

Abstract
Background
The majority of newly diagnosed prostate cancers will remain indolent, but distinguishing between aggressive and indolent disease is imprecise. This has led to the important clinical problem of overtreatment. THOC1 encodes a nuclear ribonucleoprotein whose expression is higher in some cancers than in normal tissue. The hypothesis that THOC1 may be a functionally relevant biomarker that can improve the identification of aggressive prostate cancer has not been tested.
Methods
THOC1 protein immunostaining was evaluated in a retrospective collection of more than 700 human prostate cancer specimens and the results associated with clinical variables and outcome. Thoc1 was conditionally deleted in an autochthonous mouse model (n = 22 or 23 per genotype) to test whether it is required for prostate cancer progression. All statistical tests were two-sided.
Results
THOC1 protein immunostaining increases with higher Gleason score and more advanced Tumor/Node/Metastasis stage. Time to biochemical recurrence is statistically significantly shorter for cancers with high THOC1 protein (log-rank P = .002, and it remains statistically significantly associated with biochemical recurrence after adjusting for Gleason score, clinical stage, and prostate-specific antigen levels (hazard ratio = 1.61, 95% confidence interval = 1.03 to 2.51, P = .04). Thoc1 deletion prevents prostate cancer progression in mice, but has little effect on normal tissue. Prostate cancer cells deprived of Thoc1 show gene expression defects that compromise cell growth.
Conclusions
Thoc1 is required to support the unique gene expression requirements of aggressive prostate cancer in mice. In humans, high THOC1 protein immunostaining associates with prostate cancer aggressiveness and recurrence. Thus, THOC1 protein is a functionally relevant molecular marker that may improve the identification of aggressive prostate cancers, potentially reducing overtreatment.
Prostate-specific antigen (PSA) screening has led to an increase in the diagnosis of clinically localized prostate cancer. A large proportion of diagnosed patients will not progress clinically, and few would succumb to prostate cancer even without treatment (1). This has led to the problem of overtreatment, where patients suffer the side effects of therapy like radical prostatectomy (RP) without clinically significant benefit. Active surveillance has been proposed for men with low-risk prostate cancer to reduce overtreatment. A large community practice–based clinical database indicates approximately 7% of men elect active surveillance for newly diagnosed, clinically localized prostate cancer. This is far less than the estimated 50% of newly diagnosed men who should choose active surveillance (3). Development of biomarkers that improve upon PSA, clinical stage, and Gleason score to distinguish between prostate cancers that can be observed safely and those that require immediate treatment could help “right size” treatment.
THOC1 encodes an essential subunit of the THO ribonucleoprotein complex that is cotranscriptionally assembled on nascent RNA transcripts, facilitating their interaction with RNA processing and export factors (4–6). THO deficiency is known to cause defects in transcript processing and transport (7–12), thus limiting the expression of some genes. RNA transcripts directly regulated by THOC1 are not extensively characterized, but appear to be context dependent (5,11,13,14). Thoc1 deficiency also induces R-loop accumulation, causing DNA damage and genome instability (11).
THOC1 expression is elevated in some human cancers (15–17), and THOC1 gene silencing inhibits the proliferation of some cancer cell lines (15,18,19). Normal fibroblasts are less sensitive to THOC1 deficiency than their oncogene transformed derivatives (19). These observations suggest high THOC1 expression supports cancer progression, but this hypothesis has not been tested. Here, Thoc1 has been conditionally deleted in an autochthonous mouse model of prostate cancer to test whether it is required for cancer progression in vivo. Human prostate cancer specimens have been examined to ascertain whether THOC1 expression associates with cancer aggressiveness and progression.
Methods
Mouse Model of Prostate Cancer
The mouse model was described previously (20). The construction and genotyping of floxed and hypomorphic mouse Thoc1 alleles (Thoc1 F and Thoc1 H, respectively) has been reported (21). Experimental mice were on a mixed C57BL/6:129/Sv:FVB genetic background. Randomly selected mice of the appropriate genotype were killed by CO2 inhalation when moribund or as stated in results. All mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Roswell Park Cancer Institute.
Histology, Immunohistochemical Staining, and Antibodies
Prostate lesions developing in the mouse model are identified and quantitated as described (20,22) (see the Supplementary Methods, available online). The THOC1-specific polyclonal antibody used for human tissue array immunostaining is raised by immunizing rabbits with the previously described GST-N5 fusion protein (23). All other antibodies, including the pThoc1 antibody used to stain mouse tissue, are listed in Supplementary Table 1 (available online).
Molecular Analysis
Prostate tissue from eight- to 12-week-old mice was snap-frozen and RNA extracted with TRIzol reagent (Invitrogen, Carlsbad, CA). Cell line pellets were snap-frozen and RNA extracted with the Qiagen RNeasy kit (Qiagen, Germantown, MD) as recommended. cDNA is synthesized using Superscript III reverse transcriptase and random primers as recommended (Invitrogen). Relative RNA levels are compared by the SYBR green ddCt relative quantitation method. Polymerase chain reaction (PCR) primers are listed in Supplementary Table 2 (available online). RNA immunoprecipitation, western blotting, and gene expression profiling are described in the Supplementary Methods (available online).
Cell Culture and Cell Assays
Prostate cancer cells are isolated from mouse tumors by digesting with 2.5mg/mL Collagenase II, 2.5mg/mL Collagenase IV, 1mg/mL DNase I, in Hank’s balanced salt solution with 2% NuSerum (BD Biosciences, San Jose, CA). Dissociated cells are cultured using Dulbecco’s modification of Eagle’s medium (Invitrogen) containing 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 1% penicillin/streptomycin (10000 IU/mL and 10000 μg/mL, respectively, Mediatech, Manassas, VA). Culture of human prostate cancer cell lines, shRNA, siRNA, BrdU incorporation, protein synthesis, clonogenic growth, transplantation, and apoptosis assays are described in the Supplementary Methods (available online).
Clinical Specimens and Tissue Microarrays
Tissue specimens were collected from patients who underwent RP between 1993 and 2005 at Roswell Park Cancer Institute. For histological analysis, tissue was formalin-fixed, paraffin-embedded, and tissue cores were incorporated into a prostatic tissue microarray (TMA). Clinical characteristics of this patient cohort are summarized in Supplementary Table 3 (available online). Clinical data is collected from each patient with a median follow-up time of 101.3 months. Samples lacking relevant clinical data, cores missing from the TMA, or cores lacking tumor cells were excluded from the analysis. The Roswell Park Cancer Institute (RPCI) Institutional Review Board approved the use of human patient specimens for this study. Patient samples are collected for research at RPCI only from patients that give written informed consent. The samples used in this study were obtained as deidentified.
