

Abstract
Hepatocyte (HPC) apoptosis occurs in association with hepatotoxic responses and chronic liver disease, and is coupled to activation of the blood coagulation cascade. HPCs have been shown to express tissue factor (TF), the primary activator of blood coagulation, in a form that lacks procoagulant activity. In this study, we determined the effect of inducing HPC apoptosis on the procoagulant activity of TF. Treatment of primary mouse HPCs with the Fas death receptor agonist (anti-CD95 antibody, Jo2) triggered apoptosis as shown by cleavage of caspase-3, increased caspase-3 proteolytic activity, and cell surface exposure of phosphatidylserine (PS). Jo2-induced apoptosis significantly increased TF-dependent factor Xa generation by HPCs. Moreover, Jo2 treatment was associated with increased levels of microparticle-associated TF procoagulant activity in the culture medium. Pretreatment with a caspase-3 inhibitor significantly reduced Jo2-induced HPC TF activity and prevented the increase in microparticle-associated TF procoagulant activity. Application of the high-affinity PS-binding protein lactadherin inhibited TF-dependent factor Xa generation by Jo2-treated HPCs and dramatically reduced microparticle-associated TF procoagulant activity. Treatment of wild-type mice with a sublethal dose of Jo2 was associated with a robust increase in the activation of coagulation as measured by plasma thrombin-antithrombin (TAT) levels; whereas mice with liver-specific TF deficiency had significantly lower TAT levels. Overall, the results indicate that Fas-initiated, caspase-3-dependent HPC apoptosis increases TF procoagulant activity through a mechanism involving PS externalization. This suggests that activation of liver TF likely contributes to the procoagulant state associated with HPC apoptosis in liver toxicity and disease.
Keywords: coagulation, apoptosis, tissue factor, hepatotoxicity, microparticles
Acute liver toxicity and chronic liver diseases are associated with activation of the blood coagulation cascade (Lisman et al., 2010). Persistent liver damage modifies the synthesis of coagulation factors by the liver, contributing to an altered hemostatic balance. However, the specific mechanisms whereby liver damage triggers coagulation are not known. Evidence from patients and animal models suggests that tissue factor (TF) contributes to the procoagulant state accompanying liver toxicity and disease (Kassel et al., 2011; Kato et al., 2013; Sullivan et al., 2013; Tacke et al., 2001). Although the liver expresses very low TF levels compared with other organs, liver parenchymal cells (i.e., hepatocytes, HPCs) express TF in an “encrypted” form that lacks procoagulant activity and is unable to convert factor X to active factor Xa (Sullivan et al., 2013). Liver-specific TF deficiency significantly reduced thrombin generation in a mouse model of acetaminophen-induced HPC necrosis (Sullivan et al., 2013). However, whether non-necrotic HPC injury increases HPC TF procoagulant activity and contributes to coagulation is not known.
Apoptosis of HPCs is a conspicuous feature of multiple liver diseases including hepatocellular carcinoma, chronic and acute viral hepatitis, Wilson's disease, alcoholic hepatitis, non-alcoholic steatohepatitis, and drug-induced toxicity (reviewed in Guicciardi and Gores, 2005). Apoptosis in liver disease is initiated by multiple stimuli, including binding of the death receptor ligand FasL/CD95L to its cognate receptor Fas/CD95, which initiates intracellular signaling culminating in the activation of effector caspase-3 (Guicciardi and Gores, 2005; Malhi et al., 2006). Administration of the Fas agonist, hamster monoclonal anti-CD95 antibody (Jo2) (Ogasawara et al., 1993), induces apoptosis in multiple tissues (Kakinuma et al., 1999), particularly in the liver owing to high expression of Fas by HPCs (Müschen et al., 1998; Pinkoski et al., 2000). Notably, HPC Fas expression is increased in multiple disease settings, including liver and gastrointestinal diseases (Pinkoski et al., 2000). A recent study demonstrated that Jo2-induced liver injury/apoptosis in mice was associated with a marked procoagulant response, including intravascular coagulation in the liver (Weerasinghe et al., 2011). Notably, administration of heparin moderately attenuated and delayed liver injury in this model (Weerasinghe et al., 2011). This suggests that Fas-induced liver apoptosis is associated with coagulation cascade activation. However, the role of TF in this procoagulant response has not been investigated.
