalluri & Weinberg, 2009; Thiery, 2002). In vitro studies suggest that ZEB1 and TWIST also regulate tumor invasion, chemoresistance and stemness in GBM cell lines (Mikheeva et al., 2010; Siebzehnrubl et al., 2013). In an attempt to identify other drivers of the mesenchymal phenotype in GBMs, Carro et al. (2009) used a bioinformatics approach to contrast gene expression signatures between GBM subgroups (Carro et al., 2009). The top six transcription factors that distinguished GBM subgroups (signal transducer and activator of transcription 3 (STAT3), C/EBPβ, bHLH-B2, RUNX1, FOSL2 and ZNF238) regulated >74% of the mesenchymal gene expression signature. They identified C/EBPβ and STAT3 as the main master regulators of mesenchymal transcription networks. During development, C/EBPβ and STAT3 have opposing roles on neurogenesis (Gu et al., 2005; Ménard et al., 2002). While C/EBPβ promotes neurogenesis and opposes gliogenesis, STAT3 promotes astrocytic differentiation and inhibits neurogenesis (Ménard et al., 2002; Nakashima, 1999; Paquin, 2005). As C/EBPβ and STAT3 were transduced into human fetal NSCs, the authors observed reduced neurogenesis and induction of a program towards a mesenchymal phenotype. In primary human GBM cells, C/EBPβ and STAT3 were essential for the mesenchymal phenotype and tumorigenicity when xenografted into immunocompromised mice. Other studies show that C/EBPβ and STAT3 can individually regulate glioma biology. For example, downregulation of C/EBPβ in glioma cells inhibited proliferation, invasion and tumorigenicity in mice (Aguilar-Morante, Cortes-Canteli, Sanz-Sancristobal, Santos, & Perez-Castillo, 2011; Homma et al., 2006), consistent with findings demonstrating that increased mRNA and protein levels of C/EBPβ are associated with a worse prognosis (Homma et al., 2006). Similarly, STAT3 is known to promote tumor growth in glioma. Recent findings suggest that STAT3 is required for maintenance of GSCs (Garner et al., 2013; Priester et al., 2013; Sherry, Reeves, Wu, & Cochran, 2009).
To identify additional master regulators of the mesenchymal phenotype in glioma, Bhat et al. employed a regulatory network analysis of GBM microarray data sets (Bhat et al., 2011). Compared to mesenchymal GBMs, the authors found lower expression of the transcriptional coactivator with PDZ-binding motif (TAZ) in proneural GBMs and lower-grade gliomas. Expression of TAZ correlated well with the degree of CpG island hypermethylation of its promoter. Silencing of TAZ in mesenchymal GSCs decreased mesenchymal marker expression, invasion, self-renewal, and tumor formation. Conversely, overexpression of TAZ in proneural GSCs as well as murine NSCs induced expression of mesenchymal markers, through a binding with co-activator TEAD. Interestingly, TAZ cooperates with PDGF-B to induce high-grade mesenchymal gliomas in the RCAS/Nestin-tv-a model, suggesting that aberrant RAS activation may be a prerequisite for mesenchymal transition by TAZ (Bhat et al., 2011). In other cancers, the TAZ-TEAD nuclear complex has previously been shown to play important roles in EMT, cell growth, and organ development (Hong & Yaffe, 2006; Q.-Y. Lei et al., 2008; Heng Zhang et al., 2009; B. Zhao, Li, Lei, & Guan, 2010).
As master regulators of the mesenchymal phenotype, STAT3, C/EBPβ and TAZ represent future therapeutic targets. The ability to inhibit these transcriptional nodes may lead to a collapse of the mesenchymal phenotype, reduced treatment-resistance, and improved survival in GBM patients. Pharmacological STAT3 inhibitors are currently being evaluated in clinical trials against solid tumors. A future challenge will be to identify therapeutics that target C/EBPβ and TAZ. A small molecule inhibitor screen identified the porphyrin family such as verteporfin (a macular degeneration drug) as inhibitors of YAP/TEAD-dependent transcription (Liu-Chittenden et al., 2012). As mediators of the Hippo pathway, YAP and TAZ are paralogs and display 50% homology (Q.-Y. Lei et al., 2008). Furthermore, Rho, ROCK, and WNT inhibitors inhibit YAP/TAZ activities (Piccolo, Cordenonsi, & Dupont, 2013). Lastly, since increasing data supports that proneural gliomas arise from OPCs rather than NSCs, future studies should address if introduction of TAZ or other mesenchymal master regulators into OPCs can give rise to mesenchymal gliomas.
Future studies should confirm STAT3, C/EBPβ and TAZ as master regulators of PMT in gliomas. Interestingly, gene networks regulated by TAZ were non-overlapping compared to those regulated by STAT3 or C/EBPβ (Bhat et al., 2011). Is it possible that an up-stream effector gene regulates TAZ, STAT3, and C/EBPβ in gliomas? TCGA analyses revealed that the mesenchymal GBM subgroup were enriched in nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and tumor necrosis factor (TNF) superfamily of genes (Verhaak et al., 2010). Supporting data show that TNFα treatment induced NFκB activation in patient-derived proneural human GBM cells, leading to PMT and increased radioresistance (Bhat et al., 2013). Interestingly, activated NFκB signaling increased the activation of all three master regulators: STAT3, C/EBPβ and TAZ. Inhibition of NFκB activation via a mutant IκB blunted the mesenchymal switch, and the authors also showed that minocycline, as an anti-inflammatory inhibitor that targets NFκB pathway, effectively reduce tumor proliferation of mesenchymal GBMs. This elegant study suggests that NFκB blockade can block the mesenchymal gene network. Future studies should demonstrate if NFκB inhibition combined with standard of care can improve outcome in human glioma patients.
Pathway enrichment analysis of proneural and mesenchymal GBMs revealed that genes involved in glycolysis and gluconeogenesis were enriched in mesenchymal GBM cells. At the protein level, glycolytic activity was also increased in mesenchymal cells. The authors identified aldehyde dehydrogenase ALDH1A3 as the most highly expressed metabolic enzyme in mesenchymal GBM cells. Blockade of ALDH1 activity reduced proliferation of mesenchymal, but not proneural, GBM cells (Mao et al., 2013). Interestingly, radiation of proneural GBM cells induced an increased expression of mesenchymal markers and a concomitant down-regulation of proneural genes, a shift that was reversed by ALDH1A3 inhibition. Results from this study suggest that an upregulation of the glycolysis pathway occurs when glioma cells need to maintain their energy requirement in low nutrient conditions, also observed in mesenchymal tumors in a harsh microenvironment, or in proneural tumors that undergo a stress such as radiotherapy. The importance of glycolysis in tumorigenesis was confirmed by another study that associated expression of glucose transporter, type 3 (GLUT3) with tumorigenicity of GSCs (Flavahan et al., 2013). This highlights the adaptation of GSCs to survive in a nutrient-depleted environment and respond to stress following therapy.
Other genes that are associated with the mesenchymal phenotype in gliomas include the inhibitor of differentiation (ID) genes ID1 and ID2, two bHLH factors that promote tumorigenicity in mesenchymal high-grade murine gliomas. In this mouse model, murine hippocampal NSCs were transduced with lentiviruses for H-RasV12 and short-hairpin RNAs against TP53 (Niola et al., 2013). RNA interference experiments showed that down-regulation of ID proteins reduced stem cell-associated markers such as ITGA6, Nestin and SSEA-1 in vitro and the number of perivascular glioma stem-like cells expressing SSEA-1 in vivo (Niola et al., 2013). Furthermore, c-MET has been linked to the mesenchymal phenotype in epithelial cancers. When overexpressed c-MET induced an EMT-like transition and stemness in GSCs (De Bacco et al., 2012). Similarly, incubation of GBM cells with the c-MET ligand HGF induced a mesenchymal phenotype (Yunqing Li, 2011). It is still unclear if these genes only play a role in existing mesenchymal tumor cells, or alternatively can induce PMT in proneural GBM cells
6.3 Influence of the tumor microenvironment on the mesenchymal phenotype
A tight relationship exists between tumor microenvironment and EMT during tumor progression (Finger & Giaccia, 2010; Kalluri & Weinberg, 2009; Polyak & Weinberg, 2009). As mentioned above, mesenchymal GBMs were found to be enriched in genes of the TNF superfamily and NFκB, reflecting a high level of necrosis and a prominent immune infiltration compared to proneural GBMs (H. S. Phillips et al., 2006; Verhaak et al., 2010). This was confirmed with histopathological analyses reflecting high levels of necrosis, hypoxia, and inflammation in mesenchymal specimens compared to other transcriptional subtypes (L. A. D. Cooper et al., 2012; Engler et al., 2012). Further, several mesenchymal network genes identified by Carro et al. (e.g. C/EBPβ, C/EBPδ, FOSL2 and STAT3) were amongst the highest ranked genes that positively correlated with the extent of necrosis, and C/EBPβ and C/EBPδ proteins were expressed in hypoxic perinecrotic regions in GBMs (L A. D. Cooper et al., 2012). As GBMs, in contrast to lower-grade gliomas, are associated with necrosis and more extensive inflammation, it is plausible that necrosis contributes to the mesenchymal phenotype (L. a D. Cooper et al., 2010).
In 2009, bevacizumab was approved for use in recurrent GBM patients. However, a number of studies in patients and preclinical models have demonstrated that the beneficial effects of anti-VEGF therapy are transient as the tumors become highly invasive in nature to circumvent the blockade of the vasculature (Keunen et al., 2011; K. V Lu et al., 2012; Pàez-Ribes et al., 2009). Anti-VEGF therapy and radiotherapy are known to induce HIF1α levels, leading to increased transcription of VEGF and stromal-cell derived factor 1 (SDF-1), and recruitment of myeloid cells. Ultimately, this cascade of events lead to increased vasculogenesis and recurrent tumors displaying a high degree of stemness and expression of mesenchymal markers (Kioi et al., 2010; Piao et al., 2012, 2013). Inhibition of C-X-C chemokine receptor 4 (CXCR4) activity or blockade of HIF1α effectively depleted influx of inflammatory myeloid cells following radiation of mice xenografted with human GBM cells (Kioi et al., 2010). Future studies should identify more specific immune targets and employ GEMM of glioma, displaying an intact immune system, to effectively study changes in the tumor microenvironment following radiotherapy or anti-angiogenic treatment.
As an important target in GBM therapy, recent studies have identified molecular interactions between VEGF and other RTKs. In a GEMM model of glioma, Lu et al. (2012), found that VEGF recruits tyrosine phosphatase PTP1B to prevent c-MET activation (K. V Lu et al., 2012). Hence, anti-VEGF therapy induced VEGFR2 and c-MET heterodimerization followed by HGF-induced phosphorylation of c-MET. The VEGFR2/c-MET complex produced a switch from T-cadherin to N-cadherin, increased invasion and induction of a mesenchymal phenotype. Surprisingly, these effects were independent on hypoxia, suggesting that recruitment of CXCR4-expressing myeloid cells did not contribute to the mesenchymal phenotype in this mouse model.
Targeting inflammatory cells in the tumor microenvironment has yielded some success in preclinical models. As mentioned previously, NFκB activation induces a PMT in proneural glioma cells. The authors also demonstrated that TNFα from microglia/macrophages in the tumor microenvironment is a contributing source of NFκB activation, and dual targeting of NFκB in tumors and immune cell activation via minocycline was effective in decreasing tumor growth and radioresistance only in the mesenchymal and not proneural tumors (Bhat et al., 2013). In another recent study, a colony stimulating factor 1 receptor (CSF1R) inhibitor BLZ945 that targets the tumor-associated myeloid cells was effective in causing the regression of tumors in a proneural model of GBM and increased survival of tumor-bearing mice (Pyonteck et al., 2013). Analyses of tumors revealed that myeloid numbers was unchanged, but the expression of tumor-promoting M2 markers was significantly reduced in these cells. This suggests a reeducation of the immune cells that was sufficient to cut off growth support of tumor cells. It will be interesting if CSF1R inhibition will be effective in gliomas of other subclasses, especially mesenchymal tumors that have high inflammatory signature, and in conjunction with standard of care such as irradiation or chemotherapy.
Reactive astrocytes represent another stromal component of the tumor microenvironment in gliomas. Similar to peripheral tumors where fibroblasts respond to tumor growth and injury, astrocytes become reactive in response to pathological conditions. In glioma, the reactive state and the abundance of reactive astrocytes increase with grade. A recent report show that reactive astrocytes is a major component of the tumor microenvironment in a PDGF-driven GEMM of glioma, surrounding the tumor at the periphery and in the perivascular niche, a region suggested to harbor GSCs (Katz et al., 2012). Less is known about the role of reactive astrocytes propagating tumor growth and promoting treatment-resistance in the GSC compartment.
It becomes increasingly clear that radiotherapy and anti-angiogenic treatments effectively target the tumor bulk, but leave behind subpopulations of tumor cells that become treatment-resistant and form invasive recurrent tumors. Additional investigations are needed to fully understand mechanisms underlying PMT in human glioma. As future studies try to prevent PMT or target the mesenchymal phenotype, it is important to appreciate the extensive intratumoral heterogeneity found in human GBMs. Sottoriva et al. (2013) found that multiple regions form a single GBM show proneural, classical, and mesenchymal gene expression signatures (Sottoriva et al., 2013). This finding parallels recent progress demonstrating intratumoral heterogeneity for RTK amplifications in human GBMs. Intratumoral GBM heterogeneity in the tumor microenvironment and genetic alterations represent a major challenge for future therapies.
7. Relationship Between Glioma Stem Cells and Glial Progentiors
All tumor cells within a tumor tissue must be eliminated to cure the disease. Traditional cancer therapies have been directed to eliminating tumor cells based on their susceptibility to genotoxic therapies such as radiation and DNA alkylating agents. However, cancer stem cells, also referred to as tumor-initiating or tumor-propagating cells, represent small subpopulations of cells within the bulk tumor that are resistant to genotoxic chemotherapies (Dick, 2008; Reya, Morrison, Clarke, & Weissman, 2001). Considerable research effort is being directed to the identification and characterization of cancer stem cells for determining properties that can be exploited for therapeutic eradication. To date, the identification and isolation of cancer stem cells have relied on differential uptake of cell-permeable dyes or expression of cell-surface antigens.