Statistical Analysis
Univariate statistical tests and Kaplan-Meier analysis were performed and plotted in GraphPad Prism version 6.00. Jonckheere–Terpstra tests were calculated using contingency tables and an approximation to the normal distribution. The Cox regression model was fit using Firth’s penalized likelihood. The model assumption of proportionality was verified graphically using log-cumulative hazard and Anderson plots, while model fit was assessed with a Cox-Snell residual plot. All statistical tests were two-sided, and a P value of less than .05 was considered statistically significant.
Results
THOC1 Expression and Human Prostate Cancer Progression
THOC1 protein (pThoc1) immunostaining is markedly increased in cancerous human prostate epithelium compared with adjacent benign epithelium (Figure 1A). Analysis of TMAs containing more than 700 prostate cancer specimens confirmed this observation. Immunostaining was detectable in most cancer cores, but was difficult to detect in benign tissue. When detected, pThoc1 immunostaining in benign tissue was confined to basal epithelium (Figure 1B). The purified protein used to raise pThoc1 antibody was able to block immunostaining, demonstrating specificity (Supplementary Figure 1A, available online). An independent antibody raised against a distinct pThoc1 epitope gave a similar immunostaining pattern, demonstrating reproducibility (Supplementary Figure 1B, available online). Ten independent patient samples available as frozen tissue were also analyzed for pThoc1 by western blotting. Tumor tissue had more pThoc1 than matched benign tissue for nine samples (Figure 1C).
Figure 1.
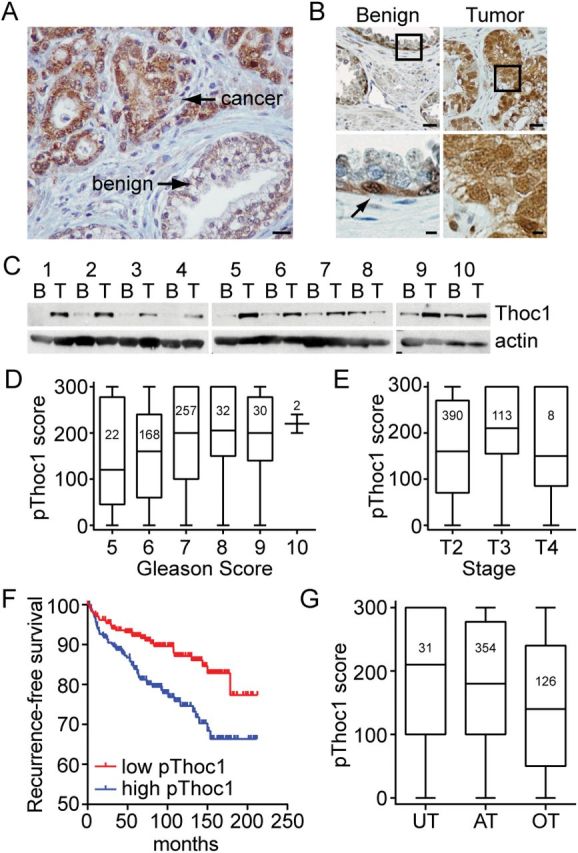
Thoc1 protein expression in human prostate cancer. A) Tissue sections from resected human prostate cancer specimens were immunostained for pThoc1. A representative image showing malignant and adjacent benign epithelium is shown. The scale bar represents 50 microns. B) A human tissue array containing 707 matched tumor and benign prostate tissue cores was immunostained for pThoc1. A representative pair of matched specimens is shown. The boxes represent the area magnified in the lower panels. The arrow represents an immunopositive cell. Scale bars are 50 (upper panel) or 10 microns (lower panel). C) Matched human tumor (T) or benign (B) prostate tissue specimens were analyzed for pThoc1 levels by western blotting. Actin is the protein loading control. D) Patient samples were stratified by Gleason score and pThoc1 immunostaining scores compared. The graph shows a box plot of the data with whiskers defining the range of scores. The numbers inside the boxes indicate sample size. E) Patient samples were stratified by TNM stage and pThoc1 immunostaining scores compared as in (D). F) 511 evaluable prostate cancer patient cores were stratified by pThoc1 immunostaining (above and below median) and biochemical recurrence (time to PSA > 0.4ng/mL) was assessed. Number of patients at risk were: 0 months low = 245, high = 245; 50 months low = 204, high = 182; 100 months low = 117, high = 84; 150 months low = 50, high = 37; 200 months low = 6, high = 7. G) Evaluable patient specimens were stratified as overtreated, appropriately treated, or undertreated and pThoc1 immunostaining analyzed as in (D). AT = appropriately treated; OT = overtreated; UT = undertreated.
To measure variation in pThoc1 levels, immunostaining was quantified using a metric accounting for stain intensity and frequency (% immunopositive multiplied by stain intensity on a 0–3 scale). Samples were scored by two pathologists in blinded fashion, and their scoring was similar (Spearman’s correlation coefficient = 0.89). pThoc1 immunostaining increased with higher pathology Gleason score (24) (Jonckheere–Terpstra trend test z = 3.4, P < .001) (Figure 1D) or with more advanced pathology TNM stage (25) (Jonckheere–Terpstra test z = 4.3, P < .001) (Figure 1E). pThoc1 immunostaining also inversely associated with biochemical recurrence, a commonly used measure of prostate cancer progression. Recurrence-free survival was statistically significantly shorter (log-rank P = .002) (Figure 1F) for cancers with high pThoc1 (>median). The estimated 60 month recurrence-free survival rate was 91.9% (95% confidence interval [CI] = 87.8% to 94.7%) for patients with low pThoc1 and 83.9% (95% CI = 78.3% to 88.1%) for patients with high pThoc1.
A Cox regression model was used to evaluate the association between pThoc1 and biochemical recurrence adjusting for biopsy Gleason score, clinical stage, and pretreatment PSA levels. pThoc1 remains statistically significantly associated with biochemical recurrence (hazard ratio [HR] = 1.61, 95% CI = 1.03 to 2.51, P = .04) (Table 1). We also used a Cox regression model to evaluate whether pThoc1 modified the association between clinical predictors and recurrence. The hazard ratios for the predictors, particularly Gleason score, were altered by pThoc1 status (Supplementary Table 4, available online). These differences did not achieve statistical significance because of the small number of events. pThoc1 immunostaining was also examined after retrospectively stratifying the patient cohort as undertreated (biochemical recurrence within two years of RP), overtreated (RP patients found to have clinically insignificant disease as defined by Gleason score of less than 7, organ-confined, margin-negative, and undetectable PSA for at least two years post-RP), or appropriately treated (all others). pThoc1 immunostaining was statistically significantly different among these classifications (Kruskal-Wallis P = .005) (Figure 1G) with overtreated patients distinguishable from appropriately treated patients (Mann Whitney P = .003). pThoc1 immunostaining, therefore, might improve the identification of prostate cancers likely to progress clinically.