Previous studies in other cell types have noted a connection between induction of apoptosis and increased procoagulant activity of cell-surface TF (Cheang et al., 2002; Greeno et al., 1996; Wolberg et al., 1999). Indeed, molecular events associated with apoptosis have the potential to directly increase the procoagulant activity of TF. Apoptotic cells undergo a change in asymmetry of the outer cell membrane, including externalization of negatively charged aminophospholipids, such as phosphatidylserine (PS), as a mechanism to signal a requirement for phagocytosis (Brouckaert et al., 2004; Fadok et al., 1998). Notably, interaction of externalized PS with the TF/factor VIIa complex increases cell surface TF-dependent procoagulant activity (Rao et al., 2012; Wolberg et al., 1999). In addition to potential increases in cell-associated TF procoagulant activity, apoptotic cell death can result in the release of microparticles (MPs), heterogeneous spherical structures between 100 and 1000 nm in size comprised of outer cell membrane and its complement of antigens (Mause and Weber, 2010). TF-bearing MPs are highly procoagulant given the unrestricted potential for TF to engage PS and are postulated to contribute to coagulation in patients with chronic liver disease (Rautou et al., 2014; Stravitz et al., 2013).
In this study, we tested the hypothesis that Fas-induced HPC apoptosis increases procoagulant activity of HPC and MP-associated TF-dependent procoagulant activity in vitro. Moreover, utilizing mice with liver-specific TF deficiency, we hypothesized that the procoagulant activity of liver TF would exacerbate injury in an in vivo model of Fas-induced liver injury.
MATERIALS AND METHODS
Mice
Wild-type mice, HPC TF(+) (TFflox/flox) mice (Pawlinski et al., 2007), and mice with liver-specific TF-deficiency HPC TF(−) (TFflox/flox/AlbCre) (Sullivan et al., 2013), all on an equivalent C57Bl/6 background, were used. Mice were housed at an ambient temperature of approximately 22°C with alternating 12/12-h light/dark cycles and provided water and rodent chow ad libitum (Teklad 8940; Harlan, Indianapolis, IN) in Association for Assessment and Accreditation of Laboratory Animal Care International–accredited facilities at Michigan State University. All animal procedures were approved by the Michigan State University Institutional Animal Care and Use Committee.
HPC isolation
HPCs were isolated by collagenase digestion, as previously described (Sullivan et al., 2013). Cell viability was determined by trypan blue exclusion and HPCs for which initial viability after isolation was at least 80% were used. HPCs were plated on 6-well culture plates (Becton Dickinson and Company, Franklin Lakes, NJ) at a density of 500,000 cells/well in Williams E medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Sigma-Aldrich). Viable HPCs were further purified by removal of non-adherent cells 2 h later by washing with PBS. The cells were then cultured at 37°C at 5% CO2 overnight prior to treatment.
Fas-induced apoptosis in vitro
To induce cell death, we utilized an established protocol for Fas-induced apoptosis, where HPCs are pre-treated with a transcriptional inhibitor, actinomycin D (ActD), rendering them more sensitive to Fas-induced apoptosis (Donthamsetty et al., 2011; Ni et al., 1994). HPCs were treated in serum-free conditions with ActD (0.2 μg/ml; Sigma-Aldrich) or its vehicle (DMSO, 0.1%) 30 min prior to treatment with 0.5 μg/ml anti-CD95 antibody (clone Jo2) (BD; Franklin Lakes, NJ) or its vehicle (PBS) for 4 or 8 h. In select experiments, cells were pre-treated with the caspase-3/7 inhibitor Ac-DEVD-CHO (50μM; Merck/EMD Millipore, Billerica, MA) or DMSO (0.1%) vehicle for 30 min prior to Jo2 treatment. To evaluate the contribution of PS in independent experiments, cells were pre-treated with bovine lactadherin (5.5 μg/ml; Haematologic Technologies Inc., Essex Junction, VT), a high-affinity PS-binding glycoprotein, or a PBS vehicle 15 min prior to sample collection after ActD/Jo2 co-treatment.