The first report identifying cancer stem cells in glioma, was based on expression of glycosylated CD133 protein on the cell surface of GBM cells (S. K. Singh et al., 2003). CD133-expressing GSCs co-expressed NSC proteins, showed high self-renewal capacity and, in contrast to CD133 negative tumor cells, established tumors in xenografted mice (S. K. Singh et al., 2003). Subsequent reports showed that CD133-expressing GBM cells undergo asymmetric cell division (a prerequisite for tumor regrowth), are resistant to treatment with the alkylating agent temozolomide, and display increased radioresistance (Bao et al., 2006; Deleyrolle et al., 2011; J D Lathia et al., 2011; G. Liu et al., 2006). Additional, and perhaps more provocative findings suggest that CD133+ GSCs regenerate endothelial cells, as well as pericytes, allowing tumors to reestablish the microenvironmental niche required for regrowth (Ricci-Vitiani et al., 2010; R. Wang et al., 2010). More recent reports suggest a large number of alternative GSC markers, many of which show partial or no overlap with CD133+ cells (Anido et al., 2010; Bao et al., 2008; He et al., 2011; Kim et al., 2012; Justin D Lathia et al., 2010; Y. Li et al., 2011; Mazzoleni et al., 2010; Rasper et al., 2010; S. K. Singh et al., 2003; Son, Woolard, Nam, Lee, & Fine, 2009; Tchoghandjian et al., 2010) (Table 2). Interestingly, CD133, A2B5, CD44, c-Met, PDGFRβ and EGFR are also expressed on OPCs (Bouvier-Labit, Liprandi, Monti, Pellissier, & Figarella-Branger, 2002; Moransard, Sawitzky, Fontana, & Suter, 2010; Raff, Miller, & Noble, 1983; Verhaak et al., 2010; J. Wang, O'Bara, Pol, & Sim, 2013). Is it possible that GSC-rich gliomas can arise from OPCs? In fact, we and other groups have demonstrated that the proneural tumor oligodendroglioma can arise from OPCs (Lindberg et al., 2009; C. Liu et al., 2011; Persson et al., 2010). As oncogenic events were introduced into NSCs and OPCs, mosaic analysis with double markers found that transformation only occurred after NSCs had differentiated into OPCs (C. Liu et al., 2011). As A2B5-expressing GSCs become more mesenchymal with grade (Auvergne et al., 2013), it is possible that cell-surface proteins on GSCs change during the disease progression or following therapy (Bhat et al., 2011, 2013; K. V Lu et al., 2012; Mao et al., 2013). In comparison to lower-grade tumors, GBMs display a transcriptional gene expression signature reminiscent of embryonic stem (ES) cells (Ben-Porath et al., 2008). Higher expression of reprogramming factors in GBM (the same factors used by Yamanaka and colleagues to reprogram somatic cells (Takahashi & Yamanaka, 2006), suggest that dedifferentiation of OPCs or more differentiated cells can produce gliomas with a high degree of stemness. In this section, we will discuss gene families that are normally associated with stem cells, but also play important roles during OPC lineage development.
Table 1.2. Examples of cell-surface antigens expressed on GSCs and the corresponding expression in NSC and OPC populations, respectively.
| GSC marker | Reference | NSC marker | OPC marker |
|---|---|---|---|
| CD133 | Singh et al. (2003) | Yes | Yes |
| A2B5 | Tchoghandjian et al. (2010) | No | Yes |
| CD15/SSEA-1 | Son et al. (2009) | Yes | No |
| ALDH1 | Rasper et al. (2010) | Yes | No |
| CD44 | Anido et al. (2010) | Yes | Yes |
| c-Met | Li et al. (2011) | Yes | Yes |
7.1 Polycomb gene family
Proneural genes are expressed in a timely and regional fashion during neocortical development (Wilkinson, Dennis, & Schuurmans, 2013). As one example, the Polycomb group (PcG) gene family regulates cell fate during development. PcG proteins function as complexes known as polycomb repressive complexes (PRC) and work by repressing transcription with methylation altering chromatin structure (Simon & Kingston, 2009). Expression of PRCs maintain stemness in ES and repress developmental genes active during differentiation (T. I. Lee et al., 2006). The PRCs play a similar role in various cancers (Easwaran et al., 2012). As the catalytic component of PRC, Enhancer of Zeste homolog 2 (EZH2), a histone-lysine N-methyltransferase, is downregulated as NSCs differentiate into astrocytes (Sher et al., 2008). Induced overexpression of EZH2 in astrocytes partially dedifferentiates them back towards NSCs (Sher, Boddeke, & Copray, 2011). Interestingly, when NSCs differentiate into OPCs, EZH2 expression remains high (Sher, Boddeke, Olah, & Copray, 2012). Down-regulation of EZH2 in OPCs resulted in derangement of the oligodendrocytic phenotype, due to re-expression of neuronal and astrocytic genes, and ultimately apoptosis (Sher et al., 2012). EZH2 catalyzes the methylations of lysine 27 on histone H3 (H3K27) (R. Cao et al., 2002). As stated above, the K27M mutant has a higher affinity for EZH3 compared to wild type. So it is clear that oncogenic event such as the K27M mutation could disregulate EZH2 leading to cancer. Inhibition of EZH2 with drugs or shRNA has been used as a treatment and has been shown to impair the self-renewal of GBM cancer stem cells (Suvà et al., 2009).
7.2 NOTCH
NOTCH is an important pathway in CNS development and tissue patterning. However, NOTCH is also active in adult NSC keeping them in a quiescent state, and preventing differentiation (Ables, Breunig, Eisch, & Rakic, 2011). In addition, NOTCH functions in OPCs as a repressor of differentiation (Sim et al., 2011; S. Wang et al., 1998) (Figure 2). In glioma, NOTCH-1 and its ligands DELTA-LIKE-1 (DLL1) and JAGGED-1 are overexpressed (Purow et al., 2005; Hongbing Zhang et al., 2007). Furthermore, NOTCH is expressed in several GBM subgroups (Verhaak et al., 2010), where it forms transcriptional networks with GLI1, Myc, BMP2, and RUNX2 (L. a D. Cooper et al., 2010). Several studies suggest that NOTCH-1 activity is essential for survival of GSCs (Ables et al., 2011; Saito et al., 2013; Hongbing Zhang et al., 2007). For example, expression of the active form of NOTCH-1 in human glioma cell lines increase proliferation and self-renewal (Hongbing Zhang et al., 2007). RNA interference experiments showed that DLL1 and JAGGED-1 knockdown reduce survival of GSCs (Purow et al., 2005). In vitro studies suggest cooperative effects of pharmacological inhibition using the γ-secretase inhibitor and γ-irradiation as they target GSCs and non-GSCs, respectively (Ables et al., 2011; Saito et al., 2013). The NOTCH pathway represents an attractive target for future therapies trying to eliminate GSCs.
7.3 Sonic hedgehog (SHH)
Sonic hedgehog is essential for survival of postnatal NSCs (Machold et al., 2003). In addition, As SHH down-stream targets, expression of members of the GLI family is associated with proliferative regions during neural development (Dahmane et al., 2001). In GBMs, GLI-1 is necessary for survival of GSCs and induced by oncogenic drivers (Clement, Sanchez, De Tribolet, Radovanovic, & Ruiz I Altaba, 2007; Dahmane et al., 2001; Santoni et al., 2013). Treatment with the pharmacological SHH inhibitor cyclopamine effectively depleted CD133+ GSCs (Clement et al., 2007). Interestingly, similar to NOTCH-1 inhibition, cyclopamine and temozolomide cooperates to target GSCs and non-GSCs, respectively (Clement et al., 2007). As a downstream target of GLI genes, NANOG is known to promote stemness and is expressed in GBMs (Clement et al., 2007; Mitsui et al., 2003). Interestingly, NANOG is able to reprogram TP53-deficient mouse astrocytes into high-grade gliomas (Moon et al., 2011). SHH also stimulates proliferation of OPCs and is necessary during OPC lineage development (Ferent, Zimmer, Durbec, Ruat, & Traiffort, 2013; Lelievre et al., 2006; Tekki-Kessaris et al., 2001), suggesting that SHH-GLI1 signaling may also drive tumor growth in more progenitor-like gliomas (Figure 2).
7.4 Wingless (WNT)
Wingless (WNT) has been extensively studied as a developmental pathway regulating cell fate specification, migration, and proliferation. The WNT pathway also plays a role in self-renewal of adult NSCs (Kalani et al., 2008). A WNT antagonists increased the numbers of immature OPCs in spinal cord explants (Shimizu et al., 2005), demonstrating that endogenous WNT signaling controls oligodendrocyte development. Isolation of OPCs from the human fetal brain based on CD140a (PDGFRA) expression showed that 12/15 WNT target genes were highly expressed in CD140a+ versus CD140- cell fractions (Sim et al., 2011). The authors concluded that WNT and NOTCH are essential for survival of OPCs. A comparison of development pathways, show that the WNT pathway is more dysregulated than NOTCH or SHH in GSCs compared to human adult NSCs (Sandberg et al., 2013). The authors show that the WNT inhibitor secreted frizzled-related protein 1 (SFRP1) effectively reduced proliferation and self-renewal of GSCs (Sandberg et al., 2013). Furthermore, WNT components are more prominently expressed in A2B5+ cells isolated from GBM versus lower-grade gliomas (Auvergne et al., 2013). During normal development, WNT influence the timing and efficacy of OPC generation in the telencephalon (Langseth et al., 2010). As a target of WNT transcriptional activation, AXIN2 is expressed in OPCs and is essential for normal kinetics of remyelination (Fancy et al., 2011). Interestingly, SOX17 is a down-stream target of the WNT pathway in both OPCs and human oligodendroglioma cells (H.-L. Chen, Chew, Packer, & Gallo, 2013; Chew et al., 2011), exemplifying how in-depth knowledge of OPCs can increase our understanding of glioma biology (Figure 2).
8. Targeted Therapy in Glioma
During development, receptor tyrosine kinases (RTKs) signaling mediate effects of growth factors and regulate expansion and differentiation programs in specific neural precursor populations (Forsberg-Nilsson, Behar, Afrakhte, Barker, & McKay, 1998; Hébert & Fishell, 2008; Kilpatrick & Bartlett, 1995). In the brain, members of the fibroblast growth factor receptor (FGFR), epidermal growth factor (EGFR), and platelet-derived growth factor (PDGFR) families are expressed in distinct NSCs and more differentiated progenitor populations. In GBM, amplifications or somatic mutations in EGFR, PDGFRA, FGFR1, or c-MET often correlate with transcriptomal subgroups (Verhaak et al., 2010). Approximately 88% of GBMs display genetic amplifications of RTKs and genetic alterations of down-stream effector genes. Towards developing a personalized medicine approach and improve outcome for GBM patients, clinical studies increasingly include only subsets of GBM patients that show distinct genetic alterations (Noushmehr et al., 2010; H. S. Phillips et al., 2006; Verhaak et al., 2010). Intra-tumoral heterogeneity of EGFR, PDGFRA, and c-MET in GBMs suggest that future approaches should target several RTKs or common down-stream effectors to benefit GBM patients (Snuderl et al., 2011; Stommel, Kimmelman, & Ying, 2007; Szerlip et al., 2012). Furthermore, pharmacological studies show that monotherapy with either EGFR or PDGFR inhibitors is not sufficient to prevent recurrence in patients (Lo, 2010; Morris & Abrey, 2010). In this section, we will discuss the expression of RTKs in glioma, why inhibition of RTKs or down-stream targets has failed in patients, and their roles in OPC lineage development.
8.1 Epidermal growth factor gene family
As the most commonly amplified RTK in GBMs, EGFR is a member of the ErbB family of membrane-bound receptor tyrosine kinases, of which the other members are ErbB2/HER2/Neu, ErbB3/HER3, and ErbB4/HER4 (Mineo et al., 2007; Verhaak et al., 2010). In addition, approximately 50% of EGFR amplified GBMs express a constitutively active mutation, called EGFRvIII, lacking the extracellular domain (Gan, Kaye, & Luwor, 2009). Other point mutations in the extracellular ligand binding domain of EGFR have been described (J. C. Lee et al., 2006; Vivanco et al., 2012). Over the years, much effort has focused on development of small molecule inhibitors against EGFR inhibitors or vaccines against EGFRvIII (Sampson et al., 2010). To date, EGFR inhibition in patients has largely failed (Table 3). Association of poor response in patients displaying EGFRvIII mutation and PTEN loss suggest that improved stratification of enrolled patients will improve future response rates (Haas-Kogan et al., 2005; Mellinghoff et al., 2005). Furthermore, development of type II that also bind to the inactive conformation of EGFR or irreversible inhibitors may more effectively block down-stream signaling and produce a better response in patients (Barkovich et al., 2012; Vivanco et al., 2012). However, redundant expression of EGFR family members that also contributes to treatment resistance. For example, proneural GBMs express high levels of ErbB3, functionally active in heterodimers with other ErbB family members (Verhaak et al., 2010).
Table 1.3. Association of pediatric H3F3A (G24 and K27) mutant high-grade gliomas to distinct brain regions.
| Subgroup | Target | Inhibitor | Other Targets | Clinical Trials | Reference |
|---|---|---|---|---|---|
| Classical | |||||
| EGFR | Gefitinib (lressa/ZD1839) | – | Yes | Reardon et al. (2006) | |
| Erlotinib (Tarceva/OSI-779) | – | Yes | Raizer and Abrey (2010) | ||
| Cetuximab (Erbitux, C225) | – | Yes | Eller, Longo, Kyle, and Bassano (2005) and Combs et al. (2006) | ||
| Panitumumab (Vectibix) | – | Yes | Gajadhar, Bogdanovic, Munñoz, and Guha (2012) | ||
| AG1478 | – | No | Nagane et al. (2001) | ||
| BIBU-1361 | – | No | Ghildiyal, Dixit, and Sen (2013) | ||
| EKI-785 | – | No | Rao et al. (2005) | ||
| EtDHC | – | No | Han (1997) | ||
| F90 | – | No | Liu et al. (2008) | ||
| NSC56452 | – | No | Yang, Yang, Pike, and Marshall (2010) | ||
| Lapantinb (GW572016) | HER2 | Yes | Vivanco et al. (2012) | ||
| AEE788 | VEGF | Yes | Reardon et al. (2012) | ||
| Vandetanib (Zactima/ZD6474) | VEGFR, RET | Yes | Drappatz et al. (2010) and Shen et al. (2013) | ||
| GW2947 | HER2 | No | Wang, Wei, et al. (2013) | ||
| Proneural | |||||
| PDFGR | α/β: Imatinib (Gleevec, imatinib mesylate, STI571 | VEGF, bcr-abl | Yes | Reardon et al. (2009) and Dong et al. (2011) | |
| α: Sunitinib (Sutent, SU11248, sunitinib malate) | VEGFR2, c-KIT | Yes | Neyns et al. (2011) | ||
| Vatalanib (PTK787, ZK222584) | VEGFR, c-kit | Yes | Gerstner et al. (2011) | ||
| Pazopanib (GW786034) | VEGFR, c-Kit | Yes | Iwamoto (2010) | ||
| β: Dasatinib (Sprycel, BMS-354825) | Src. bcr-abl, c-KIT, EPHA2 | Yes | Lu-Emerson et al. (2011) | ||
| β: Sorafenib (Nexavar,) | VEGFR2, Raf | Yes | Lee et al. (2012) | ||
| α/β: Tandutinib (MLN0518) | FLT3, c-KIT | Yes | Boult, Terkelsen, Walker-Samuel, Bradley, and Robinson (2013) | ||
| Leflunomide (SU101) | – | Yes | Vlassenko, Thiessen, Beattie, Malkin, and Blasberg (2000) and Shawver et al. (1997) | ||
| Nintedanib (BIBF-1120) | VEGFR, FGFR | Yes | Muhic, Poulsen, Sorensen, Grunnet, and Lassen (2013) | ||
| CP-673,451 | No | Roberts et al. (2005) | |||
| IDH1R132H | AGI-5198 | IDH1R132H | No | Rohle et al. (2013) | |
| Mesenchymal | |||||
| VEGF | Bevacizumab | – | Yes | Kreisl, Kim, et al. (2009) | |
| Cediranib (Recentin, AZD2171) | – | Yes | Batchelor et al. (2010) and Wachsberger et al. (2011) | ||
| CT322 (BMS-844203) | – | Yes | Waters et al. (2012) | ||
| Sunitinib (Sutent, SU11248, sunitinib malate) | PDGFRα, c-KIT | Yes | Neyns et al. (2011) | ||
| Vatalanib (PTK787, ZK222584) | PDGFR, c-Kit | Yes | Gerstner et al. (2011) | ||
| Pazopanib (GW786034) | PDGFR, c-Kit | Yes | Iwamoto (2010) | ||
| Vandetanib (Zactima/ZD6474) | EGFR, RET | Yes | Broniscer et al. (2013) | ||
| Sorafenib (Nexavar) | PDGFRβ, Raf | Yes | Lee et al. (2012) | ||
| MET | Cabozantinib (XL184) | RET, KIT, VEGFR2 | Yes | Yakes et al. (2011) and De Groot et al. (2009) | |
| SU11274 | – | No | Stommel et al. (2007) and Joshi et al. (2012) | ||
Receptor tyrosine kinases have been a major therapeutic target in GBMs. As the most commonly amplified RTK, EGFR represent has been an attractive target in clinical trials. Since subsets of GBMs show PDGFRA amplifications and overexpression, Imatinib and other PDGFR inhibitors have been evaluated in patients. Other clinical trials have inhibited VEGF and c-MET signaling in GBMs. For future clinical trials, whole-genome sequencing and transcriptomal studies will be valuable to stratify glioma patients into defined subgroups.