Table 1.
Multivariable analysis of pThoc1 immunostaining and biochemical recurrence*
| Variable | Hazard ratio (95% CI) | P† |
|---|---|---|
| pThoc1 | ||
| High vs low | 1.61 (1.03 to 2.51) | .04 |
| Clinical stage | ||
| T>T3a vs T<T1c | 5.24 (1.90 to 14.46) | .001 |
| T2b,T2c vs <T1c | 1.27 (0.66 to 2.43) | .47 |
| T2a vs <T1c | 1.30 (0.78 to 2.16) | .31 |
| Biopsy Gleason score | ||
| >7 vs <7 | 4.99 (2.65 to 9.40) | <.001 |
| =7 vs <7 | 2.64 (1.62 to 4.32) | <.001 |
| Pretreatment PSA, ng/mL | ||
| >20 vs <10 | 2.98 (1.62 to 5.48) | <.001 |
| 10–20 vs <10 | 1.11 (0.63 to 1.98) | .72 |
* The Cox regression model was fit using Firth’s penalized likelihood. CI = confidence interval; PSA = prostate-specific antigen.
† P value is calculated as a two-sided Type III test.
Requirements for Thoc1 in a Mouse Model of Prostate Cancer
Immunostaining data suggests high pThoc1 levels support prostate cancer progression. To test this, Thoc1 has been conditionally deleted in an autochthonous mouse model of prostate cancer initiated by Rb1 and Trp53 deletion (20), two tumor suppressor genes frequently mutated in human prostate cancer (26–28). Tumors arising in these mice progress from focal atypia to intraepithelial neoplasia (mPIN), invasive tumors, and metastatic disease. This mouse model mimics aggressive human prostate cancers that exhibit neuroendocrine features. Human prostate cancers with neuroendocrine features have been estimated to account for 30% of all prostate cancer lethality (28). pThoc1 immunostaining increases with cancer progression in this model (Figure 2, A and B). Antibody specificity for mouse tissue immunostaining has been verified (Supplementary Figure 1C, available online).
Figure 2.
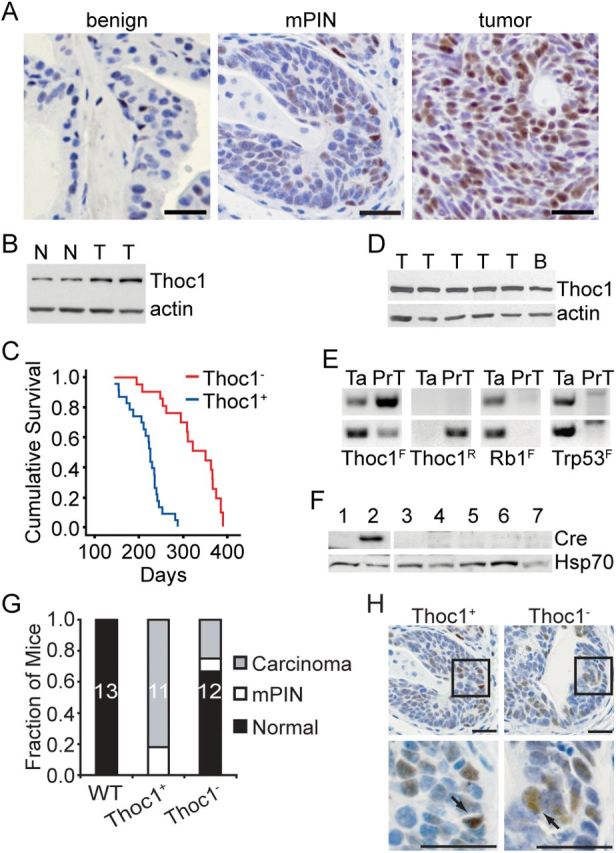
Thoc1 deletion in a mouse model of prostate cancer. A) Tissue sections containing benign epithelium, mPIN lesions, or prostate tumors were immunostained for pThoc1. Scale bars represent 50 microns. B) Thoc1 protein levels were analyzed by western blotting in normal (N) or tumor-bearing prostate tissue (T) in the Rb1/Trp53 mouse model of prostate cancer. Actin is the protein loading control. C) The cumulative survival of mice of the indicated genotypes is shown. The Thoc1 - cohort includes 23 mice while the Thoc1+ cohort includes 22 mice. D) Western blot analysis of pThoc1 in representative prostate tumors (T) from Thoc1 - mice. Normal brain tissue (B) serves as a positive control. Actin is the protein loading control. E) Polymerase chain reaction genotyping of the unrecombined (F) or recombined (R) floxed alleles for the indicated genes is shown. The DNA was extracted from the tail (Ta) or from prostate tumor cells (PrT). The upper and lower panels are samples from two different Thoc1 - mice. The prostate tumor cells retain at least one unrecombined floxed Thoc1 allele. F) Western blot analysis of Cre recombinase levels in normal ventral prostate tissue from one wild-type mouse lacking the PB-Cre4 transgene (lane 1, negative control), uninvolved ventral prostate tissue from one Thoc1 - mouse (lane 2, positive control), prostate tumors from two Thoc1 + mice (lanes 3, 7) or three Thoc1 - mice (lanes 4, 5, 6). Hsp70 is the protein loading control. G) A cohort of 20-week-old mice with the indicated genotypes was sacrificed and prostate histopathology examined by serially sectioning through the entire gland. Each mouse was scored based on the most advanced neoplastic lesion detected. The fraction of mice scored in each category is indicated. The sample size is shown within the bar. H) Prostate tissue sections from mice of the indicated genotypes containing mPIN lesions were immunostained for pThoc1. The boxes highlight the region magnified in the lower panels. Arrows indicate pThoc1 positive cells. The scale bars represent 50 microns.
Tumorigenesis was compared in mice with Rb1 F/F :Trp53 F/F: Thoc1 F/F:PB-Cre4 (Thoc1 -) and Rb1 F/F :Trp53 F/F:Thoc1 +/+:PB-Cre4 (Thoc1 +) genotypes. Prostate-specific deletion of the floxed alleles was verified by PCR analysis of genomic DNA (Supplementary Figure 2, available online). Consistent with previous reports (20), Thoc1 + mice die of prostate cancer with a median survival of 226 days. The median survival for Thoc1 - test mice was statistically significantly longer, at 351 days (log-rank P < .001) (Figure 2C). Thoc1 - mice survived longer, but succumbed to prostate cancer eventually. However, all tumors from Thoc1 - mice that have been examined (n = 29) expressed pThoc1 (Figure 2D). Since Cre-mediated recombination occurred with less than 100% efficiency, tumors in Thoc1 - mice originated from cells escaping Thoc1 deletion (Figure 2E). The low probability of generating prostate epithelial cells lacking all four Rb1 F /Trp53 F alleles to initiate tumorigenesis, but retaining an intact Thoc1 F allele, probably explained the extended survival of Thoc1 - mice. The stability of the unrecombined Thoc1 F alleles was because of silencing of Cre transgene expression during neoplastic transformation (Figure 2F).