TF activity assay
Determination of TF-dependent factor Xa generation was performed as described previously (Sullivan et al., 2013). Briefly, the media were aspirated and replaced with 750 μl of pre-warmed (37°C) sterile HBSA (137mM NaCl, 5.38mM KCl, 5.55mM glucose, 10mM HEPES, 0.1% bovine serum albumin), and immediately 250 μl of 600nM factor X ([150nM final]; Enzyme Research Laboratories, South Bend, IN) in HBSA with 20mM CaCl2 [5mM final] was added and allowed to incubate at 37°C for 15 min. The reaction was stopped by the addition of 250 μl of 25mM EDTA (pH 7.4; [5mM final]). To determine if Jo2-treated HPCs released procoagulant MPs, culture medium was collected and spun at 4000 × g for 10 min at 4°C. The supernatant was spun at 20,000 × g for 15 min at 4°C and resulting pellet washed with 1 ml of HBSA and briefly mixed on a vortex mixer. This was spun at 20,000 × g for 15 min at 4°C and the pellet was resuspended in 100 μl of HBSA. Factor Xa generation was performed as above for 2 h at 37°C. In select experiments, either HPCs or MPs were incubated in HBSA with 5.5 μg/ml bovine lactadherin (Haematologic Technologies Inc., Essex Junction, VT) for 15 min prior to factor Xa generation. To quantify factor Xa activity, 125 μl of each sample was added to a 96-well plate in the presence of 25 μl of 4mM Pefachrome FXa 8595 ([0.667mM final] Pentapharm, Norwalk, CT) and the change in absorbance at 405nM was evaluated for 20 min at 37°C using Infinity M200 plate reader (Tecan, Durham, NC). The data for each sample were then compared with a standard curve generated using human factor Xa (Enzyme Research Laboratories) and factor Xa activity was expressed as pM/min.
MP PS equivalent assay and flow cytometry
The levels of PS-positive MPs in pre-cleared (3200 × g for 5 min) cell culture supernatant were measured using the Zymuphen MP-activity kit (Hyphen Biomed, West Chester, OH). To determine if induction of HPC apoptosis increased PS on the cell surface, 5 × 105 HPCs were washed with PBS, collected by centrifugation, and resuspended in 0.5 ml of FACS buffer (PBS, 1% FBS) or annexin V binding buffer (Biotium, Hayward, CA). Two hundred microliter of this cell suspension was stained with annexin V-alexa 488 (0.25 μg/ml) for 15 min at room temperature, washed, and resuspended in either FACS buffer or annexin V binding buffer. For detection of caspase-3 catalytic activity, HPCs were incubated with NucView 488 caspase-3 substrate (Biotium) for 15 min at room temperature, washed and resuspended in either FACS buffer or annexin V binding buffer. The fluorescence was then detected and quantified with a BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). The data were analyzed using CFlow software (BD Accuri, San Jose, CA).