ErbB3 is highly expressed on both human OPCs and proneural glioma cells expressing A2B5 (Auvergne et al., 2013; Sim et al., 2011; Verhaak et al., 2010). ErbB3 and EGFR regulate survival in OPCs (Flores et al., 2000; Ivkovic, Canoll, & Goldman, 2008) (Figure 2). In fact, we and others have shown that proneural gliomas can arise from OPCs (Lai et al., 2011; Lindberg et al., 2009; Persson et al., 2010). Introduction of human EGFRvIII into OPCs produced hyperplasia in postnatal white matter in mice (Ivkovic et al., 2008). Moreover, we showed that a constitutively active avian form of EGFRvIII, v-erbB, generates oligodendroglioma in mice (Persson et al., 2010). In conclusion, EGFR signaling remains an important therapeutic target as research generates better pharmacological tools and identifies molecular characteristics in glioma subgroups for stratification of patients.
8.2 Targeting the proneural subgroup by PDGFR inhibition
Platelet-derived growth factor isoform α and β forms homo- or heterodimers upon ligand binding. Amplification of PDGFRA was identified in 11% of patients (The Cancer Genome Atlas Research Network, 2008). Increased PDGF pathway activity, however, has been reported in up to 33% of adult GBM (Brennan et al., 2009). However, a more recent paper suggests that not only focal, but also low-level, amplifications of PDGFRA are frequent in pediatric (29%) and adult (20%) GBM patients (J. J. Phillips et al., 2013). The authors show that in GBM patients displaying IDH1R132H mutations, PDGFRA amplification was a negative prognostic marker (J. J. Phillips et al., 2013). Recent studies suggest that PDGFRB is expressed on subsets of GSCs and can cross-talk with EGFR in GBMs (Akhavan et al., 2013; Kim et al., 2012).
As one of the first successful small molecule kinase inhibitors, Imatinib effectively inhibited kinase activity of the fusion gene BCR-ABL (Druker et al., 2001). However, Imatinib also inhibits kinase activity for PDGFRA, tyrosine-protein kinase Kit (c-KIT), and fms-like tyrosine kinase 3 (FLT-3). To inhibit PDGFRA signaling in human GBMs, Imatinib reduced phosphorylated AKT levels in 4/11 patients but had no effect on overall survival benefit (Razis et al., 2009) (Table 3). However, since enrolled patients were not stratified based on PDGFA amplification, it is unclear if Imatinib will increase the survival in subsets of GBM patients (Figure 2).
In patients, approximately 35% of proneural GBMs display focal amplifications of PDGFRA (Verhaak et al., 2010). These proneural GBMs express genes associated with OPCs, including PDGFRA commonly used to isolate murine or human OPCs (Sim et al., 2011). PDGFB ligand transforms murine OPCs into gliomas (Lindberg et al., 2009). In postnatal mice, PDGF ligand increases proliferation of NG2+ OPCs in white matter, but not grey matter, an effect that was largely dependent on WNT and PI3K signaling (Hill et al., 2013). In summary, PDGFRA represents a relevant future therapeutic target in proneural gliomas, in particular PDGFRA amplified gliomas.
8.3 Targeting the mesenchymal phenotype through c-MET inhibition
As hepatocyte growth factor (HGF) binds to the c-mesenchymal-epithelial transition factor (c-MET) it can stimulate proliferation of NSCs and OPCs in the postnatal brain (Ohya, Funakoshi, Kurosawa, & Nakamura, 2007; T.-W. Wang, Zhang, Gyetko, & Parent, 2011). Expression of HGF and c-MET at the boundaries between epithelial and mesenchymal cells regulate normal organogenesis; epithelial cells expressing c-MET, and mesenchymal cells secreting the HGF ligand. In glioma, c-MET and HGF expression levels correlate with grade (Koochekpour et al., 1997). Amplification of c-MET is present in approximately 5% of GBMs (Dunn et al., 2012). In a study comparing 19 paired primary and recurrent patient biopsies, overexpression of c-MET has was reported in over 30% of newly diagnosed primary GBM and was much more common (>75%) in relapse cases (W. Liu et al., 2011). Importantly, c-MET overexpression is a key feature of the mesenchymal subclass (H. S. Phillips et al., 2006; Verhaak et al., 2010) and associated with worse prognosis across GBM subgroups (Abounader & Laterra, 2005). Mesenchymal GBMs are associated with high expression of immune-related genes and dense vasculature (Verhaak et al., 2010).
Pre-clinical and clinical studies show that anti-angiogenic therapy in GBMs results in a more invasive phenotype (Gerstner et al., 2010; K. V Lu et al., 2012) (Table 3). In an elegant study by Lu et al., HGF-dependent MET phosphorylation and tumor cell migration were suppressed by a direct interaction between the protein tyrosine phosphatase 1B (PTP1B) and a MET/VEGFR2 heterocomplex, implicating VEGF as a negative regulator of tumor cell invasion (K. V Lu et al., 2012). Concordantly, the dual c-MET and VEGFR inhibitor Cabozantinib (XL184) reduced proliferation GBM cells in vitro and in vivo, potently reducing phosphorylated c-MET, pAKT, and pERK1/2 levels (Navis et al., 2013; Yakes et al., 2011). Cabozantinib is currently used in clinical trials against newly diagnosed or recurrent GBMs (Table 3).
Several studies show that c-MET signaling induces a reprogramming network and support the GSC phenotype (Joo et al., 2012; Y. Li et al., 2011). Expression of c-MET has even been suggested as a functional marker of GSCs (De Bacco et al., 2012). Experiments in an orthotopic xenograft model show that c-MET inhibition reduces growth by selectively targeting the GSC compartment in GBMs (Rath et al., 2013). Future studies should characterize the response of rare GSCs expressing c-MET to radiotherapy and temozolomide treatment in glioma patients.
8.4 Treatment-resistance associated with RTK inhibition
RTK Cooperativity
Cooperativity between multiple RTKs drive tumor growth in GBMs (Stommel et al., 2007). Combined inhibition of MET, PDGFRA, and EGFR was needed to effectively inhibit proliferation in human primary GBM cultures. This is consistent with findings demonstrating intra-tumoral GBM heterogeneity of RTKs in vivo (Snuderl et al., 2011). Furthermore, significant cross-talk occurs between different RTKs. For example, cross-talk has been observed between c-MET and EGFR (Jo et al., 2000; Reznik et al., 2008), c-MET and VEGFR2 (K. V Lu et al., 2012) IGFR1 and PDGFRA (Bielen et al., 2011), and EGFR and PDGFRB (Akhavan et al., 2013).
Redundant Activation of PI3K/mTOR
Ligand binding of the EGFR, PDGFR and c-MET promotes activation of down-stream phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin (mTOR) signaling. Activating mutations in PI3K, observed in 15% of GBMs and missense mutation or deletion of PTEN, seen in more than a third of GBMs, effectively uncouple this pathway from upstream RTK control (The Cancer Genome Atlas Research Network, 2008). Monotherapy with either PI3K inhibitors alone are unlikely to yield any clinical benefit, as PI3K inhibitors yield a cytostatic effect (Cheng, Fan, & Weiss, 2009). As mTOR kinase is a key effector molecule downstream of PI3K, much effort has focused on development of specific mTOR kinase inhibitors. However, the canonical allosteric mTOR kinase; rapamycin/sirolimus, increased phosphorylation of AKT through a feedback loop (Akhavan, Cloughesy, & Mischel, 2010). Furthermore, rapamycin/sirolimus and subsequent “rapalogs”, including temsirolimus/CCI-779 and everolimus/RAD-001, showed limited efficacy in clinical trials. Instead, second generation mTOR inhibitors bind the active site of mTOR kinase, block downstream signaling from both mTORC1 and mTORC2, and are currently being evaluated in clinical trials.
Combined inhibition of PI3K and mTOR potently reduce survival and proliferation of cancer cells. PI-103 is a p110α isoform (catalytic subunit of PI3K) and mTOR kinase dual inhibitor that potently blocks phosphorylation of mTOR targets and induce a robust G0G1 arrest (Fan et al., 2006). The clinical compound, NVP-BEZ235 (Maira et al., 2008) demonstrated robust anti-proliferative activity in many cancer types (Baumann, Mandl-Weber, Oduncu, & Schmidmaier, 2009; P. Cao, Maira, García-Echeverría, & Hedley, 2009; Chapuis et al., 2010; Chiarini et al., 2010; Santiskulvong et al., 2011), including glioma (T.-J. Liu et al., 2009). However, NVP-BEZ235 fails to cross the BBB and therefore is of limited use in GBM patients. Combined inhibition of EGFR effectively cooperated with PI3K/mTOR inhibitors (Fan et al., 2007). These studies exemplify that often combined inhibition of key survival pathways is needed.
Feedback Loops
Blockade of mTOR can paradoxically lead to activation of MAPK (Carracedo et al., 2008) and AKT (O'Reilly et al., 2006; Hongbing Zhang et al., 2007). Similarly, blockade of MAPK (MEK-ERK pathway) can increase phosphorylation of EGFR (X. Li, Huang, Jiang, & Frank, 2008). To avoid the negative effects of feedback loops, researchers use a strategy based on combined inhibition of RTKs and down-stream effectors (Rao et al., 2005; Ronellenfitsch, Steinbach, & Wick, 2010; M. Y. Wang et al., 2006). This strategy has also been evaluated in a clinical setting in GBM patients (Doherty et al., 2006; Kreisl, Lassman, et al., 2009; Nghiemphu, Lai, Green, Reardon, & Cloughesy, 2012; David A Reardon et al., 2006, 2010). Combined treatment regimens were well-tolerated and patients receiving dual inhibitors showed a slight survival benefit (De Witt Hamer, 2010).
Activation of Alternative Survival Pathways
Combined or single treatment with PI3K and mTOR inhibitors induces autophagy in human GBM cells as a survival mechanism (Fan & Weiss, 2010). Interestingly, blockade of autophagy sensitizes GBM cells to radiotherapy, chemotherapy, and RTK inhibition (Kanzawa et al., 2004; Lin et al., 2012; Paglin et al., 2001; Shingu et al., 2009). Preliminary studies in GBM patients suggest that blockade of autophagy in combination with standard of care is a promising strategy (Briceño, Calderon, & Sotelo, 2007; Sotelo, Briceño, & López-González, 2006).
8.5 Therapeutic targeting of IDH-mutant gliomas
Several studies have shown that IDH1R132H mutations in glioma are associated with better survival (Gravendeel et al., 2010; Ichimura et al., 2009; Labussière et al., 2010; Nobusawa, Watanabe, Kleihues, & Ohgaki, 2009; Parsons et al., 2008; Sanson et al., 2009; S. Zhao et al., 2009). However, there is still controversy over the use of IDH mutations as a prognostic indicator. In low-grade glioma patients, Houillier et al. (2010) found that IDH1R132H mutation and 1p-19q co-deletion were associated with prolonged overall survival and a better response to temozolomide treatment (Houillier et al., 2010). Unlike 1p-19q co-deletion, IDH1R132H mutation was not associated with prolonged progression-free (PFS) survival. The data indicate that IDH1R132H mutation may be a significant prognostic indicator independently of 1p-19q status. Similarly, Hartman and colleagues found that IDH status was the most important predictor of progression-free and overall survival only in patients receiving adjuvant therapy (Hartmann et al., 2009). In contrast, van den Bent et al. reported that the presence of IDH1R132H mutation was an indicator of improved prognosis independent of adjuvant therapy (van den Bent et al., 2010). Taken together, these results emphasize the need for more in-depth studies in order to determine the prognostic versus predictive role of IDH status in human glioma.
Reduced expression of IDH1R132H inhibited cell proliferation and clone formation in vitro (Jin et al., 2012), suggesting that direct targeting of the mutation is a viable strategy. Rohle and colleages identified a selective IDH1R132H inhibitor (AGI-5198) through a high throughput screen that effectively reduced tumor growth in a GBM xenograft model (Rohle et al., 2013). The inhibitor blocked the ability of IDH1R132H mutation to produce 2-HG, demethylation of histone H3K9me3, and changes in global methylation signatures. The SHH pathway has been proposed as a possible target downstream of IDH1R132H mutation (Valadez et al., 2013). Since IDH1R132H mutation is associated with a CIMP phenotype, researchers are evaluating hypomethylating agents or epigenetic modifiers (Fathi & Abdel-Wahab, 2012).
In summary, the discovery of IDH mutations has been pivotal for furthering our understanding of how tumorigenesis may occur in gliomas. These findings have led scientists to identify molecular targets in glioma patients diagnosed with IDH-mutant tumors. Future studies should aim to extend histological identification of IDH-mutant tumors to non-invasive imaging correlates in patients.