Twenty-week-old Thoc1 - and Thoc1 + mice were examined to test whether Thoc1 was required at earlier stages of tumorigenesis. Consistent with prior work (20,29), all Thoc1 + mice at this age contained histologically detectable mPIN lesions and 82% (nine of 11) had early invasive carcinoma (Figure 2G). In contrast, 33% of Thoc1 - mice had detectable neoplasia and only 25% (three of 12) showed evidence of invasive carcinoma. An examination of 30 mPIN lesions from Thoc1 - mice indicated that all retained pThoc1 immunopositive cells (Figure 2H), suggesting Thoc1 was required for the progression of initiated cells to mPIN.
Requirements for Thoc1 in Normal Mouse Prostate Tissue
Thoc1 - mice did not exhibit detectable defects in benign prostate tissue, suggesting Thoc1 was not required for normal prostate homeostasis. Thoc1 F/F:PB-Cre4 mice at 19 to 20 months of age have been examined to test this. Male Thoc1 F/F:PB-Cre4 mice (Thoc1F/F) were viable and did not exhibit phenotypes distinguishable from wild type. Mice had normal prostate histology, genitourinary tract weights, and fertility (Figure 3, A and B), despite extensive Cre expression (Supplementary Figure 3, available online) and reduced Thoc1 RNA and protein (Figure 3, C-E).
Figure 3.
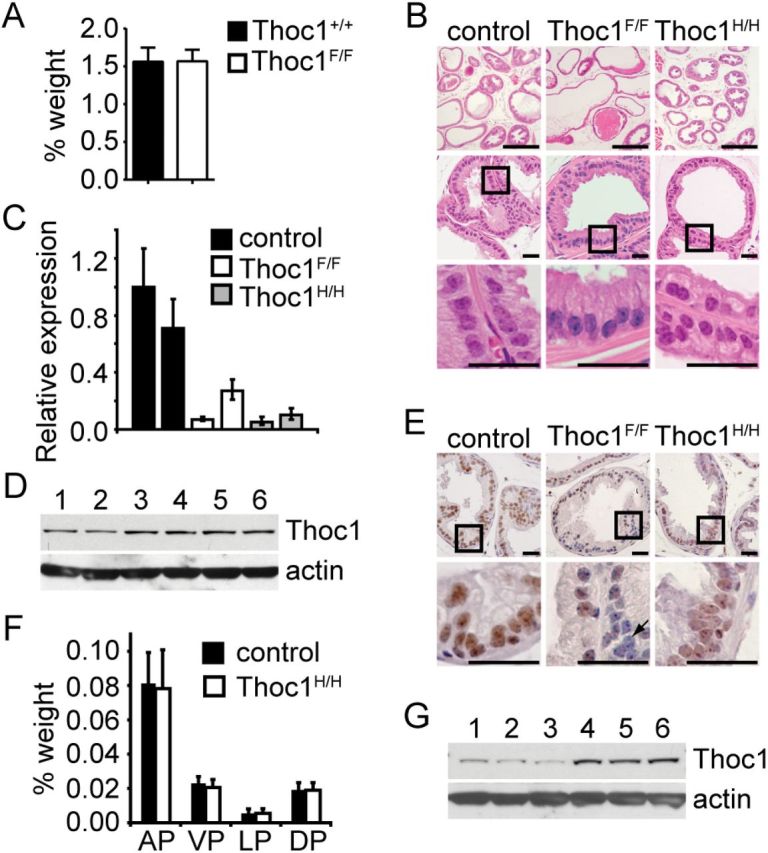
Effects of Thoc1 deficiency on normal prostate tissue. A) The mean genitourinary tract wet weight, relative to total body mass, from five mice for each of the indicated genotypes (Thoc1F/F= Thoc1 F/F:PB-Cre4) was measured. The error bars represent the standard deviation. B) Tissue sections from the lateral prostate of 36-week-old mice of the indicated genotypes were stained with hematoxylin and eosin. The boxes represent the area magnified in the lower panels. Scale bars represent 500 (upper panels) or 50 (middle and lower panels) microns. Similar results were observed for all lobes of the prostate. C) Thoc1 RNA expression was assayed in prostate tissue from mice of the indicated genotypes by quantitative reverse transcription–polymerase chain reaction. The data represent the signal from a primer pair targeting the Cre deleted region of Thoc1 transcript relative to a primer pair targeting a region outside the deleted region. Each bar represents the result from a single mouse analyzed in triplicate with error bars as the standard deviation. The results are normalized to the first control mouse. D) Western blotting was used to analyze pThoc1 levels in Thoc1- (lanes 1–3) or Thoc1+ (lanes 4–6) mice. Each lane represents prostate tissue extract from a single mouse. Actin serves as a protein loading control. E) Prostate tissue sections from mice of the indicated genotypes were immunostained for pThoc1. The boxes represent the area magnified in the lower panels. The arrow highlights a cell with no detectable pThoc1 immunostaining. Scale bars represent 50 microns. F) The wet weight of dissected anterior, ventral, lateral, or dorsal prostate lobes, relative to total body mass, from mice of the indicated genotype is shown. Each bar represents the mean and standard deviation from three to four mice. G) Prostate tissue from Thoc1H/H (lanes 1–3) and control (lanes 4–6) mice were analyzed by western blotting as in D. AP = anterior prostate; DP = dorsal prostate; LP = lateral prostate; VP = ventral prostate.
Lack of a detectable phenotype in Thoc1 F/F:PB-Cre4 mice demonstrates that normal prostate tissue is relatively insensitive to Thoc1 deficiency, but we cannot exclude the possibility that Cre is not expressed in cells that would otherwise be sensitive to pThoc1 loss. To address this, six-month-old mice homozygous for a hypomorphic Thoc1 allele (Thoc1 H), which express reduced pThoc1 in all cells, have been examined (13,21). Prostate histology (Figure 3B), lobe weights (Figure 3F), and ductal branching (Supplementary Figure 4, available online) are normal in Thoc1 H/H mice relative to their smaller size, despite reduced Thoc1 RNA and protein (Figure 3, C,E, and G).