Western blot
Cleaved caspase-3 levels were determined by western blotting, involving SDS-PAGE separation using Criterion XT pre-cast 4–12% Bis-Tris gels and XT MOPS running buffer (Bio-Rad, Hercules, CA) and semi-dry transfer to Immobilon PVDF membrane (Millipore, Billerica, MA). Membranes were blocked for 1 h with 3% BSA in TBST (50mM Tris, 150mM NaCl, 0.1% Tween-20, pH 7.4) and then incubated overnight with rabbit anti-cleaved caspase-3 (Asp175, clone 5A1E, 1:5000, Cell Signaling Technology, Danvers, MA) or mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (clone 6C5, Millipore) (1:10,000) in 1% BSA in TBST at 4°C. Membranes were washed three times in TBST and then incubated with goat anti-rabbit or anti-mouse HRP-conjugated secondary antibodies, as appropriate, diluted 1:10,000 in 1% BSA in TBST for 1 h at room temperature. Membranes were washed three additional times and incubated with Clarity Western enhanced chemiluminescence substrate solution (Bio-Rad) and exposed to blue autoradiography film (ISC BioExpress, Kaysville, UT).
Jo2-induced liver apoptosis in mice
Mice were treated with a sublethal or lethal dose of Jo2 (0.16 mg/kg or 0.4 mg/kg, respectively) (Donthamsetty et al., 2011) or PBS via intraperitoneal injection (ip). Four hours after Jo2 administration, mice were anesthetized using isoflurane, and blood was collected from the caudal vena cava into a syringe containing sodium citrate [0.38% final] or an empty syringe for the collection of plasma and serum, respectively. The liver was excised and washed in PBS and multiple sections from the left lateral lobe were fixed in 10% neutral-buffered formalin for approximately 48 h and then embedded in paraffin. The remaining liver was snap frozen in liquid nitrogen.
Serum alanine aminotransferase and plasma thrombin-antithrombin determination
Serum alanine aminotransferase (ALT) activity was measured using a commercially available reagent (Infinity ALT/GPT, Thermo Fisher, Waltham, MA). Plasma thrombin-antithrombin (TAT) levels were determined using a commercial enzyme-linked immunosorbent assay kit (Enzygnost TAT micro, Siemens Healthcare Diagnostics, Deerfield, IL).
Histopathology and immunohistochemistry
Paraffin-embedded livers were sectioned at 5 μm and stained with hematoxylin and eosin (H&E) by the Michigan State University Investigative HistoPathology Laboratory, a division of Human Pathology. At least 2 to 3 sections of liver from the left lateral lobe from each animal were evaluated by light microscopy to qualitatively determine the severity of hepatic injury. Fibrin(ogen) immunohistochemistry was performed on de-paraffinized formalin-fixed sections after antigen retrieval with proteinase K, using a rabbit anti-human fibrinogen antibody (1:200) (Dako North America, Carpinteria, CA). Caspase-3 immunohistochemistry was performed on de-paraffinized formalin-fixed sections after antigen retrieval with boiling citrate buffer, using a rabbit anti-cleaved caspase-3 antibody (1:800) (Asp175, clone 5A1E, 1:5000, Cell Signaling Technology). The primary antibodies were detected utilizing a biotinylated goat anti-rabbit antibody [1:600 for fibrin(ogen) and 1:2000 for caspase-3] (Jackson ImmunoResearch Laboratories, West Grove, PA) and Vectastain Elite ABC kit and ImmPACT DAB substrate (Vector Laboratories, Burlingame, CA). H&E, fibrin(ogen), and caspase-3 stained sections were evaluated qualitatively.
RNA Isolation, cDNA synthesis, and Real-Time PCR
Total RNA was isolated from approximately 50 mg of snap-frozen liver using TRI Reagent (Molecular Research Center, Cincinnati, OH). One microgram of total RNA was utilized for the synthesis of cDNA, accomplished using a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) and a C1000 Thermal Cycler (Bio-Rad). Hepatic levels of mRNAs encoding TF were determined using SYBR Green PCR, iTaq (Bio-Rad), and a CFX Connect thermal cycler (Bio-Rad). Primers were purchased from IDT (Coralville, IA). The expression of TF was normalized to the geometric Ct mean of two housekeeper genes (Vandesompele et al., 2002), hypoxanthine guanine phosphoribosyl transferase (HPRT) and GAPDH and the relative levels of each gene were evaluated using the ΔΔCt method. Mouse TF primer sequences were 5’-ACAATTTTGGAGTGGCAACC-3’ (forward), 5’-TCACGATCTCGTCTGTGAGG-3’ (reverse). Mouse HPRT primer sequences were 5’-AAGCCTAAGATGAGCGCAAG-3’ (forward), 5’-TTACTAGGCAGATGGCCACA-3’ (reverse). Mouse GAPDH primer sequences were 5’-GTGGACCTCATGGCCTACAT-3’ (forward), 5’-TGTGAGGGAGATGCTCAGTG-3’ (reverse).