9. Concluding Remarks and Future Perspectives
The wide-spread distribution and life-long proliferative capacity of OPCs match the temporal and regional occurrence of gliomas, and therefore represent targets for transformation (Figure 6). Genetically distinct gliomas displaying PDGFRA amplifications, IDH1R132H mutations, and H3F3A mutations express the OPC-related genes OLIG2, NKX2.2, PDGFRα and SOX10, implicating a common cell of origin (Sturm et al., 2012; Verhaak et al., 2010). Even mesenchymal gliomas may arise from OPCs as novel data suggest that defined intrinsic factors and influence from the tumor microenvironment enable gliomas to toggle between proneural and mesenchymal phenotypes (Bhat et al., 2013; Mao et al., 2013). An emerging focus on OPCs in gliomagenesis will provide critical information to researchers studying etiology, tumor microenvironment, and therapeutic intervention in glioma.
Figure 6.
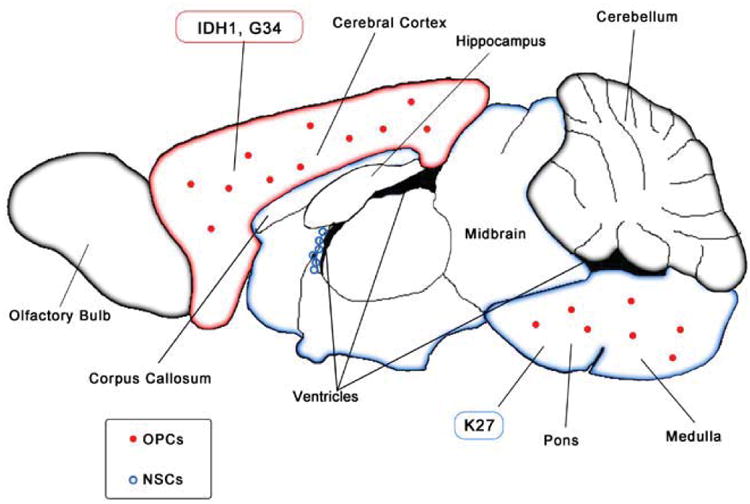
OPCs, but not NSCs, are highly abundant in regions where IDH1R132H mutant and H3F3A K27 mutant tumors arise in GBM patients, implicating their role in gliomagenesis.
Acknowledgments
AIP was supported by research grants from the TDC Foundation, the Guggenhime Endowment Fund, National Brain Tumor Society, and NIH/U54CA163155-01. SI was supported by the Swedish Childhood Cancer Foundation and the Swedish Brain Foundation.
References
- Abel TW, Clark C, Bierie B, Chytil A, Aakre M, Gorska A, Moses HL. GFAP-Cre-mediated activation of oncogenic K-ras results in expansion of the subventricular zone and infiltrating glioma. Molecular cancer research MCR. 2009;7:645–653. doi: 10.1158/1541-7786.MCR-08-0477. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19435821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ables J, Breunig J, Eisch A, Rakic P. Not(ch) just development: Notch signalling in the adult brain. Nature Reviews Neuroscience. 2011;12:269–283. doi: 10.1038/nrn3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abounader R, Laterra J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro-oncology. 2005;7(4):436–51. doi: 10.1215/S1152851705000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar-Morante D, Cortes-Canteli M, Sanz-Sancristobal M, Santos A, Perez-Castillo A. Decreased CCAAT/enhancer binding protein β expression inhibits the growth of glioblastoma cells. Neuroscience. 2011;176:110–119. doi: 10.1016/j.neuroscience.2010.12.025. [DOI] [PubMed] [Google Scholar]
- Akhavan D, Cloughesy TF, Mischel PS. mTOR signaling in glioblastoma: lessons learned from bench to bedside. Neuro-oncology. 2010;12(8):882–9. doi: 10.1093/neuonc/noq052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhavan D, Pourzia AL, Nourian Aa, Williams KJ, Nathanson D, Babic I, et al. Mischel PS. De-repression of PDGFRβ transcription promotes acquired resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients. Cancer discovery. 2013;3(5):534–47. doi: 10.1158/2159-8290.CD-12-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, et al. Parada LF. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15:45–56. doi: 10.1016/j.ccr.2008.12.006. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19111880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anido J, Sáez-Borderías A, Gonzàlez-Juncà A, Rodón L, Folch G, Carmona Ma, et al. Seoane J. TGF-β Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer cell. 2010;18(6):655–68. doi: 10.1016/j.ccr.2010.10.023. [DOI] [PubMed] [Google Scholar]
- Atai NA, Renkema-Mills NA, Bosman J, Schmidt N, Rijkeboer D, Tigchelaar W, et al. Van Noorden CJF. Differential activity of NADPH-producing dehydrogenases renders rodents unsuitable models to study IDH1R132 mutation effects in human glioblastoma. The journal of histochemistry and cytochemistry official journal of the Histochemistry Society. 2011;59:489–503. doi: 10.1369/0022155411400606. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3201175&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auvergne RM, Sim FJ, Wang S, Chandler-Militello D, Burch J, Al Fanek Y, et al. Goldman SA. Transcriptional Differences between Normal and Glioma-Derived Glial Progenitor Cells Identify a Core Set of Dysregulated Genes. Cell reports. 2013:1–15. doi: 10.1016/j.celrep.2013.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, et al. DePinho RA. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12086863. [DOI] [PubMed] [Google Scholar]
- Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta neuropathologica. 2008;116(6):597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, McLendon RE, et al. Rich JN. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Research. 2008;68:6043–6048. doi: 10.1158/0008-5472.CAN-08-1079. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2739001&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/17051156. [DOI] [PubMed] [Google Scholar]
- Barkovich KJ, Hariono S, Garske AL, Zhang J, Blair Ja, Fan QW, et al. Weiss Wa. Kinetics of inhibitor cycling underlie therapeutic disparities between EGFR-driven lung and brain cancers. Cancer discovery. 2012;2(5):450–457. doi: 10.1158/2159-8290.CD-11-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor TT, Duda DG, Di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, et al. Jain RK. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. Journal of Clinical Oncology. 2010;28:2817–2823. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann P, Mandl-Weber S, Oduncu F, Schmidmaier R. The novel orally bioavailable inhibitor of phosphoinositol-3-kinase and mammalian target of rapamycin, NVP-BEZ235, inhibits growth and proliferation in multiple myeloma. Experimental cell research. 2009;315(3):485–97. doi: 10.1016/j.yexcr.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nature Genetics. 2008;40:499–507. doi: 10.1038/ng.127. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/18443585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KPL, Balasubramaniyan V, Vaillant B, Ezhilarasan R, Hummelink K, Hollingsworth F, Aldape K. Mesenchymal Differentiation Mediated by NF-κB Promotes Radiation Resistance in Glioblastoma. Cancer cell. 2013 doi: 10.1016/j.ccr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KPL, Salazar KL, Balasubramaniyan V, Wani K, Heathcock L, Hollingsworth F, et al. Aldape KD. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes & Development. 2011;25:2594–609. doi: 10.1101/gad.176800.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielen A, Perryman L, Box GM, Valenti M, de Haven Brandon A, Martins V, et al. Jones C. Enhanced efficacy of IGF1R inhibition in pediatric glioblastoma by combinatorial targeting of PDGFRα/β. Molecular cancer therapeutics. 2011;10(8):1407–18. doi: 10.1158/1535-7163.MCT-11-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerke L, Mackay A, Nandhabalan M, Burford A, Jury A, Popov S, et al. Jones C. Histone H3.3 Mutations Drive Pediatric Glioblastoma through Upregulation of MYCN. Cancer discovery. 2013 doi: 10.1158/2159-8290.CD-12-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, et al. Bardelli A. IDH1mutations at residue p.R132 (IDH1 R132) occur frequently in high-grade gliomas but not in other solid tumors. Human mutation. 2009;30(1):7–11. doi: 10.1002/humu.20937. [DOI] [PubMed] [Google Scholar]
- Boult JKR, Terkelsen J, Walker-Samuel S, Bradley DP, Robinson SP. A multi-parametric imaging investigation of the response of C6 glioma xenografts to MLN0518 (tandutinib) treatment. PloS one. 2013;8(4):e63024. doi: 10.1371/journal.pone.0063024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier-Labit C, Liprandi A, Monti G, Pellissier JF, Figarella-Branger D. CD44H is expressed by cells of the oligodendrocyte lineage and by oligodendrogliomas in humans. Journal of neurooncology. 2002;60:127–134. doi: 10.1023/a:1020630732625. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12635659. [DOI] [PubMed] [Google Scholar]
- Bralten LBC, Kloosterhof NK, Balvers R, Sacchetti A, Lapre L, Lamfers M, et al. French PJ. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Annals of neurology. 2011;69(3):455–463. doi: 10.1002/ana.22390. [DOI] [PubMed] [Google Scholar]
- Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, Holland E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. In: Creighton C, editor. PloS one. 11. Vol. 4. 2009. p. e7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briceño E, Calderon A, Sotelo J. Institutional experience with chloroquine as an adjuvant to the therapy for glioblastoma multiforme. Surgical neurology. 2007;67(4):388–91. doi: 10.1016/j.surneu.2006.08.080. [DOI] [PubMed] [Google Scholar]
- Broniscer A, Baker SD, Wetmore C, Pai Panandiker AS, Huang J, Davidoff AM, et al. Stewart CF. Phase I trial, pharmacokinetics, and pharmacodynamics of vandetanib and dasatinib in children with newly diagnosed diffuse intrinsic pontine glioma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(11):3050–3058. doi: 10.1158/1078-0432.CCR-13-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Maira SM, García-Echeverría C, Hedley DW. Activity of a novel, dual PI3-kinase/mTor inhibitor NVP-BEZ235 against primary human pancreatic cancers grown as orthotopic xenografts. British journal of cancer. 2009;100(8):1267–76. doi: 10.1038/sj.bjc.6604995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12351676. [DOI] [PubMed] [Google Scholar]
- Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Pandolfi PP. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. The Journal of clinical …. 2008;118(9):3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, et al. Iavarone A. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2009;463(7279):318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, et al. Zhang Z. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes & development. 2013;27(9):985–990. doi: 10.1101/gad.217778.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuis N, Tamburini J, Green AS, Vignon C, Bardet V, Neyret A, et al. Bouscary D. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(22):5424–35. doi: 10.1158/1078-0432.CCR-10-1102. [DOI] [PubMed] [Google Scholar]
- Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Görlich K, et al. Heuser M. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood. 2013 doi: 10.1182/blood-2013-03-491571. [DOI] [PubMed] [Google Scholar]
- Chaumeil MM, Larson PEZ, Yoshihara HAI, Danforth OM, Vigneron DB, Nelson SJ, et al. Ronen SM. Non-invasive in vivo assessment of IDH1 mutational status in glioma. Nature communications. 2013;4:2429. doi: 10.1038/ncomms3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HL, Chew LJ, Packer RJ, Gallo V. Modulation of the Wnt/beta-catenin pathway in human oligodendroglioma cells by Sox17 regulates proliferation and differentiation. Cancer letters. 2013;335(2):361–371. doi: 10.1016/j.canlet.2013.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488(7412):522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CK, Fan QW, Weiss WA. PI3K signaling in glioma--animal models and therapeutic challenges. Brain pathology. 2009;19(1):112–20. doi: 10.1111/j.1750-3639.2008.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler L, Goldenberg DD, Collins R, Grimmer M, Kim GE, Tihan T, et al. Weiss WA. Chemotherapy-induced apoptosis in a transgenic model of neuroblastoma proceeds through p53 induction. Neoplasia. 2008;10(11):1268–1274. doi: 10.1593/neo.08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew LJ, Shen W, Ming X, Senatorov VV, Chen HL, Cheng Y, et al. Gallo V. SRY-box containing gene 17 regulates the Wnt/β-catenin signaling pathway in oligodendrocyte progenitor cells. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31(39):13921–13935. doi: 10.1523/JNEUROSCI.3343-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nature reviews Cancer. 2010;10(7):457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A, et al. Martelli AM. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer research. 2010;70(20):8097–107. doi: 10.1158/0008-5472.CAN-10-1814. [DOI] [PubMed] [Google Scholar]
- Chow LML, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, et al. Baker SJ. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell. 2011;19:305–316. doi: 10.1016/j.ccr.2011.01.039. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3060664&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement V, Sanchez P, De Tribolet N, Radovanovic I, Ruiz I Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Current Biology. 2007;17:165–172. doi: 10.1016/j.cub.2006.11.033. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/17196391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs SE, Heeger S, Haselmann R, Edler L, Debus J, Schulz-Ertner D. Treatment of primary glioblastoma multiforme with cetuximab, radiotherapy and temozolomide (GERT)--phase I/II trial: study protocol. BMC cancer. 2006;6:133. doi: 10.1186/1471-2407-6-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway GD, O'Bara MA, Vedia BH, Pol SU, Sim FJ. Histone deacetylase activity is required for human oligodendrocyte progenitor differentiation. Glia. 2012;000 doi: 10.1002/glia.22410. [DOI] [PubMed] [Google Scholar]
- Cooper LaD, Gutman Da, Long Q, Johnson Ba, Cholleti SR, Kurc T, et al. Moreno CS. The proneural molecular signature is enriched in oligodendrogliomas and predicts improved survival among diffuse gliomas. PloS one. 2010;5(9):e12548. doi: 10.1371/journal.pone.0012548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper LAD, Gutman DA, Chisolm C, Appin C, Kong J, Rong Y, et al. Brat DJ. The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. American Journal Of Pathology. 2012;180(5):2108–2119. doi: 10.1016/j.ajpath.2012.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craene B De, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nature Reviews Cancer. 2013;13(2):97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmane N, Sánchez P, Gitton Y, Palma V, Sun T, Beyna M, et al. Ruiz I Altaba A. The Sonic Hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development Cambridge England. 2001;128:5201–5212. doi: 10.1242/dev.128.24.5201. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11748155. [DOI] [PubMed] [Google Scholar]
- Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes & Development. 2001;15:1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Lyustikman Y, Shih A, Hu X, Fuller GN, Rosenblum M, Holland EC. The Characteristics of Astrocytomas and Oligodendrogliomas Are Caused by Two Distinct and Interchangeable Signaling Formats. Neoplasia. 2005;7:397–406. doi: 10.1593/neo.04691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MRL, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Molecular And Cellular Neurosciences. 2003;24:476–488. doi: 10.1016/s1044-7431(03)00210-0. Retrieved from http://linkinghub.elsevier.com/retrieve/pii/S1044743103002100. [DOI] [PubMed] [Google Scholar]
- De Bacco F, Casanova E, Medico E, Pellegatta S, Orzan F, Albano R, et al. Boccaccio C. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer research. 2012;72(17):4537–4550. doi: 10.1158/0008-5472.CAN-11-3490. [DOI] [PubMed] [Google Scholar]
- De Groot JF, Prados M, Urquhart T, Robertson S, Yaron Y, Sorensen AG, et al. Wen P. A phase II study of XL184 in patients (pts) with progressive glioblastoma multiforme (GBM) in first or second relapse. Journal of Clinical Oncology. 2009;27(15S) De Groot et al. 27 (15): 2047 – ASCO Meeting Abstracts. 2047. Retrieved from http://meeting.ascopubs.org/cgi/content/abstract/27/15S/2047. [Google Scholar]
- De Witt Hamer P. Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro-oncology. 2010;12(3):304–316. doi: 10.1093/neuonc/nop068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleyrolle LP, Harding A, Cato K, Siebzehnrubl FA, Rahman M, Azari H, et al. Reynolds BA. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain: A journal of neurology. 2011;134:1331–1343. doi: 10.1093/brain/awr081. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3097894&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793–4807. doi: 10.1182/blood-2008-08-077941. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19064739. [DOI] [PubMed] [Google Scholar]
- Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Clarke MF. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Shannon P, Lau N, Wu X, Roncari L, Baldwin RL, et al. Guha A. Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer Research. 2003;55:1106–1113. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12615729. [PubMed] [Google Scholar]
- Doetsch F. The glial identity of neural stem cells. Nature Neuroscience. 2003;6:1127–1134. doi: 10.1038/nn1144. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/14583753. [DOI] [PubMed] [Google Scholar]
- Doherty L, Gigas DC, Kesari S, Drappatz J, Kim R, Zimmerman J, et al. Wen PY. Pilot study of the combination of EGFR and mTOR inhibitors in recurrent malignant gliomas. Neurology. 2006;67(1):156–8. doi: 10.1212/01.wnl.0000223844.77636.29. [DOI] [PubMed] [Google Scholar]
- Dong Y, Jia L, Wang X, Tan X, Xu J, Deng Z, et al. Ren H. Selective inhibition of PDGFR by imatinib elicits the sustained activation of ERK and downstream receptor signaling in malignant glioma cells. International journal of oncology. 2011;38(2):555–569. doi: 10.3892/ijo.2010.861. [DOI] [PubMed] [Google Scholar]
- Drappatz J, Norden AD, Wong ET, Doherty LM, LaFrankie DC, Ciampa A, et al. Wen PY. Phase I Study of Vandetanib With Radiotherapy and Temozolomide for Newly Diagnosed Glioblastoma. International Journal of Radiation Oncology, Biology, Physics. 2010;78(1):85–90. doi: 10.1016/j.ijrobp.2009.07.1741. Retrieved from http://www.sciencedirect.com/science/article/pii/S0360301609029319. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. The New England journal of medicine. 2001;344(14):1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, et al. Hahn WC. Emerging insights into the molecular and cellular basis of glioblastoma. Genes & Development. 2012;26(8):756–784. doi: 10.1101/gad.187922.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easwaran H, Johnstone S, Vanneste L, Ohm J, Mosbruger T, Wang Q, et al. Baylin SB. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Research. 2012:837–849. doi: 10.1101/gr.131169.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eller JL, Longo SL, Kyle MM, Bassano D. Anti-epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery. 2005;56(1):155–162. doi: 10.1227/01.NEU.0000145865.25689.55. [DOI] [PubMed] [Google Scholar]
- Elson A, Deng C, Campos-Torres J, Donehower LA, Leder P. The MMTV/c-myc transgene and p53 null alleles collaborate to induce T-cell lymphomas, but not mammary carcinomas in transgenic mice. Oncogene. 1995;11(1):181–190. [PubMed] [Google Scholar]
- Engler JR, Robinson AE, Smirnov I, Hodgson JG, Berger MS, Gupta N, et al. Phillips JJ. Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PloS one. 2012;7(8):e43339. doi: 10.1371/journal.pone.0043339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nature Medicine. 1998;4:1313–1317. doi: 10.1038/3305. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9809557. [DOI] [PubMed] [Google Scholar]
- Fan QW, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM, Weiss WA. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer research. 2007;67(17):7960–5. doi: 10.1158/0008-5472.CAN-07-2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan QW, Knight Za, Goldenberg DD, Yu W, Mostov KE, Stokoe D, et al. Weiss Wa. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer cell. 2006;9(5):341–9. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan QW, Weiss WA. Targeting the RTK-PI3K-mTOR axis in malignant glioma: overcoming resistance. Current topics in microbiology and immunology. 2010;347:279–296. doi: 10.1007/82_2010_67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SPJ, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, et al. Rowitch DH. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nature Neuroscience. 2011;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fathi AT, Abdel-Wahab O. Mutations in Epigenetic Modifiers in Myeloid Malignancies and the Prospect of Novel Epigenetic-Targeted Therapy. Advances in hematology. 2012;2012(12):1–12. doi: 10.1155/2012/469592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferent J, Zimmer C, Durbec P, Ruat M, Traiffort E. Sonic Hedgehog Signaling Is a Positive Oligodendrocyte Regulator during Demyelination. The Journal of neuroscience the official journal of the Society for Neuroscience. 2013;33:1759–72. doi: 10.1523/JNEUROSCI.3334-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell. 2010;18(6):553–67. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer and Metastasis Reviews. 2010;29(2):285–293. doi: 10.1007/s10555-010-9224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, et al. Hjelmeland AB. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nature neuroscience. 2013 doi: 10.1038/nn.3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores aI, Mallon BS, Matsui T, Ogawa W, Rosenzweig a, Okamoto T, Macklin WB. Akt-mediated survival of oligodendrocytes induced by neuregulins. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20(20):7622–7630. doi: 10.1523/JNEUROSCI.20-20-07622.2000. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11027222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg-Nilsson K, Behar TN, Afrakhte M, Barker JL, McKay RD. Platelet-derived growth factor induces chemotaxis of neuroepithelial stem cells. Journal of Neuroscience Research. 1998;53:521–530. doi: 10.1002/(SICI)1097-4547(19980901)53:5<521::AID-JNR2>3.0.CO;2-B. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9726423. [DOI] [PubMed] [Google Scholar]
- Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, et al. Verma IM. Dedifferentiation of Neurons and Astrocytes by Oncogenes Can Induce Gliomas in Mice. Science. 2012 doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajadhar AS, Bogdanovic E, Muñoz DM, Guha A. In situ analysis of mutant EGFRs prevalent in glioblastoma multiforme reveals aberrant dimerization, activation, and differential response to anti-EGFR targeted therapy. Molecular cancer research : MCR. 2012;10(3):428–440. doi: 10.1158/1541-7786.MCR-11-0531. [DOI] [PubMed] [Google Scholar]
- Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2009;16(6):748–754. doi: 10.1016/j.jocn.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Garner JM, Fan M, Yang CH, Du Z, Sims M, Davidoff AM, Pfeffer LM. Constitutive Activation of Signal Transducer and Activator of Transcription 3 (STAT3) and Nuclear Factor B Signaling in Glioblastoma Cancer Stem Cells Regulates the Notch Pathway. Journal of Biological Chemistry. 2013;288(36):26167–26176. doi: 10.1074/jbc.M113.477950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavalas a, Trainor P, Ariza-McNaughton L, Krumlauf R. Synergy between Hoxa1 and Hoxb1: the relationship between arch patterning and the generation of cranial neural crest. Development (Cambridge, England) 2001;128(15):3017–27. doi: 10.1242/dev.128.15.3017. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11532923. [DOI] [PubMed] [Google Scholar]
- Gerstner ER, Chen PJ, Wen PY, Jain RK, Batchelor TT, Sorensen G. Infiltrative patterns of glioblastoma spread detected via diffusion MRI after treatment with cediranib. Neuro-oncology. 2010;12(5):466–472. doi: 10.1093/neuonc/nop051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner ER, Eichler AF, Plotkin SR, Drappatz J, Doyle CL, Xu L, et al. Batchelor TT. Phase I trial with biomarker studies of vatalanib (PTK787) in patients with newly diagnosed glioblastoma treated with enzyme inducing anti-epileptic drugs and standard radiation and temozolomide. Journal of neuro-oncology. 2011;103(2):325–332. doi: 10.1007/s11060-010-0390-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal R, Dixit D, Sen E. EGFR inhibitor BIBU induces apoptosis and defective autophagy in glioma cells. Molecular carcinogenesis. 2012 doi: 10.1002/mc.21938. [DOI] [PubMed] [Google Scholar]
- Godard S, Getz G, Delorenzi M, Farmer P, Kobayashi H, Desbaillets I, et al. Hegi ME. Classification of human astrocytic gliomas on the basis of gene expression: a correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes. Cancer research. 2003;63(20):6613–6625. [PubMed] [Google Scholar]
- Gravendeel LAM, Kloosterhof NK, Bralten LBC, van Marion R, Dubbink HJ, Dinjens W, et al. French PJ. Segregation of non-p.R132H mutations in IDH1 in distinct molecular subtypes of glioma. Human mutation. 2010;31(3):E1186–99. doi: 10.1002/humu.21201. [DOI] [PubMed] [Google Scholar]
- Gu F, Hata R, Ma YJ, Tanaka J, Mitsuda N, Kumon Y, et al. Sakanaka M. Suppression of Stat3 promotes neurogenesis in cultured neural stem cells. Journal of neuroscience research. 2005;81(2):163–171. doi: 10.1002/jnr.20561. [DOI] [PubMed] [Google Scholar]
- Gutman DA, Cooper LAD, Hwang SN, Holder CA, Gao J, Aurora TD, et al. Brat DJ. MR Imaging Predictors of Molecular Profile and Survival: Multi-institutional Study of the TCGA Glioblastoma Data Set. Radiology. 2013;267(2):560–569. doi: 10.1148/radiol.13120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas-Kogan DA, Prados MD, Tihan T, Eberhard DA, Jelluma N, Arvold ND, et al. Stokoe D. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. Journal of the National Cancer Institute. 2005;97(12):880–7. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- Hall A, Giese NA, Richardson WD. Spinal cord oligodendrocytes develop from ventrally derived progenitor cells that express PDGF alpha-receptors. Development Cambridge England. 1996;122:4085–4094. doi: 10.1242/dev.122.12.4085. Retrieved from http://discovery.ucl.ac.uk/109515/ [DOI] [PubMed] [Google Scholar]
- Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, et al. Deimling A. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta neuropathologica. 2009;118(4):469–474. doi: 10.1007/s00401-009-0561-9. [DOI] [PubMed] [Google Scholar]
- He J, Liu Y, Zhu T, Zhu J, Dimeco F, Vescovi AL, et al. Lubman DM. CD90 is identified as a marker for cancer stem cells in primary high-grade gliomas using tissue microarrays. Molecular cellular proteomics MCP. 2011:1–24. doi: 10.1074/mcp.M111.010744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert JM, Fishell G. The genetics of early telencephalon patterning: some assembly required. Nature Reviews Neuroscience. 2008;9:678–685. doi: 10.1038/nrn2463. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2669317&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hede SM, Hansson I, Afink GB, Eriksson A, Nazarenko I, Andrae J, et al. Nistér M. GFAP promoter driven transgenic expression of PDGFB in the mouse brain leads to glioblastoma in a Trp53 null background. Glia. 2009;57:1143–1153. doi: 10.1002/glia.20837. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19115382. [DOI] [PubMed] [Google Scholar]
- Hill Ra, Patel KD, Medved J, Reiss AM, Nishiyama A. NG2 Cells in White Matter But Not Gray Matter Proliferate in Response to PDGF. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33(36):14558–14566. doi: 10.1523/JNEUROSCI.2001-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstim C, Deneen B, Lukaszewicz A, Zhou Q, Anderson DJ. Identification of positionally distinct astrocyte subtypes whose identities are specified by a homeodomain code. Cell. 2008;133:510–522. doi: 10.1016/j.cell.2008.02.046. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/18455991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson JG, Yeh RF, Ray A, Wang NJ, Smirnov I, Yu M, et al. James CD. Comparative analyses of gene copy number and mRNA expression in glioblastoma multiforme tumors and xenografts. Neuro-oncology. 2009;11(5):477–487. doi: 10.1215/15228517-2008-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nature Genetics. 2000;25:55–57. doi: 10.1038/75596. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10802656. [DOI] [PubMed] [Google Scholar]
- Holland Eric C, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes & Development. 1998;12:3675–3685. doi: 10.1101/gad.12.23.3675. Retrieved from http://www.genesdev.org/cgi/doi/10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma J, Yamanaka R, Yajima N, Tsuchiya N, Genkai N, Sano M, Tanaka R. Increased expression of CCAAT/enhancer binding protein β correlates with prognosis in glioma patients. Oncology reports. 2006;15(3):595–601. [PubMed] [Google Scholar]
- Hong JH, Yaffe MB. TAZ: a beta-catenin-like molecule that regulates mesenchymal stem cell differentiation. Cell cycle. 2006;5(2):176–179. doi: 10.4161/cc.5.2.2362. [DOI] [PubMed] [Google Scholar]
- Houillier C, Wang X, Kaloshi G, Mokhtari K, Guillevin R, Laffaire J, et al. Delattre JY. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75(17):1560–1566. doi: 10.1212/WNL.0b013e3181f96282. [DOI] [PubMed] [Google Scholar]
- Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR Promotes Survival and Astrocytic Characteristics Induced by Pten/Akt Signaling in Glioblastoma1. Neoplasia. 2005;7:356–368. doi: 10.1593/neo.04595. Retrieved from http://www.ingentaselect.com/rpsv/cgi-bin/cgi?ini=xref&body=linker&reqdoi=10.1593/neo.04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XL, Wang Y, Shen Q. Epigenetic control on cell fate choice in neural stem cells. Protein & cell. 2012;3(4):278–290. doi: 10.1007/s13238-012-2916-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Kang SH, Fukaya M, Bergles DE. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nature Neuroscience. 2013;16:668–676. doi: 10.1038/nn.3390. Retrieved from http://www.nature.com/doifinder/10.1038/nn.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nature Reviews Cancer. 2010;10:319–331. doi: 10.1038/nrc2818. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/20414201. [DOI] [PubMed] [Google Scholar]
- Ichimura K, Pearson DM, Kocialkowski S, Bäcklund LM, Chan R, Jones DTW, Collins VP. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro-Oncology. 2009;11(4):341–347. doi: 10.1215/15228517-2009-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Furusho M, Bansal R. Sustained activation of ERK1/2 MAPK in oligodendrocytes and schwann cells enhances myelin growth and stimulates oligodendrocyte progenitor expansion. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33(1):175–86. doi: 10.1523/JNEUROSCI.4403-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivkovic S, Canoll P, Goldman JE. Constitutive EGFR signaling in oligodendrocyte progenitors leads to diffuse hyperplasia in postnatal white matter. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28(4):914–922. doi: 10.1523/JNEUROSCI.4327-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto FM. trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06. Neuro-oncology. 2010;12(8):855–861. doi: 10.1093/neuonc/noq025. Retrieved from http://neuro-oncology.oxfordjournals.org/content/12/8/855.short. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques TS, Swales A, Brzozowski MJ, Henriquez NV, Linehan JM, Mirzadeh Z, et al. Brandner S. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. the The European Molecular Biology Organization Journal. 2010;29:222–235. doi: 10.1038/emboj.2009.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen NA, Pedersen KM, Lihme F, Rask L, Nielsen JV, Rasmussen TE, Mitchelmore C. Astroglial c-Myc overexpression predisposes mice to primary malignant gliomas. The Journal of Biological Chemistry. 2003;278:8300–8308. doi: 10.1074/jbc.M211195200. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12501251. [DOI] [PubMed] [Google Scholar]
- Jin G, Pirozzi CJ, Chen LH, Lopez GY, Duncan CG, Feng J, et al. Yan H. Mutant IDH1 is required for IDH1 mutated tumor cell growth. Oncotarget. 2012;3(8):774–782. doi: 10.18632/oncotarget.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. The Journal of biological chemistry. 2000;275(12):8806–11. doi: 10.1074/jbc.275.12.8806. [DOI] [PubMed] [Google Scholar]
- Jones Pa, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo KM, Jin J, Kim E, Ho Kim K, Kim Y, Gu Kang B, et al. Nam DH. MET Signaling Regulates Glioblastoma Stem Cells. Cancer Research. 2012 doi: 10.1158/0008-5472.CAN-11-3760. [DOI] [PubMed] [Google Scholar]