Effects of Thoc1 Loss on Prostate Tumor Cell Growth and Viability
Prostate tumor cells have been isolated from Rb1 F/F :Trp53 F/F:Thoc1 F/F:PB-Cre4 (PrT-) or Rb1 F/F :Trp53 F/F:Thoc1 +/+:PB-Cre4 (PrT+) mice and cultured in vitro. The origin of PrT lines was validated by androgen receptor expression (Supplementary Figure 5A, available online), the presence of recombined Rb1 F and Trp53 F alleles (Figure 2E), and their tumorigenic potential upon transplantation into immunodeficient mice (Supplementary Figure 5B, available online). Thoc1 was deleted in PrT- by forced Cre expression using recombinant adenovirus (AdCre). Infection with AdCre, but not a green fluorescent protein (GFP)-expressing adenovirus (AdGFP), caused near complete depletion of Thoc1 RNA and protein three days postinfection (Fig. 4, A and B). AdCre infection had no effect on Thoc1 RNA or protein in PrT+ cells as expected, because these cells lack floxed Thoc1 alleles.
Figure 4.
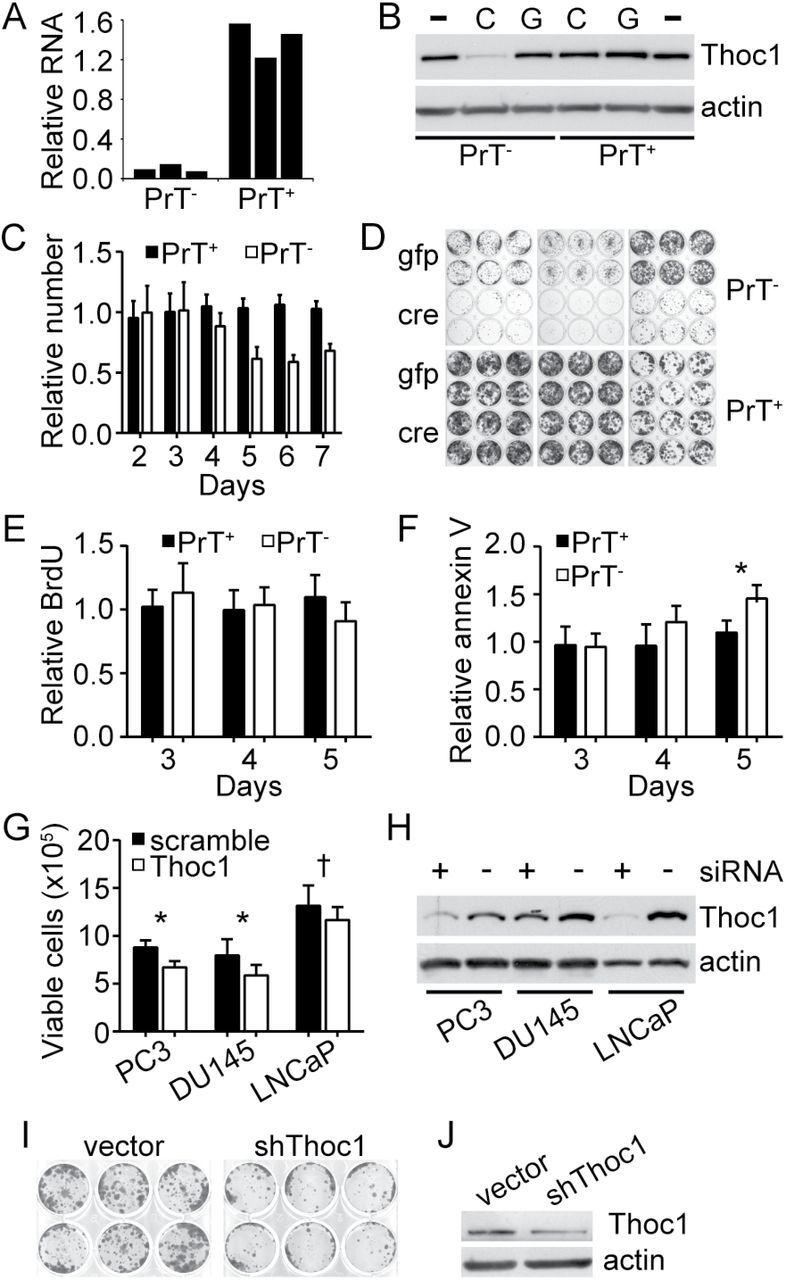
Effects of Thoc1 deficiency on prostate tumor cell viability in vitro. A) PrT+ or PrT- were treated with AdCre or AdGFP and Thoc1 RNA levels measured by quantitative reverse transcription–polymerase chain reaction as in Figure 3. The graph shows RNA levels of AdCre-treated cells relative to AdGFP-treated control cells. Each bar represents data from one biological replicate for a different PrT cell line. B) Thoc1 protein levels were measured by western blotting. PrT+ or PrT- were untreated (-), treated with AdCre (C), or treated with AdGFP (G). Actin is the protein loading control. The results are representative of three independent PrT cell lines for each genotype. C) PrT+ or PrT- were treated with AdCre or AdGFP and cell numbers counted over time. The graph shows the number of AdCre-treated cells relative to the number of AdGFP-treated cells. Each column represents the mean and standard deviation for six independent experiments with two independent PrT cell lines. D) Equal numbers of viable PrT+ or PrT- treated with either AdGFP (gfp) or AdCre (cre) were plated at low density. Cells were fixed and colonies counted nine days later. Each twelve-well plate shows results of an independent PrT cell line. The results are representative of two or more independent experiments. E) PrT+ or PrT- were treated with AdCre or AdGFP and pulse labeled with BrdU at the indicated time subsequent to infection. The graph shows the percentage of BrdU-positive AdCre-treated cells relative to AdGFP-treated cells. Data for each bar is the mean and standard deviation from four to five independent experiments. F) PrT+ or PrT- were treated as in (E) and the percentage of annexin V–positive AdCre-treated cells relative to AdGFP-treated cells is shown. Each bar represents the mean and standard deviation for three to four independent experiments. The asterisk denotes a statistically significant difference (t test P = .02). G) The indicated cell lines were transfected with siRNA targeting THOC1 or a scrambled control. The number of viable cells was counted three days later. The graph shows the mean and standard error for three biological replicates, each performed in triplicate. The P value of the Student’s t test is indicated for each line (*<.005, †<.05). H) The indicated cell lines treated as in (G) with Thoc1 targeted siRNA (+) or scrambled control siRNA (-) were analyzed for pThoc1 by western blotting. Actin is the protein loading control. I) PC-3 cultures were infected with a pool of lentivirus expressing three shRNA targeting different regions of the THOC1 transcript (shThoc1), or with an empty vector control virus and were plated at low density to assess clonogenic growth as in (D). J) PC-3 cells treated as in (I) and pThoc1 levels measured by western blotting. Actin is the protein loading control. All statistical tests were two-sided.