Statistics
Comparison of two groups was performed using Student's t-test. Comparison of three or more groups was performed using one- or two-way analysis of variance, as appropriate, and the Student-Newman-Keuls post hoc test. The criterion for statistical significance was p < 0.05.
RESULTS
Fas-Induced HPC Apoptosis Increases Cell-Associated TF-Dependent Procoagulant Activity
Jo2 treatment of HPCs increased cleaved caspase-3 protein levels at 4 and 8 h, indicating induction of apoptosis (Fig. 1A). Compared with vehicle-treated HPCs, Jo2 treatment increased factor Xa generation by HPCs (Fig. 1B) in a time-dependent manner, increasing ∼3-fold at 8 h. To assess the contribution of TF to factor Xa generation by apoptotic HPCs, we utilized HPCs isolated from HPC TF(−) mice, in which TF expression is reduced by >95% (Sullivan et al., 2013). Importantly, a lower level of HPC-driven factor Xa generation was observed in Jo2-treated HPCs isolated from HPC TF(−) mice (Fig. 1C). Levels of cleaved caspase-3 were similar 8 h after Jo2 treatment in HPCs isolated from control HPC TF(+) mice and HPC TF(−) mice, indicating that TF deficiency did not affect Jo2-mediated induction of apoptosis at this time (Fig. 1D). Overall, the results indicate that Fas-induced apoptosis increases TF-dependent factor Xa generation by HPCs.
FIG. 1.

Effect of Fas-induced HPC apoptosis on TF procoagulant activity. Primary HPCs from HPC TF(+) (TFflox/flox) mice or HPC TF(−) (TFflox/flox/AlbCre) mice were treated with ActD (0.2 μg/ml) or its vehicle (DMSO, 0.1%), then 30 min later with Jo2 (0.5 μg/ml) or its vehicle (PBS) for 4 or 8 h. (A and D) Western blot showing cleaved caspase-3 and GAPDH levels. (B and C) TF procoagulant activity was assessed in HPCs by measuring factor Xa generation (see Materials and Methods). Data are represented as mean + SEM for six independent experiments. *p < 0.05 versus time-matched vehicle-treated HPCs.
Fas-Induced HPC Apoptosis Increases Levels of TF-Positive Procoagulant MPs
Next, we determined whether Jo2-induced HPC apoptosis was associated with the release of procoagulant MPs. Compared with vehicle-treated HPCs, Jo2 treatment increased MP-driven factor Xa generation in a time-dependent manner, increasing ∼9-fold at 8 h (Fig. 2A). Jo2-induced apoptosis triggered similar increases in total MPs in the culture medium of HPCs from control HPC TF(+) mice and HPC TF(−) mice (Fig. 2B). In contrast, Jo2 treatment of TF(+) HPCs significantly increased MP-driven factor Xa generation, whereas MP-driven factor Xa generation was abolished in vehicle- and Jo2-treated HPCs from HPC TF(−) mice (Fig. 2C). The results indicate that Jo2-induced apoptosis triggers the release of TF(+) procoagulant MPs.
FIG. 2.