- Joshi AD, Loilome W, Siu IM, Tyler B, Gallia GL, Riggins GJ. Evaluation of tyrosine kinase inhibitor combinations for glioblastoma therapy. PloS one. 2012;7(10):e44372. doi: 10.1371/journal.pone.0044372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalani MYS, Cheshier SH, Cord BJ, Bababeygy SR, Vogel H, Weissman IL, et al. Nusse R. Wnt-mediated self-renewal of neural stem/progenitor cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16970–16975. doi: 10.1073/pnas.0808616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of clinical investigation. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SIl, et al. Lee SH. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. International journal of cancer Journal international du cancer. 2009;125(2):353–355. doi: 10.1002/ijc.24379. [DOI] [PubMed] [Google Scholar]
- Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell death and differentiation. 2004;11(4):448–57. doi: 10.1038/sj.cdd.4401359. [DOI] [PubMed] [Google Scholar]
- Katz AM, Amankulor NM, Pitter K, Helmy K, Squatrito M, Holland EC. Astrocyte-specific expression patterns associated with the PDGF-induced glioma microenvironment. PloS one. 2012;7(2):e32453. doi: 10.1371/journal.pone.0032453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, et al. Roussel MF. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer cell. 2012;21(2):168–180. doi: 10.1016/j.ccr.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nature Neuroscience. 2006;9:173–9. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keunen O, Johansson M, Oudin A, Sanzey M, Rahim SAA, Fack F, et al. Niclou SP. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3749–3754. doi: 10.1073/pnas.1014480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, et al. Hawkins C. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta neuropathologica. 2012;124(3):439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick TJ, Bartlett PF. Cloned multipotential precursors from the mouse cerebrum require FGF-2, whereas glial restricted precursors are stimulated with either FGF-2 or EGF. Journal of Neuroscience. 1995;15:3653–3661. doi: 10.1523/JNEUROSCI.15-05-03653.1995. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/7751935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Kim E, Wu Q, Guryanova O, Hitomi M, Lathia JD, et al. Rich JN. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes & Development. 2012;26:1247–62. doi: 10.1101/gad.193565.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. The Journal of clinical investigation. 2010;120(3):694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala S, Corfas G. Identification of Novel Glial Genes by Single-Cell Transcriptional Profiling of Bergmann Glial Cells from Mouse Cerebellum. PLoS ONE. 2010;5:15. doi: 10.1371/journal.pone.0009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koochekpour S, Jeffers M, Rulong S, Taylor G, Klineberg E, Hudson EA, et al. Vande Woude GF. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer research. 1997;57(23):5391–8. [PubMed] [Google Scholar]
- Kraus AC, Ferber I, Bachmann SO, Specht H, Wimmel A, Gross MW, et al. Schuermann M. In vitro chemo- and radio-resistance in small cell lung cancer correlates with cell adhesion and constitutive activation of AKT and MAP kinase pathways. Oncogene. 2002;21(57):8683–8695. doi: 10.1038/sj.onc.1205939. [DOI] [PubMed] [Google Scholar]
- Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, et al. Fine HA. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(5):740–5. doi: 10.1200/JCO.2008.16.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisl TN, Lassman AB, Mischel PS, Rosen N, Scher HI, Teruya-Feldstein J, et al. Abrey LE. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM) Journal of neuro-oncology. 2009;92(1):99–105. doi: 10.1007/s11060-008-9741-z. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, et al. Parada LF. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Research. 2008;68:3286–3294. doi: 10.1158/0008-5472.CAN-07-6867. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2760841&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labussière M, Idbaih A, Wang XW, Marie Y, Boisselier B, Falet C, et al. Sanson M. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology. 2010;74(23):1886–1890. doi: 10.1212/WNL.0b013e3181e1cf3a. [DOI] [PubMed] [Google Scholar]
- Lai A, Kharbanda S, Pope WB, Tran A, Solis OE, Peale F, et al. Phillips HS. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29(34):4482–90. doi: 10.1200/JCO.2010.33.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langseth AJ, Munji RN, Choe Y, Huynh T, Pozniak CD, Pleasure SJ. Wnts influence the timing and efficiency of oligodendrocyte precursor cell generation in the telencephalon. Journal of Neuroscience. 2010;30:13367–13372. doi: 10.1523/JNEUROSCI.1934-10.2010. Retrieved from http://www.jneurosci.org/cgi/doi/10.1523/JNEUROSCI.1934-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasiene J, Matsui A, Sawa Y, Wong F, Horner PJ. Age-related myelin dynamics revealed by increased oligodendrogenesis and short internodes. Aging Cell. 2009;8:201–213. doi: 10.1111/j.1474-9726.2009.00462.x. Retrieved from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2703583/?tool=pubmed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Hitomi M, Gallagher J, Gadani SP, Adkins J, Vasanji A, et al. Rich JN. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell death disease. 2011;2:e200. doi: 10.1038/cddis.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia Justin D, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, et al. Rich JN. Integrin alpha 6 regulates glioblastoma stem cells. Cell stem cell. 2010;6:421–432. doi: 10.1016/j.stem.2010.02.018. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2884275&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EQ, Kuhn J, Lamborn KR, Abrey L, DeAngelis LM, Lieberman F, et al. Wen PY. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro-oncology. 2012;14(12):1511–1518. doi: 10.1093/neuonc/nos264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Vivanco I, Beroukhim R, Huang JHY, Feng WL, DeBiasi RM, et al. Mellinghoff IK. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. In: Lassman A, editor. PLoS medicine. 12. Vol. 3. 2006. p. e485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, et al. Young RA. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. Retrieved from http://discovery.ucl.ac.uk/7048/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S, et al. Canoll P. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PLoS ONE. 2011;6:e20041. doi: 10.1371/journal.pone.0020041. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3100315&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, et al. Guan KL. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Molecular and cellular biology. 2008;28(7):2426–2436. doi: 10.1128/MCB.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelievre V, Ghiani CA, Seksenyan A, Gressens P, De Vellis J, Waschek JA. Growth factor-dependent actions of PACAP on oligodendrocyte progenitor proliferation. Regulatory Peptides. 2006;137:58–66. doi: 10.1016/j.regpep.2006.04.024. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16989910. [DOI] [PubMed] [Google Scholar]
- Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, et al. Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340(6134):857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Huang Y, Jiang J, Frank SJ. ERK-dependent threonine phosphorylation of EGF receptor modulates receptor downregulation and signaling. Cellular Signalling. 2008;20:2145–2155. doi: 10.1016/j.cellsig.2008.08.006. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2613789&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li A, Glas M, Lal B, Ying M, Sang Y, et al. Laterra J. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:9951–9956. doi: 10.1073/pnas.1016912108. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3116406&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon KL, Kesari S, Kitada M, Sun T, Arnett HA, Alberta JA, et al. Rowitch DH. Development of NG2 neural progenitor cells requires Olig gene function. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7853–7858. doi: 10.1073/pnas.0511001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CJ, Lee CC, Shih YL, Lin CH, Wang SH, Chen TH, Shih CM. Inhibition of mitochondria- and endoplasmic reticulum stress-mediated autophagy augments temozolomide-induced apoptosis in glioma cells. PloS one. 2012;7(6):e38706. doi: 10.1371/journal.pone.0038706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene. 2009;28:2266–2275. doi: 10.1038/onc.2009.76. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19421151. [DOI] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RGW, Hippenmeyer S, Vogel H, et al. Zong H. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146(2):209–221. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu FJ, Gui SB, Li CZ, Sun ZL, Zhang YZ, Academy C. Antitumor activity of F90, an epidermal growth factor receptor tyrosine kinase inhibitor, on glioblastoma cell line SHG-44. Chinese medical journal. 2008;121(17):1702–1706. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19024103. [PubMed] [Google Scholar]
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, et al. Yu JS. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Molecular Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TJ, Koul D, LaFortune T, Tiao N, Shen RJ, Maira SM, et al. Yung WKA. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Molecular cancer therapeutics. 2009;8(8):2204–10. doi: 10.1158/1535-7163.MCT-09-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Fu Y, Xu S, Ding F, Zhao G, Zhang K, et al. Pang Q. c-Met expression is associated with time to recurrence in patients with glioblastoma multiforme. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2011;18(1):119–21. doi: 10.1016/j.jocn.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes & Development. 2012;26(12):1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HW. EGFR-targeted therapy in malignant glioma: novel aspects and mechanisms of drug resistance. Current molecular pharmacology. 2010;3(1):37–52. doi: 10.2174/1874467211003010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta neuropathologica. 2007;114(2):97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. Thompson CB. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2013;483(7390):474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KV, Chang JP, Parachoniak Ca, Pandika MM, Aghi MK, Meyronet D, et al. Bergers G. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer cell. 2012;22(1):21–35. doi: 10.1016/j.ccr.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchman HA, Stechishin OD, Dang NH, Blough MD, Chesnelong C, Kelly JJ, et al. Weiss S. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro-Oncology. 2012;14(2):184–191. doi: 10.1093/neuonc/nor207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu-Emerson C, Norden AD, Drappatz J, Quant EC, Beroukhim R, Ciampa AS, et al. Wen PY. Retrospective study of dasatinib for recurrent glioblastoma after bevacizumab failure. Journal of neuro-oncology. 2011;104(1):287–291. doi: 10.1007/s11060-010-0489-x. [DOI] [PubMed] [Google Scholar]
- Lumsden a, Krumlauf R. Patterning the vertebrate neuraxis. Science. 1996;274(5290):1109–15. doi: 10.1126/science.274.5290.1109. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8895453. [DOI] [PubMed] [Google Scholar]
- Machold R, Hayashi S, Rutlin M, Muzumdar MD, Nery S, Corbin JG, et al. Fishell G. Sonic hedgehog is required for progenitor cell telencephalic stem cell niches. Neuron. 2003;39:937–50. doi: 10.1016/s0896-6273(03)00561-0. [DOI] [PubMed] [Google Scholar]
- Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. García-Echeverría C. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Molecular cancer therapeutics. 2008;7(7):1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- Mallini P, Lennard T, Kirby J, Meeson A. Epithelial-to-mesenchymal transition: What is the impact on breast cancer stem cells and drug resistance. Cancer Treatment Reviews. 2013 doi: 10.1016/j.ctrv.2013.09.008. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. Weinberg RA. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, et al. Nakano I. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(21):8644–9. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, Verma IM. Development of a novel mouse glioma model using lentiviral vectors. Nature Medicine. 2009;15:110–116. doi: 10.1038/nm.1863. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19122659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoleni S, Politi LS, Pala M, Cominelli M, Franzin A, Sergi Sergi L, et al. Galli R. Epidermal growth factor receptor expression identifies functionally and molecularly distinct tumor-initiating cells in human glioblastoma multiforme and is required for gliomagenesis. Cancer research. 2010;70(19):7500–13. doi: 10.1158/0008-5472.CAN-10-2353. [DOI] [PubMed] [Google Scholar]
- Mehta S, Huillard E, Kesari S, Maire CL, Golebiowski D, Harrington EP, et al. Stiles CD. The central nervous system-restricted transcription factor Olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer cell. 2011;19(3):359–371. doi: 10.1016/j.ccr.2011.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Mischel PS. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. The New England journal of medicine. 2005;353(19):2012–24. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- Ménard C, Hein P, Paquin A, Savelson A, Yang XM, Lederfein D, et al. Miller FD. An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron. 2002;36(4):597–610. doi: 10.1016/s0896-6273(02)01026-7. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Mirzadeh Z, Alvarez-Buylla A. Mosaic organization of neural stem cells in the adult brain. Science. 2007;317:381–384. doi: 10.1126/science.1144914. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/17615304. [DOI] [PubMed] [Google Scholar]
- Mikheeva SA, Mikheev AM, Petit A, Beyer R, Oxford RG, Khorasani L, et al. Rostomily RC. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Molecular cancer. 2010;9:194. doi: 10.1186/1476-4598-9-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineo JF, Bordron A, Baroncini M, Maurage CA, Ramirez C, Siminski RM, et al. Dam Hieu P. Low HER2-expressing glioblastomas are more often secondary to anaplastic transformation of low-grade glioma. Journal of neuro-oncology. 2007;85(3):281–287. doi: 10.1007/s11060-007-9424-1. [DOI] [PubMed] [Google Scholar]
- Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, et al. Yamanaka S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113:631–642. doi: 10.1016/s0092-8674(03)00393-3. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12787504. [DOI] [PubMed] [Google Scholar]
- Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, et al. Beachy PA. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(11):4453–4458. doi: 10.1073/pnas.1101657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JH, Kwon S, Jun EK, Kim A, Whang KY, Kim H, et al. You S. Nanog-induced dedifferentiation of p53-deficient mouse astrocytes into brain cancer stem-like cells. Biochemical and biophysical research communications. 2011;412:175–181. doi: 10.1016/j.bbrc.2011.07.070. [DOI] [PubMed] [Google Scholar]
- Moransard M, Sawitzky M, Fontana A, Suter T. Expression of the HGF receptor c-met by macrophages in experimental autoimmune encephalomyelitis. Glia. 2010;58:559–571. doi: 10.1002/glia.20945. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19941340. [DOI] [PubMed] [Google Scholar]
- Morris PG, Abrey LE. Novel targeted agents for platelet-derived growth factor receptor and c-KIT in malignant gliomas. Targeted oncology. 2010;5(3):193–200. doi: 10.1007/s11523-010-0160-7. [DOI] [PubMed] [Google Scholar]
- Muhic A, Poulsen HS, Sorensen M, Grunnet K, Lassen U. Phase II open-label study of nintedanib in patients with recurrent glioblastoma multiforme. Journal of neuro-oncology. 2013;111(2):205–12. doi: 10.1007/s11060-012-1009-y. [DOI] [PubMed] [Google Scholar]
- Nagane M, Narita Y, Mishima K, Levitzki A, Burgess AW, Cavenee WK, Huang HJ. Human glioblastoma xenografts overexpressing a tumor-specific mutant epidermal growth factor receptor sensitized to cisplatin by the AG1478 tyrosine kinase inhibitor. Journal of neurosurgery. 2001;95(3):472–479. doi: 10.3171/jns.2001.95.3.0472. [DOI] [PubMed] [Google Scholar]