Proliferation of AdCre-infected PrT- cells were similar to AdGFP-infected cells for up to four days postinfection, but then declined (Figure 4C). These differences were statistically significant (analysis of variance P < .001). AdCre-infected PrT- also exhibited reduced clonogenic growth (Figure 4D). Despite these effects, the fraction of PrT- cells incorporating BrdU during a pulse label was similar after AdCre or AdGFP infection (Figure 4E). The cell cycle phase distribution was also similar (Supplementary Figure 6, available online). Thoc1-depleted PrT- did show an increased fraction of cells staining positive for the apoptosis marker annexin V beginning five days post–AdCre infection (Figure 4F). PC-3, DU145, and LNCaP human prostate cancer cell lines were depleted of pThoc1 via siRNA. All three showed statistically significantly reduced accumulation of viable cells compared with control siRNA transfections (Figure 4, G and H). Stable depletion of pThoc1 in PC-3 cells using shRNA reduced their clonogenic potential (Figure 4, I and J). These observations suggested that both human and mouse prostate cancer cells require high levels of pThoc1 to maintain growth and viability.
RNA transcript levels were profiled in six independent PrT- or PrT+ lines treated with AdCre or AdGFP to explore underlying mechanisms (Supplementary Table 5, available online). Most transcripts altered by pThoc1 depletion were highly expressed normally (Supplementary Figure 7, available online), consistent with observations in yeast (30). We did not observe statistically significant differences in the expression of androgen receptor or androgen-responsive genes. Gene set enrichment analysis (31) indicated that 31 of 2445 curated gene sets were statistically significantly enriched for genes whose transcript levels were highly dependent on pThoc1 (false discovery rate q < .05) (Supplementary Table 6, available online). The 31 gene sets shared many overlapping members involved in ribosome function. The KEGG Ribosome gene set, for example, was the most enriched KEGG gene set among genes highly ranked for pThoc1 dependency (Figure 5A). The transcript profiling results were verified for a subset of ribosomal genes by quantitative RT-PCR analysis (Figure 5B). RNA immunoprecipitation analysis indicated that these ribosomal gene transcripts were bound by pThoc1 (Figure 5C), and depletion of pThoc1 reduced the RNA immunoprecipitation signal demonstrating specificity (Figure 5D). This indicates some ribosomal gene transcripts were regulated directly by pThoc1.
Figure 5.

Effects of Thoc1 deficiency on ribosomal gene expression. A) The graph shows the results of gene set enrichment analysis for the KEGG Ribosome gene set. The X axis is the ranked list of genes with those to the left decreasing most upon pThoc1 depletion. The Y axis shows the running enrichment score. The tick marks show the position of gene set members within the ranked list. B) Microarray results were verified by quantitative reverse transcription–polymerase chain reaction (RT-PCR) for the indicated genes. Each data point measures the relative fold change between AdCre- and AdGFP-treated PrT of the indicated genotype from an independent experiment. Cd44 transcript levels are not affected by pThoc1 depletion and were used as the endogenous control. Bars represent the mean and standard deviation. Relative expression is statistically significantly different between PrT+ and PrT- where indicated by asterisks (t test P < .001). C) The graph shows the results of RNA immunoprecipitation analysis of pThoc1. Extracts from PrT+ were immunoprecipitated with Thoc1 or nonimmune (IgG) antibody, and the copurifying RNA analyzed for the presence of the indicated gene transcripts by quantitative RT-PCR. The columns show the mean and standard deviation of signal as % input for five independent experiments. Asterisks indicate statistically significant differences between Thoc1 and IgG signal (t test P < .05). RNA polymerase I 47S preribosomal RNA serves as a negative control. D) The graph shows RNA immunoprecipitation analysis as in (C) for AdCre (Cre)- or AdGFP (GFP)-treated PrT- for three independent experiments. All statistical tests were two-sided.
35S-methionine protein incorporation was measured to test whether changes in ribosomal gene expression affected protein synthesis. 35S-methionine protein incorporation declined by about 25% in AdCre-infected PrT- relative to AdGFP-infected cells, but was unchanged in AdCre- or AdGFP-infected PrT+ (t test P < .001) (Figure 6A). Decreased protein synthesis was expected to induce additional stress like autophagy (32). Autophagy increased in pThoc1-depleted prostate cancer cells, as indicated by the LC3B-II marker (Figure 6B) (33). The rate of conversion to LC3B-II increased, as revealed by a pulse treatment with chloroquine that blocks autophagosome processing. Since PrT lack Trp53- and Rb1-dependent cell cycle checkpoints triggered normally in response to reduced biosynthesis (34–36), pThoc1-depleted PrT were predicted to decline in size as they divided. Consistent with this prediction, the diameter of PrT cells decreased upon Thoc1 loss (t test P = .01) (Figure 6C).
Figure 6.

Thoc1 deficiency compromises protein synthesis and cell growth. A) The effect of pThoc1 depletion on the rate of protein translation was assayed in viable AdCre (cre)- or AdGFP (gfp)-infected PrT of the indicated genotypes by 35S-methionine incorporation. The data show the ratio of 35S incorporated into trichloroacetic acid-precipitated protein vs total cellular 35S during a pulse label. Each data point is from an independent experiment using two different PrT isolates for each genotype. The bars represent the mean and standard deviation. The asterisk indicates a statistically significant difference (t test P < .001). B) Autophagy in PrT was characterized by western blot analysis of LC3B. PrT of the indicated genotypes were infected with AdCre (cre), AdGFP (gfp), or treated with chloroquine (cq) for one hour prior to extraction. Increased conversion of the LC3B-I to LC3B-II is apparent in pThoc1-depleted PrT. The blot is representative of multiple experiments with independent PrT isolates. Actin is the protein loading control. C) PrT of the indicated genotypes were treated with AdCre (cre) or AdGFP (gfp) and the diameter of viable cells measured using a Vi-cell Coulter counter. The histogram shows cumulative data for three to four independent experiments using two to three different PrT isolates for each genotype. All statistical tests were two-sided.