Effect of Fas-induced HPC apoptosis on MP release and TF procoagulant activity. Primary HPCs from HPC TF(+) (TFflox/flox) mice or HPC TF(−) (TFflox/flox/AlbCre) mice were treated with ActD (0.2 μg/ml) or its vehicle (DMSO, 0.1%), then 30 min later with Jo2 (0.5 μg/ml) or its vehicle (PBS) for 4 or 8 h. (A and C) Factor Xa generation by MPs released into the cell culture medium was determined (see Materials and Methods). (B) MP levels in the cell culture medium were quantified and data expressed as PS equivalents. Data are represented as mean + SEM for at least three independent experiments. *p < 0.05 versus time-matched vehicle-treated HPCs.
Fas-Induced Increase in HPC and MP TF Procoagulant Activity is Caspase-Dependent
To determine if caspase-3 activity contributed to Jo2-induced increases in HPC and MP TF procoagulant activity, HPCs were pretreated with the competitive caspase 3/7 inhibitor Ac-DEVD-CHO. Pretreatment with Ac-DEVD-CHO abolished caspase-3 activity in Jo2-treated HPCs, as indicated by flow cytometric detection of caspase-3 activity using a caspase-3 sensitive substrate which when cleaved releases a high-affinity fluorescent DNA dye that stains the nuclei of apoptotic cells (Fig. 3A). Compared with vehicle-treated cells, Ac-DEVD-CHO treatment significantly reduced Jo2-induced factor Xa generation by HPCs (Fig. 3B) and abolished Jo2-induced increase in MP TF procoagulant activity (Fig. 3C). The results suggest that Jo2-induced increases in HPC and MP TF activity are caspase-3 dependent.
FIG. 3.

Caspase-3 inhibition effects on Fas-induced increases in HPC and MP TF procoagulant activity. Primary HPCs from wild-type mice were treated with ActD (0.2 μg/ml) or its vehicle (DMSO, 0.1%), then 30 min later with Jo2 (0.5 μg/ml) or its vehicle (PBS) for 8 h. Thirty minutes prior to Jo2 treatment HPCs were pre-treated with caspase-3 inhibitor, Ac-DEVD-CHO (50μM) or DMSO (0.1%) vehicle. (A) Caspase-3 activity was measured using a NucView 488 substrate, detected using flow cytometry and expressed as% positive cells of the live cell population. (B–C) TF procoagulant activity was assessed in HPCs and MPs by measuring factor Xa generation (see Materials and Methods). Data are represented as mean + SEM for three independent experiments. *p < 0.05 versus vehicle-treated HPCs. #p < 0.05 versus Jo2-treated HPCs pretreated with vehicle (DMSO).
Fas-Induced Increases in HPC and MP TF Procoagulant Activity Involve PS Externalization
Externalization of PS to the outer leaflet of the cell membrane is a hallmark of apoptotic cell death (Fadok et al., 1998; Vermes et al., 1995) and also supports an increase in the procoagulant activity of the TF/factor VIIa complex (Bach, 2006). In agreement with previous studies (Sangwan et al., 2006; Sukocheva and Carpenter, 2006), compared with vehicle-treated HPCs, Jo2 treatment significantly increased annexin V staining of HPCs, indicating an increase in PS externalization associated with Fas-induced HPC apoptosis (Fig. 4A). Increases in annexin V staining were not affected by TF deficiency (Fig. 4A). Interestingly, addition of the high-affinity PS-binding protein lactadherin 15 min prior to assessment of factor Xa generation significantly reduced factor Xa generation by Jo2-treated HPCs (Fig. 4B). Moreover, lactadherin greatly reduced factor Xa generation by MPs from Jo2-treated HPCs (Fig. 4C). The results suggest that increased PS on the outer membrane of HPCs and on MPs contributes to increases in TF-dependent procoagulant activity during apoptosis.
FIG. 4.