- Nakashima K. Synergistic Signaling in Fetal Brain by STAT3-Smad1 Complex Bridged by p300. Science. 1999;284(5413):479–482. doi: 10.1126/science.284.5413.479. [DOI] [PubMed] [Google Scholar]
- Navis AC, Bourgonje A, Wesseling P, Wright A, Hendriks W, Verrijp K, et al. Leenders WPJ. Effects of dual targeting of tumor cells and stroma in human glioblastoma xenografts with a tyrosine kinase inhibitor against c-MET and VEGFR2. PloS one. 2013;8(3):e58262. doi: 10.1371/journal.pone.0058262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarenko I, Hedrén A, Sjödin H, Orrego A, Andrae J, Afink GB, et al. Lindström MS. Brain Abnormalities and Glioma-Like Lesions in Mice Overexpressing the Long Isoform of PDGF-A in Astrocytic Cells. PLoS ONE. 2011;6:14. doi: 10.1371/journal.pone.0018303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyns B, Sadones J, Chaskis C, Dujardin M, Everaert H, Lv S, et al. De Greve J. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. Journal of neuro-oncology. 2011;103(3):491–501. doi: 10.1007/s11060-010-0402-7. [DOI] [PubMed] [Google Scholar]
- Nghiemphu PL, Lai A, Green RM, Reardon DA, Cloughesy T. A dose escalation trial for the combination of erlotinib and sirolimus for recurrent malignant gliomas. Journal of neuro-oncology. 2012;110(2):245–50. doi: 10.1007/s11060-012-0960-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niola F, Zhao X, Singh D, Sullivan R, Castano A, Verrico A, et al. Lasorella A. Mesenchymal high-grade glioma is maintained by the ID-RAP1 axis. The Journal of clinical investigation. 2013;123(1):405–17. doi: 10.1172/JCI63811DS1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB. Co-localization of NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells in the developing rat brain. Journal of Neuroscience Research. 1996;43:299–314. doi: 10.1002/(SICI)1097-4547(19960201)43:3<;299∷AID-JNR5>;3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Nobusawa S, Watanabe T, Kleihues P, Ohgaki H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(19):6002–6007. doi: 10.1158/1078-0432.CCR-09-0715. [DOI] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Network TCGAR. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell. 2010;17(5):510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer research. 2006;66(3):1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya W, Funakoshi H, Kurosawa T, Nakamura T. Hepatocyte growth factor (HGF) promotes oligodendrocyte progenitor cell proliferation and inhibits its differentiation during postnatal development in the rat. Brain Research. 2007;1147:51–65. doi: 10.1016/j.brainres.2007.02.045. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/17382307. [DOI] [PubMed] [Google Scholar]
- Olar A, Aldape KD. Biomarkers Classification and Therapeutic Decision-Making for Malignant Gliomas. Current Treatment Options in Oncology. 2012:1–20. doi: 10.1007/s11864-012-0210-8. [DOI] [PubMed] [Google Scholar]
- Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, et al. Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, et al. Yahalom J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer research. 2001;61(2):439–44. [PubMed] [Google Scholar]
- Paquin A. CCAAT/Enhancer-Binding Protein Phosphorylation Biases Cortical Precursors to Generate Neurons Rather Than Astrocytes In Vivo. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25(46):10747–10758. doi: 10.1523/JNEUROSCI.2662-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JCH, Leary RJ, Angenendt P, et al. Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/18772396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, et al. Wechsler-Reya RJ. An animal model of MYC-driven medulloblastoma. Cancer cell. 2012;21(2):155–167. doi: 10.1016/j.ccr.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R, et al. Weiss WA. Non-stem cell origin for oligodendroglioma. Cancer Cell. 2010;18:669–682. doi: 10.1016/j.ccr.2010.10.033. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3031116&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryniak MA, Potter GB, Rowitch DH, Rubenstein JLR. Dlx1 and Dlx2 control neuronal versus oligodendroglial cell fate acquisition in the developing forebrain. Neuron. 2007;55:417–433. doi: 10.1016/j.neuron.2007.06.036. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2039927&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Aldape K. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer cell. 2006;9(3):157–73. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Phillips JJ, Aranda D, Ellison DW, Judkins AR, Croul SE, Brat DJ, et al. Perry A. PDGFRA Amplification is Common in Pediatric and Adult High-Grade Astrocytomas and Identifies a Poor Prognostic Group in IDH1 Mutant Glioblastoma. Brain pathology. 2013;23(5):565–573. doi: 10.1111/bpa.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao Y, Liang J, Holmes L, Henry V, Sulman E, de Groot JF. Acquired Resistance to Anti-VEGF Therapy in Glioblastoma Is Associated with a Mesenchymal Transition. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(16):4392–403. doi: 10.1158/1078-0432.CCR-12-1557. [DOI] [PubMed] [Google Scholar]
- Piao Y, Liang J, Holmes L, Zurita AJ, Henry V, Heymach JV, de Groot JF. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro-oncology. 2012;14(11):1379–1392. doi: 10.1093/neuonc/nos158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piaskowski S, Bienkowski M, Stoczynska-Fidelus E, Stawski R, Sieruta M, Szybka M, et al. Rieske P. Glioma cells showing IDH1 mutation cannot be propagated in standard cell culture conditions. British journal of cancer. 2011;104(6):968–970. doi: 10.1038/bjc.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo S, Cordenonsi M, Dupont S. Molecular Pathways: YAP and TAZ Take Center Stage in Organ Growth and Tumorigenesis. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(18):4925–4930. doi: 10.1158/1078-0432.CCR-12-3172. [DOI] [PubMed] [Google Scholar]
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nature Reviews Cancer. 2009;9(4):265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- Priester M, Copanaki E, Vafaizadeh V, Hensel S, Bernreuther C, Glatzel M, et al. Weissenberger J. STAT3 silencing inhibits glioma single cell infiltration and tumor growth. Neuro-oncology. 2013;15(7):840–852. doi: 10.1093/neuonc/not025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J, et al. Fine HA. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Research. 2005;65:2353–2363. doi: 10.1158/0008-5472.CAN-04-1890. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15781650. [DOI] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. Joyce Ja. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nature Medicine. 2013 Aug;:1–12. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Miller RH, Noble M. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature. 1983;303:390–396. doi: 10.1038/303390a0. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/6304520. [DOI] [PubMed] [Google Scholar]
- Raizer JJ, Abrey LE. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-oncology. 2010;12(1):95–103. doi: 10.1093/neuonc/nop015. Retrieved from http://neuro-oncology.oxfordjournals.org/content/12/1/95.short. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. Principles of neural cell migration. Experientia. 1990;46(9):882–891. doi: 10.1007/BF01939380. [DOI] [PubMed] [Google Scholar]
- Rao RD, Mladek AC, Lamont JD, Goble JM, Erlichman C, James CD, Sarkaria JN. Disruption of Parallel and Converging Signaling Pathways Contributes to the Synergistic Antitumor Effects of Simultaneous mTOR and EGFR Inhibition in GBM Cells. Neoplasia. 2005;7(10):921–929. doi: 10.1593/neo.05361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasper M, Schäfer A, Piontek G, Teufel J, Brockhoff G, Ringel F, et al. Schlegel J. Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neurooncology. 2010;12:1024–1033. doi: 10.1093/neuonc/noq070. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3018920&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath P, Lal B, Ajala O, Li Y, Xia S, Kim J, Laterra J. In Vivo c-Met Pathway Inhibition Depletes Human Glioma Xenografts of Tumor-Propagating Stem-Like Cells. Translational oncology. 2013;6(2):104–11. doi: 10.1593/tlo.13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razis E, Selviaridis P, Labropoulos S, Norris JL, Zhu MJ, Song DD, et al. Fountzilas G. Phase II study of neoadjuvant imatinib in glioblastoma: evaluation of clinical and molecular effects of the treatment. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(19):6258–66. doi: 10.1158/1078-0432.CCR-08-1867. [DOI] [PubMed] [Google Scholar]
- Reardon DA, Dresemann G, Taillibert S, Campone M, van den Bent M, Clement P, et al. Nikolova Z. Multicentre phase II studies evaluating imatinib plus hydroxyurea in patients with progressive glioblastoma. British journal of cancer. 2009;101(12):1995–2004. doi: 10.1038/sj.bjc.6605411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon David A, Conrad CA, Cloughesy T, Prados MD, Friedman HS, Aldape KD, et al. Yung WKA. Phase I study of AEE788, a novel multitarget inhibitor of ErbB- and VEGF-receptor-family tyrosine kinases, in recurrent glioblastoma patients. Cancer chemotherapy and pharmacology. 2012;69(6):1507–1518. doi: 10.1007/s00280-012-1854-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon David A, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE, et al. Friedman HS. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. Journal of neuro-oncology. 2010;96(2):219–30. doi: 10.1007/s11060-009-9950-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon David A, Quinn JA, Vredenburgh JJ, Gururangan S, Friedman AH, Desjardins A, et al. Rich JN. Phase 1 trial of gefitinib plus sirolimus in adults with recurrent malignant glioma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(3 Pt 1):860–8. doi: 10.1158/1078-0432.CCR-05-2215. [DOI] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11689955. [DOI] [PubMed] [Google Scholar]
- Reznik TE, Sang Y, Ma Y, Abounader R, Rosen EM, Xia S, Laterra J. Transcription-dependent epidermal growth factor receptor activation by hepatocyte growth factor. Molecular cancer research : MCR. 2008;6(1):139–50. doi: 10.1158/1541-7786.MCR-07-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, et al. De Maria R. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:824–828. doi: 10.1038/nature09557. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/21102434. [DOI] [PubMed] [Google Scholar]
- Rickman DS, Bobek MP, Misek DE, Kuick R, Blaivas M, Kurnit DM, et al. Hanash SM. Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer research. 2001;61(18):6885–6891. [PubMed] [Google Scholar]
- Roberts WG, Whalen PM, Soderstrom E, Moraski G, Lyssikatos JP, Wang HF, et al. Ung E. Antiangiogenic and antitumor activity of a selective PDGFR tyrosine kinase inhibitor, CP-673,451. Cancer research. 2005;65(3):957–966. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15705896. [PubMed] [Google Scholar]
- Robinson JP, Vanbrocklin MW, Lastwika KJ, McKinney AJ, Brandner S, Holmen SL. Activated MEK cooperates with Ink4a/Arf loss or Akt activation to induce gliomas in vivo. Oncogene. 2011;30:1341–1350. doi: 10.1038/onc.2010.513. Retrieved from http://discovery.ucl.ac.uk/441249/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. Mellinghoff IK. An Inhibitor of Mutant IDH1 Delays Growth and Promotes Differentiation of Glioma Cells. Science. 2013;340(6132):626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronellenfitsch MW, Steinbach JP, Wick W. Epidermal growth factor receptor and mammalian target of rapamycin as therapeutic targets in malignant glioma: current clinical status and perspectives. Targeted oncology. 2010;5(3):183–91. doi: 10.1007/s11523-010-0154-5. [DOI] [PubMed] [Google Scholar]
- Saito N, Fu J, Zheng S, Yao J, Wang S, Liu DD, et al. Koul D. A high Notch pathway activation predicts response to γ secretase inhibitors in proneural subtype of glioma tumor initiating cells. Stem cells. 2013 doi: 10.1002/stem.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Bigner DD. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(31):4722–4729. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanai N, Nguyen T, Ihrie RA, Mirzadeh Z, Tsai HH, Wong M, et al. Alvarez-Buylla A. Corridors of migrating neurons in the human brain and their decline during infancy. Nature. 2011;478:1–6. doi: 10.1038/nature10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg CJ, Altschuler G, Jeong J, Strømme KK, Stangeland B, Murrell W, et al. Langmoen IA. Comparison of glioma stem cells to neural stem cells from the adult human brain identifies dysregulated Wnt- signaling and a fingerprint associated with clinical outcome. Experimental cell research. 2013:1–14. doi: 10.1016/j.yexcr.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, et al. Delattre JY. Isocitrate Dehydrogenase 1 Codon 132 Mutation Is an Important Prognostic Biomarker in Gliomas. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(25):4150–4154. doi: 10.1200/JCO.2009.21.9832. [DOI] [PubMed] [Google Scholar]
- Santiskulvong C, Konecny GE, Fekete M, Chen KYM, Karam A, Mulholland D, et al. Dorigo O. Dual targeting of phosphoinositide 3-kinase and mammalian target of rapamycin using NVP-BEZ235 as a novel therapeutic approach in human ovarian carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(8):2373–84. doi: 10.1158/1078-0432.CCR-10-2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoni M, Burattini L, Nabissi M, Beatrice Morelli M, Berardi R, Santoni G, Cascinu S. Essential Role of Gli Proteins in Glioblastoma Multiforme. Current Protein and Peptide Science. 2013;14:8. doi: 10.2174/1389203711314020005. Retrieved from http://www.ingentaconnect.com.sci-hub.org/content/ben/cpps/2013/00000014/00000002/art00005. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio IIC, et al. Mak TW. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes & Development. 2012;26(18):2038–2049. doi: 10.1101/gad.198200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brüstle A, et al. Mak TW. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2013;488(7413):656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW. Dissecting p53 tumor suppressor functions in vivo. Cancer cell. 2002;1(3):289–298. doi: 10.1016/s1535-6108(02)00047-8. [DOI] [PubMed] [Google Scholar]
- Schwartzentruber J, Korshunov A, Liu XY, Jones DTW, Pfaff E, Jacob K, et al. Tonjes M. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- Shannon P, Sabha N, Lau N, Kamnasaran D, Gutmann DH, Guha A. Pathological and molecular progression of astrocytomas in a GFAP:12 V-Ha-Ras mouse astrocytoma model. The American journal of pathology. 2005;167:859–867. doi: 10.1016/S0002-9440(10)62057-3. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1698742&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shawver LK, Schwartz DP, Mann E, Chen H, Tsai J, Chu L, et al. Powell TJ. Inhibition of platelet-derived growth factor-mediated signal transduction and tumor growth by N-[4-(trifluoromethyl)-phenyl]5-methylisoxazole-4-carboxamide. Clinical cancer research : an official journal of the American Association for Cancer Research. 1997;3(7):1167–1177. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9815796. [PubMed] [Google Scholar]
- Shen J, Zheng H, Ruan J, Fang W, Li A, Tian G, et al. Zhao P. Autophagy inhibition induces enhanced proapoptotic effects of ZD6474 in glioblastoma. British journal of cancer. 2013;109(1):164–171. doi: 10.1038/bjc.2013.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher F, Boddeke E, Copray S. Ezh2 expression in astrocytes induces their dedifferentiation toward neural stem cells. Cellular reprogramming. 2011;13:1–6. doi: 10.1089/cell.2010.0052. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/20979531. [DOI] [PubMed] [Google Scholar]
- Sher F, Boddeke E, Olah M, Copray S. Dynamic Changes in Ezh2 Gene Occupancy Underlie Its Involvement in Neural Stem Cell Self-Renewal and Differentiation towards Oligodendrocytes. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0040399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher F, Rössler R, Brouwer N, Balasubramaniyan V, Boddeke E, Copray S. Differentiation of neural stem cells into oligodendrocytes: involvement of the polycomb group protein Ezh2. Stem Cells. 2008;26:2875–2883. doi: 10.1634/stemcells.2008-0121. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/18687996. [DOI] [PubMed] [Google Scholar]
- Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 Is Required for Proliferation and Maintenance of Multipotency in Glioblastoma Stem Cells. Stem Cells. 2009;27(10):2383–2392. doi: 10.1002/stem.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AH, Dai C, Hu X, Shih AH, Dai C, Hu X, et al. Holland EC. Dose-Dependent Effects of Platelet-Derived Growth Factor-B on Glial Tumorigenesis Dose-Dependent Effects of Platelet-Derived Growth Factor-B on Glial Tumorigenesis. Cancer research. 2004;64:4783–4789. doi: 10.1158/0008-5472.CAN-03-3831. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Kagawa T, Wada T, Muroyama Y, Takada S, Ikenaka K. Wnt signaling controls the timing of oligodendrocyte development in the spinal cord. Developmental Biology. 2005;282:397–410. doi: 10.1016/j.ydbio.2005.03.020. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15950605. [DOI] [PubMed] [Google Scholar]
- Shingu T, Fujiwara K, Bögler O, Akiyama Y, Moritake K, Shinojima N, et al. Kondo S. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. International journal of cancer Journal international du cancer. 2009;124(5):1060–71. doi: 10.1002/ijc.24030. [DOI] [PubMed] [Google Scholar]
- Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR, et al. Steindler DA. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO molecular medicine. 2013;5(8):1196–1212. doi: 10.1002/emmm.201302827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim FJ, McClain CR, Schanz SJ, Protack TL, Windrem MS, Goldman SA. CD140a identifies a population of highly myelinogenic, migration-competent and efficiently engrafting human oligodendrocyte progenitor cells. Nature Biotechnology. 2011;29:934–41. doi: 10.1038/nbt.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nature Reviews Molecular Cell Biology. 2009;10:697–708. doi: 10.1038/nrm2763. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19738629. [DOI] [PubMed] [Google Scholar]
- Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Research. 2003;63:5821–8. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, et al. Iafrate AJ. Mosaic Amplification of Multiple Receptor Tyrosine Kinase Genes in Glioblastoma. Cancer Cell. 2011;20(6):810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Son MJ, Woolard K, Nam DH, Lee J, Fine HA. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell stem cell. 2009;4:440–452. doi: 10.1016/j.stem.2009.03.003. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19427293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo J, Briceño E, López-González MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Annals of internal medicine. 2006;144(5):337–43. doi: 10.7326/0003-4819-144-5-200603070-00008. [DOI] [PubMed] [Google Scholar]
- Sottile V, Li M, Scotting PJ. Stem cell marker expression in the Bergmann glia population of the adult mouse brain. Brain research. 2006;1099(1):8–17. doi: 10.1016/j.brainres.2006.04.127. [DOI] [PubMed] [Google Scholar]
- Sottoriva A, Spiteri I, Piccirillo SGM, Touloumis A, Collins VP, Marioni JC, et al. Tavaré S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(10):4009–14. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner LA, Schulz VP, Maksimova Y, Wong C, Gallagher PG. Patterns of histone H3 lysine 27 monomethylation and erythroid cell type-specific gene expression. The Journal of biological chemistry. 2011;286(45):39457–39465. doi: 10.1074/jbc.M111.243006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes & Development. 2003;17:1677–1689. doi: 10.1101/gad.259003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt C, Schlierf A, Lommes P, Hillgartner S, Werner T, Kosian T, et al. Wegner M. SoxD proteins influence multiple stages of oligodendrocyte development and modulate SoxE protein function. Developmental Cell. 2006;11:697–709. doi: 10.1016/j.devcel.2006.08.011. Retrieved from http://discovery.ucl.ac.uk/143376/ [DOI] [PubMed] [Google Scholar]
- Stommel J, Kimmelman A, Ying H. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;287(2007) doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DTW, Konermann C, et al. Bender S. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer cell. 2012;22(4):425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- Sugiarto S, Persson AI, Munoz EG, Waldhuber M, Lamagna C, Andor N, et al. Petritsch C. Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell. 2011;20:328–340. doi: 10.1016/j.ccr.2011.08.011. Retrieved from http://linkinghub.elsevier.com/retrieve/pii/S1535610811003084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvà ML, Riggi N, Janiszewska M, Radovanovic I, Provero P, Stehle JC, et al. Stamenkovic I. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Research. 2009;69:9211–9218. doi: 10.1158/0008-5472.CAN-09-1622. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19934320. [DOI] [PubMed] [Google Scholar]
- Szerlip NJ, Pedraza A, Chakravarty D, Azim M, McGuire J, Fang Y, et al. Brennan CW. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(8):3041–3046. doi: 10.1073/pnas.1114033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tchoghandjian A, Baeza N, Colin C, Cayre M, Metellus P, Beclin C, et al. Figarella-Branger D. A2B5 cells from human glioblastoma have cancer stem cell properties. Brain pathology. 2010;20(1):211–221. doi: 10.1111/j.1750-3639.2009.00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekki-Kessaris N, Woodruff R, Hall AC, Gaffield W, Kimura S, Stiles CD, et al. Richardson WD. Hedgehog-dependent oligodendrocyte lineage specification in the telencephalon. Development Cambridge England. 2001;128:2545–2554. doi: 10.1242/dev.128.13.2545. Retrieved from http://discovery.ucl.ac.uk/10570/ [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP. Epithelial--mesenchymal transitions in tumour progression. Nature Reviews Cancer. 2002;2(6):442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- Trainor Pa, Krumlauf R. Hox genes, neural crest cells and branchial arch patterning. Current opinion in cell biology. 2001;13(6):698–705. doi: 10.1016/s0955-0674(00)00273-8. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11698185. [DOI] [PubMed] [Google Scholar]
- Tsai HH, Li H, Fuentealba LC, Molofsky AV, Taveira-Marques R, Zhuang H, et al. Rowitch DH. Regional Astrocyte Allocation Regulates CNS Synaptogenesis and Repair. Science. 2012;358:1–15. doi: 10.1126/science.1222381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. Chan TA. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2013;483(7390):479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrbom L, Dai C, Celestino JC, Rosenblum MK, Fuller GN, Holland EC. Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Research. 2002;62:5551–5558. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12359767. [PubMed] [Google Scholar]
- Valadez JG, Grover VK, Carter MD, Calcutt MW, Abiria SA, Lundberg CJ, et al. Cooper MK. Identification of Hedgehog pathway responsive glioblastomas by isocitrate dehydrogenase mutation. Cancer letters. 2013;328(2):297–306. doi: 10.1016/j.canlet.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bent MJ, Dubbink HJ, Marie Y, Brandes Aa, Taphoorn MJB, Wesseling P, et al. Sanson M. IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(5):1597–604. doi: 10.1158/1078-0432.CCR-09-2902. [DOI] [PubMed] [Google Scholar]
- Van den Boom J, Wolter M, Kuick R, Misek DE, Youkilis AS, Wechsler DS, et al. Hanash SM. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. The American journal of pathology. 2003;163(3):1033–1043. doi: 10.1016/S0002-9440(10)63463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S, Garimella MT, Sullivan LM, Martinez D, Huse JT, Heguy A, et al. Judkins AR. Evaluation of Histone 3 Lysine 27 Trimethylation (H3K27me3) and Enhancer of Zest 2 (EZH2) in Pediatric Glial and Glioneuronal Tumors Shows Decreased H3K27me3 in H3F3A K27M Mutant Glioblastomas. Brain pathology. 2013 doi: 10.1111/bpa.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RGW, Hoadley Ka, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Hayes DN. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I, Robins HI, Rohle D, Campos C, Grommes C, Nghiemphu PL, et al. Mellinghoff IK. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer discovery. 2012;2(5):458–471. doi: 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlassenko AG, Thiessen B, Beattie BJ, Malkin MG, Blasberg RG. Evaluation of early response to SU101 target-based therapy in patients with recurrent supratentorial malignant gliomas using FDG PET and Gd-DTPA MRI. Journal of neuro-oncology. 2000;46(3):249–259. doi: 10.1023/a:1006481313747. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10902856. [DOI] [PubMed] [Google Scholar]
- Wachsberger PR, Lawrence RY, Liu Y, Xia X, Andersen B, Dicker AP. Cediranib enhances control of wild type EGFR and EGFRvIII-expressing gliomas through potentiating temozolomide, but not through radiosensitization: implications for the clinic. Journal of neuro-oncology. 2011;105(2):181–190. doi: 10.1007/s11060-011-0580-y. [DOI] [PubMed] [Google Scholar]
- Wang J, O'Bara Ma, Pol SU, Sim FJ. CD133/CD140a-based isolation of distinct human multipotent neural progenitor cells and oligodendrocyte progenitor cells. Stem cells and development. 2013;22(15):2121–2131. doi: 10.1089/scd.2013.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM, et al. Mischel PS. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer research. 2006;66(16):7864–9. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
- Wang Q, Wei F, Li C, Lv G, Wang G, Liu T, et al. Hao C. Combination of mTOR and EGFR Kinase Inhibitors Blocks mTORC1 and mTORC2 Kinase Activity and Suppresses the Progression of Colorectal Carcinoma. PloS one. 2013;8(8):e73175. doi: 10.1371/journal.pone.0073175. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, et al. Tabar V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–33. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, et al. Barres BA. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron. 1998;21:63–75. doi: 10.1016/s0896-6273(00)80515-2. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9697852. [DOI] [PubMed] [Google Scholar]
- Wang TW, Zhang H, Gyetko MR, Parent JM. Hepatocyte growth factor acts as a mitogen and chemoattractant for postnatal subventricular zone-olfactory bulb neurogenesis. Molecular And Cellular Neurosciences. 2011;48:38–50. doi: 10.1016/j.mcn.2011.06.003. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3160177&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yang J, Zheng H, Tomasek GJ, Zhang P, McKeever PE, et al. Zhu Y. Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell. 2009;15:514–526. doi: 10.1016/j.ccr.2009.04.001. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/19477430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. The American journal of pathology. 2009;174(4):1149–1153. doi: 10.2353/ajpath.2009.080958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters JD, Sanchez C, Sahin A, Futalan D, Gonda DD, Scheer JK, et al. Carter BS. CT322, a VEGFR-2 antagonist, demonstrates anti-glioma efficacy in orthotopic brain tumor model as a single agent or in combination with temozolomide and radiation therapy. Journal of neuro-oncology. 2012;110(1):37–48. doi: 10.1007/s11060-012-0948-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Clarke L, Scheidenhelm DK, Qian B, Tong A, Sabha N, et al. Guha A. High-grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer Research. 2006;66:7429–7437. doi: 10.1158/0008-5472.CAN-06-0712. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/16885338. [DOI] [PubMed] [Google Scholar]
- Weiss S, Dunne C, Hewson J, Wohl C, Wheatley M, Peterson AC, Reynolds BA. Multipotent CNS stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16(23):7599–7609. doi: 10.1523/JNEUROSCI.16-23-07599.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss WA, Burns MJ, Hackett C, Aldape K, Hill JR, Kuriyama H, et al. Israel MA. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Research. 2003;63:1589–1595. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12670909. [PubMed] [Google Scholar]
- Wilkinson G, Dennis D, Schuurmans C. Proneural genes in neocortical development. Neuroscience. 2013;253C:256–273. doi: 10.1016/j.neuroscience.2013.08.029. [DOI] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, et al. St Jude Children's Research Hospital--Washington University Pediatric Cancer Genome Project. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature genetics. 2012;44(3):251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Tamamaki N, Noda T, Kimura K, Itokazu Y, Matsumoto N, et al. Ide C. Neurogenesis in the ependymal layer of the adult rat 3rd ventricle. Experimental neurology. 2005;192(2):251–264. doi: 10.1016/j.expneurol.2004.12.021. [DOI] [PubMed] [Google Scholar]
- Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, et al. Joly AH. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Molecular cancer therapeutics. 2011;10(12):2298–308. doi: 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. Bigner DD. IDH1 and IDH2 mutations in gliomas. The New England journal of medicine. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang RYC, Yang KS, Pike LJ, Marshall GR. Targeting the dimerization of epidermal growth factor receptors with small-molecule inhibitors. Chemical biology & drug design. 2010;76(1):1–9. doi: 10.1111/j.1747-0285.2010.00986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip S, Butterfield YS, Morozova O, Chittaranjan S, Blough MD, An J, et al. Marra MA. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. The Journal of pathology. 2011;226(1):7–16. doi: 10.1002/path.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Thomas-Tikhonenko A. A non-transgenic mouse model for B-cell lymphoma: in vivo infection of p53-null bone marrow progenitors by a Myc retrovirus is sufficient for tumorigenesis. Oncogene. 2002;21(12):1922–1927. doi: 10.1038/sj.onc.1205244. [DOI] [PubMed] [Google Scholar]
- Li Yunqing, L A, G M, L B, Y M, S Y, X S, T D, G-C H, E CG, Q-H A, S B, L J. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(24):9951–9956. doi: 10.1073/pnas.1016912108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Heng, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, et al. Guan KL. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. The Journal of biological chemistry. 2009;284(20):13355–13362. doi: 10.1074/jbc.M900843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Hongbing, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, et al. Kwiatkowski DJ. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. The Journal of clinical investigation. 2007;117(3):730–738. doi: 10.1172/JCI28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. St. Jude Children's Research Hospital--Washington University Pediatric Cancer Genome Project. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nature genetics. 2013;45(6):602–612. doi: 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XP, Zheng G, Zou L, Liu HL, Hou LH, Zhou P, et al. Chen JY. Notch activation promotes cell proliferation and the formation of neural stem cell-like colonies in human glioma cells. Molecular and Cellular Biochemistry. 2008;307:101–108. doi: 10.1007/s11010-007-9589-0. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/17849174. [DOI] [PubMed] [Google Scholar]
- Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes & Development. 2010;24(9):862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Xiong Y. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, et al. Parada LF. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119–130. doi: 10.1016/j.ccr.2005.07.004. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3024718&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo H, Nishiyama A. Polydendrocytes in development and myelin repair. Neuroscience bulletin. 2013:1–12. doi: 10.1007/s12264-013-1320-4. [DOI] [PMC free article] [PubMed] [Google Scholar]