Discussion
The data presented here demonstrate that Thoc1 is required for tumorigenesis in an autochthonous mouse model of aggressive prostate cancer. Strong biological selection for retention of a nonredundant function provided by pThoc1 is apparent, as mouse prostate tumors invariably retain undeleted Thoc1 alleles despite complete Cre-mediated deletion of the four Rb1/Trp53 alleles necessary to initiate tumorigenesis. Thoc1 knock down in human prostate cancer cell lines suggests requirements in humans and mice are similar. In contrast to prostate cancer, Thoc1 deletion does not detectably alter normal mouse prostate size, histology, or homeostasis.
Potential limitations of these experiments include the slow turnover of prostate tissue and that Cre expression, and thus Thoc1 deletion, does not occur in all prostate epithelial cells. Thus, we cannot exclude the possibility that Thoc1 deletion affects some normal prostate epithelial cells over a longer period of time.
The mechanistic basis for this differential effect on normal and neoplastic prostate epithelium is not entirely clear, but the data presented indicates that pThoc1 loss adversely affects the expression of some ribosomal genes and inhibits protein synthesis. Large numbers of ribosomes are required to support rapid cell growth and proliferation that is characteristic of aggressive prostate cancer (37–42). We speculate that increased pThoc1 is necessary in aggressive prostate cancer to support high-level ribosome production. Consistent with this idea, cancer cells are also uniquely affected by other proteins known to regulate protein synthesis like mTOR (43) or RNA polymerase I (44). Similarly, increased demand for pThoc1 could be exploited therapeutically by identifying drugs that inhibit it.
Of more immediate potential clinical relevance, however, is the possibility of using pThoc1 as a biomarker to predict prostate cancer phenotype. As cell turnover is slow in indolent prostate cancer, biosynthetic demand and pThoc1 levels are low. As prostate cancer cells progress toward increasingly aggressive malignancy, pThoc1 levels are predicted to rise to support the increased production of ribosomes necessary for increased cell growth. Our analysis of human prostate cancer patient specimens supports this prediction. pThoc1 immunostaining increases with increasing prostate cancer aggressiveness and is higher in prostate cancers that will eventually progress to recurrent disease. Importantly, pThoc1 levels predict recurrence even after adjusting for other prognostic criteria in current use. Based on these results, pThoc1 may provide independent prognostic information that improves the identification of aggressive prostate cancers. pThoc1 is complementary to a recently identified three-protein panel that identifies indolent prostate cancer (45). Immunohistochemical staining of prostate biopsies using such biomarkers may thus increase the accuracy of assigning patients to active surveillance vs immediate therapy, thereby reducing overtreatment. A limitation of this study is reliance on a single cohort of patients undergoing radical prostatectomy. Analysis of additional and independent cohorts of prostate cancer patients, particularly the analysis of biopsy specimens, will be required to confirm the utility of pThoc1 as a prognostic biomarker.
While the results presented here are germane to prostate cancer, a differential requirement for pThoc1 in normal and neoplastic tissue may extend to other tissues. pThoc1 depletion adversely affects growth and viability of cell lines established from other types of cancer (11,15,18,19), and we have previously shown that fibroblasts are less sensitive to pThoc1 depletion than their oncogene transformed derivative (19). The viability and normal development of hypomorphic Thoc1 mice indicates that normal tissues beyond the prostate are also relatively insensitive to pThoc1 deficiency (13). However, some normal tissues like the small intestine are clearly affected by Thoc1 deletion (46). Thus the effects of pThoc1 deficiency are context dependent. A more systematic analysis of the requirements for pThoc1 in other cancer types will be required to test whether the observations reported here are relevant more generally to cancer or whether they are unique to prostate cancer.
Cancer cells acquire genetic alterations that endow them with unwanted malignant properties, but also handicap them with unique vulnerabilities (47). Thus, cancer cells can become uniquely dependent on nononcogenes (48), genes that support the unique demands of the malignant state but that are not directly involved in initiating neoplastic transformation. Few nononcogenes have been identified to date, but this study demonstrates that Thoc1 qualifies as one. Ribonucleoproteins like pThoc1, therefore, may be a novel source of molecular targets potentially useful for the assessment and treatment of cancer.
Funding
The work was supported by grants from the National Cancer Institute (R01 CA125665 and RO1 CA70292 to DWG), the Department of Defense Prostate Cancer Research Program (W81XWH-08-1-0316 to MC), and the University at Buffalo Mark Diamond fund (F-09-02 to MC). Core facilities utilized in this work were supported by National Cancer Institute grant P30 CA016056.
Supplementary Material
The study sponsor played no role in the design of the study, the collection, analysis, or interpretation of the data, the writing of the manuscript, nor the decision to submit the manuscript for publication.
This manuscript is dedicated to the fond memory of Patricia Lorenzo, PhD, a former member of the Molecular Oncogenesis National Institutes of Health study section and the University of Hawaii Cancer Center, who brightened deliberations with her compassion, affability, and dedication. Her remarkable courage in the face of terminal cancer has been inspirational. We thank Dr. Allen Gao (University of California, Davis) for the gift of protein extracts from matched benign and prostate cancer tissue. We recognize Dr. Kristopher Attwood for performing the Cox regression analysis. We acknowledge Ellen Karasik for expert technical assistance with tissue processing and histology.