Fas-induced increases in HPC and MP TF procoagulant activity involve PS. Primary HPCs from HPC TF(+) (TFflox/flox) mice or HPC TF(−) (TFflox/flox/AlbCre) mice were treated with ActD (0.2 μg/ml) or its vehicle (DMSO, 0.1%), then 30 min later with Jo2 (0.5 μg/ml) or its vehicle (PBS) for 8 h. (A) Annexin V staining of HPCs was measured using flow cytometry. (B–C) 15 min prior to factor Xa generation, cells were pre-treated with high-affinity PS-binding protein, lactadherin (5.5 μg/ml) or vehicle (PBS). TF procoagulant activity of HPCs and HPC-derived MPs was assessed by measuring factor Xa generation (see Materials and Methods). Data are represented as mean + SEM for five independent experiments. *p < 0.05 versus genotype-matched vehicle-treated HPCs (panel A).
Fas-Induced Coagulation In Vivo Requires Liver TF
Owing to a high expression of Fas, the liver is exquisitely sensitive to Jo2-induced apoptosis in vivo (Kakinuma et al., 1999; Müschen et al., 1998; Pinkoski et al., 2000). Previous studies have shown that Jo2-induced liver apoptosis in mice is associated with a marked procoagulant response, and that heparin confers protection against Jo2-induced liver injury (Weerasinghe et al., 2011). Because our in vitro studies indicated that Jo2-induced apoptosis increased TF procoagulant activity, we determined the role of liver TF in Jo2-induced coagulation in vivo. We selected two doses of Jo2 for these studies; a sublethal dose (0.16 mg/kg), which produces modest liver damage in mice, and a lethal dose (0.4 mg/kg), which is lethal within 7–12 h after injection (Donthamsetty et al., 2011; Feng and Kaplowitz, 2000).
In wild-type HPC TF(+) mice, Jo2 treatment (0.16 mg/kg, ip) significantly increased plasma TAT levels compared with vehicle (PBS)-treated mice, whereas there was no increase in Jo2-treated HPC TF(−) mice (Fig. 5A). A corresponding increase in hepatic fibrin deposition was observed in Jo2-treated HPC TF(+) mice localizing predominantly in proximity of HPCs displaying apoptotic features (e.g., nuclear condensation, cell shrinkage) and in the hepatic sinusoids (Fig. 5B). Significantly less hepatic fibrin deposition was observed in Jo2-treated HPC TF(−) mice (Fig. 5B), consistent with the significant decrease in plasma TAT levels. HPC TF(−) mice had significantly lower levels of hepatic TF mRNA compared with HPC TF(+) mice, irrespective of Jo2 administration (Fig. 5C). Interestingly, Jo2 treatment significantly reduced hepatic TF mRNA levels in HPC TF(+) mice but not in HPC TF(−) mice (Fig. 5C).
FIG. 5.
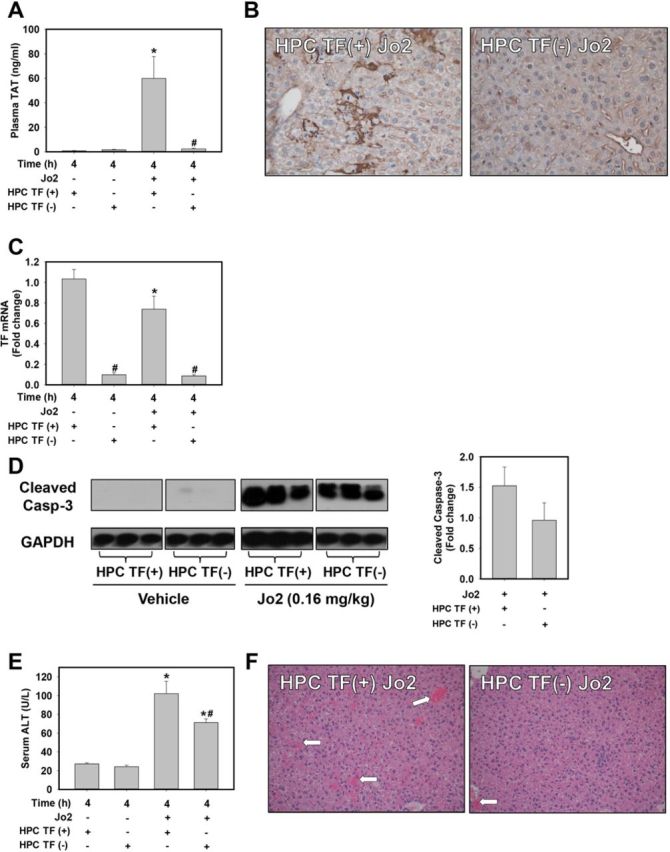