References
- 1. Sboner A, Demichelis F, Calza S, et al. Molecular sampling of prostate cancer: a dilemma for predicting disease progression. BMC Med Genomics. 2010;3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cooperberg MR, Broering JM, Carroll PR. Time trends and local variation in primary treatment of localized prostate cancer. J Clin Oncol. 2010;28(7):1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller DC, Gruber SB, Hollenbeck BK, et al. Incidence of initial local therapy among men with lower-risk prostate cancer in the United States. J Natl Cancer Inst. 2006;98(16):1134–1141. [DOI] [PubMed] [Google Scholar]
- 4. Luna R, Rondon AG, Aguilera A. New clues to understand the role of THO and other functionally related factors in mRNP biogenesis. Biochim Biophys Acta. 2012;1819(6):514–520. [DOI] [PubMed] [Google Scholar]
- 5. Rehwinkel J, Herold A, Gari K, et al. Genome-wide analysis of mRNAs regulated by the THO complex in Drosophila melanogaster. Nat Struct Mol Biol. 2004;11(6):558–566. [DOI] [PubMed] [Google Scholar]
- 6. Strasser K, Masuda S, Mason P, et al. TREX is a conserved complex coupling transcription with messenger RNA export. Nature. 2002;417(6886):304–308. [DOI] [PubMed] [Google Scholar]
- 7. Viphakone N, Hautbergue GM, Walsh M, et al. TREX exposes the RNA-binding domain of Nxf1 to enable mRNA export. Nat Commun. 2012;3:1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gwizdek C, Iglesias N, Rodriguez MS, et al. Ubiquitin-associated domain of Mex67 synchronizes recruitment of the mRNA export machinery with transcription. Proc Natl Acad Sci U S A. 2006;103(44):16376–16381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huertas P, Aguilera A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell. 2003;12(3):711–721. [DOI] [PubMed] [Google Scholar]
- 10. Li Y, Wang X, Zhang X, et al. Human hHpr1/p84/Thoc1 Regulates Transcriptional Elongation and Physically Links RNA Polymerase II and RNA Processing Factors. Mol Cell Biol. 2005;25(10):4023–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dominguez-Sanchez MS, Barroso S, Gomez-Gonzalez B, et al. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet. 2011;7(12):e1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schneiter R, Guerra CE, Lampl M, et al. The Saccharomyces cerevisiae hyperrecombination mutant hpr1Delta is synthetically lethal with two conditional alleles of the acetyl coenzyme A carboxylase gene and causes a defect in nuclear export of polyadenylated RNA. Mol Cell Biol. 1999;19(5):3415–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang X, Chinnam M, Wang J, et al. Thoc1 Deficiency Compromises Gene Expression Necessary for Normal Testis Development in the Mouse. Mol Cell Biol. 2009;29(10):2794–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mancini A, Niemann-Seyde SC, Pankow R, et al. THOC5/FMIP, an mRNA export TREX complex protein, is essential for hematopoietic primitive cell survival in vivo. BMC Biol. 2010;8:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo S, Hakimi MA, Baillat D, et al. Linking Transcriptional Elongation and Messenger RNA Export to Metastatic Breast Cancers. Cancer Res. 2005;65(8):3011–3016. [DOI] [PubMed] [Google Scholar]
- 16. Yang J, Li Y, Khoury T, et al. Relationships of hHpr1/p84/Thoc1 expression to clinicopathologic characteristics and prognosis in non-small cell lung cancer. Ann Clin Lab Sci. 2008;38(2):105–112. [PMC free article] [PubMed] [Google Scholar]
- 17. Dominguez-Sanchez MS, Saez C, Japon MA, et al. Differential expression of THOC1 and ALY mRNP biogenesis/export factors in human cancers. BMC Cancer. 2011;11:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li Y, Lin AW, Zhang X, et al. Cancer cells and normal cells differ in their requirements for Thoc1. Cancer Res. 2007;67(14):6657–6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou Z, Flesken-Nikitin A, Corney DC, et al. Synergy of p53 and Rb Deficiency in a Conditional Mouse Model for Metastatic Prostate Cancer. Cancer Res. 2006;66(16):7889–7898. [DOI] [PubMed] [Google Scholar]
- 21. Wang X, Li Y, Zhang X, et al. An allelic series for studying the mouse Thoc1 gene. Genesis. 2007;45(1):32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun H, Wang Y, Chinnam M, et al. E2f binding-deficient Rb1 protein suppresses prostate tumor progression in vivo. Proc Natl Acad Sci U S A. 2011;108(2):704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Durfee T, Mancini MA, Jones D, et al. The amino-terminal region of the retinoblastoma gene product binds a novel nuclear matrix protein that co-localizes to centers for RNA processing. J Cell Biol. 1994;127:609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Epstein JI, Allsbrook WC, Jr, Amin MB, et al. The 2005 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma. Am J Surg Pathol. 2005;29(9):1228–1242. [DOI] [PubMed] [Google Scholar]
- 25. Prostate. In: Edge SB, Byrd DR, Compton CC, et al. (eds). AJCC Cancer Staging Manual. New York, NY: Springer; 2010, 457–468. [Google Scholar]
- 26. Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Logothetis CJ, Gallick GE, Maity SN, et al. Molecular classification of prostate cancer progression: foundation for marker-driven treatment of prostate cancer. Cancer Discov. 2013;3(8):849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Z, Flesken-Nikitin A, Nikitin AY. Prostate Cancer Associated with p53 and Rb Deficiency Arises from the Stem/Progenitor Cell-Enriched Proximal Region of Prostatic Ducts. Cancer Res. 2007;67(12):5683–5690. [DOI] [PubMed] [Google Scholar]
- 30. Gomez-Gonzalez B, Garcia-Rubio M, Bermejo R, et al. Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. Embo. J 2011;30(15):3106–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. [DOI] [PubMed] [Google Scholar]
- 32. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. [DOI] [PubMed] [Google Scholar]
- 33. Kabeya Y, Mizushima N, Yamamoto A, et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117(Pt 13):2805–2812. [DOI] [PubMed] [Google Scholar]
- 34. Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009;16(5):369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Montanaro L, Mazzini G, Barbieri S, et al. Different effects of ribosome biogenesis inhibition on cell proliferation in retinoblastoma protein- and p53-deficient and proficient human osteosarcoma cell lines. Cell Prolif. 2007;40(4):532–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Donati G, Brighenti E, Vici M, et al. Selective inhibition of rRNA transcription downregulates E2F-1: a new p53-independent mechanism linking cell growth to cell proliferation. J Cell Sci. 2011;124(Pt 17):3017–3028. [DOI] [PubMed] [Google Scholar]
- 37. Rudra D, Warner JR. What better measure than ribosome synthesis? Genes Dev. 2004;18(20):2431–2436. [DOI] [PubMed] [Google Scholar]
- 38. Tomlins SA, Mehra R, Rhodes DR, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39(1):41–51. [DOI] [PubMed] [Google Scholar]
- 39. Lin CY, Loven J, Rahl PB, et al. Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell. 2012;151(1):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3(3):179–192. [DOI] [PubMed] [Google Scholar]
- 41. Uemura M, Zheng Q, Koh CM, et al. Overexpression of ribosomal RNA in prostate cancer is common but not linked to rDNA promoter hypomethylation. Oncogene. 2012;31(10):1254–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reschke M, Clohessy JG, Seitzer N, et al. Characterization and analysis of the composition and dynamics of the mammalian riboproteome. Cell Rep. 2013;4(6):1276–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nardella C, Carracedo A, Alimonti A, et al. Differential requirement of mTOR in postmitotic tissues and tumorigenesis. Sci Signal. 2009;2(55):ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bywater MJ, Poortinga G, Sanij E, et al. Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of p53. Cancer Cell. 2012;22(1):51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Irshad S, Bansal M, Castillo-Martin M, et al. A molecular signature predictive of indolent prostate cancer. Sci Transl Med. 2013;5(202):202ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pitzonka L, Wang X, Ullas S, et al. The THO Ribonucleoprotein Complex Is Required for Stem Cell Homeostasis in the Adult Mouse Small Intestine. Mol Cell Biol. 2013;33(17):3505–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5(9):689–698. [DOI] [PubMed] [Google Scholar]
- 48. Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell. 2009;136(5):823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.