Fas-induced coagulation in vivo requires liver TF. HPC TF(+) (TFflox/flox) mice or HPC TF(−) (TFflox/flox/AlbCre) mice were treated with Jo2 (0.16 mg/kg, ip) or vehicle (PBS) and samples were collected 4 h later. (A) Plasma TAT levels. (B) Representative photomicrographs showing immunohistochemical staining for fibrin (brown) (200× magnification). (C) Hepatic TF mRNA expression. (D) Representative western blot showing cleaved caspase-3 and GAPDH levels and associated densitometry. (E) Serum ALT activity. (F) Representative photomicrographs showing H&E–stained liver sections (100× magnification). Hemorrhage is indicated by white arrows. Data are represented as mean + SEM. n = 4–5 for PBS-treated mice per genotype and n = 5–9 for Jo2-treated mice per genotype. *p < 0.05 versus same genotype treated with vehicle. #p < 0.05 versus control mice given the same treatment.
To determine whether liver TF deficiency affected Jo2-induced apoptosis or associated liver pathologies (e.g., hemorrhage, secondary nec rosis), we evaluated hepatic levels of cleaved caspase-3, serum ALT activity, and liver histopathology. Cleaved caspase-3 levels, indicative of apoptosis, increased in Jo2-treated HPC TF(+) and HPC TF(−) mice (Fig. 5D). Jo2 treatment significantly increased serum ALT activity in HPC TF(+) mice, suggestive of minimal secondary necrosis (Fig. 5E). This response was significantly attenuated in Jo2-treated HPC TF(−) mice (Fig. 5E). Modest hemorrhage was evident in livers of Jo2-treated HPC TF(+) mice, and this was largely absent in livers of Jo2-treated HPC TF(−) mice (Fig. 5F).
Robust hepatic fibrin deposition was evident in HPC TF(+) mice treated with the lethal Jo2 dose (0.4 mg/kg), whereas fibrin was largely absent in livers of HPC TF(−) mice (Supplementary fig. 1A). We could not assess plasma TAT levels in mice treated with 0.4 mg/kg Jo2 at this time point due to challenges with blood collection owing to hypovolemia. Immunohistochemical staining showed similar levels of cleaved caspase-3 in Jo2 (0.4 mg/kg)-treated HPC TF(+) and HPC TF(−) mice (Supplementary fig. 1B). Similarly, gross liver histopathology, marked by severe hemorrhage and congestion, was unaffected by liver TF deficiency at this higher Jo2 dose (Supplementary fig. 1C). Of interest, although not statistically significant, serum ALT activity was modestly decreased in HPC TF(−) mice compared with HPC TF(+) mice treated with 0.4 mg/kg Jo2 (Supplementary fig. 1D).
DISCUSSION
The mechanisms of coagulation cascade activation associated with liver toxicity and disease are not completely understood. Several studies indicate that TF triggers procoagulant responses observed in models of liver injury and disease (Hammad et al., 2013; Kassel et al., 2011; Kato et al., 2013; Luyendyk et al., 2010, 2011; Sullivan et al., 2013). However, the cellular sources of procoagulant TF in mouse models and in patients with liver disease have not been fully defined. The liver itself is now emerging as a possible source, as human and mouse HPCs have been shown to express TF (Stéphenne et al., 2007; Sullivan et al., 2013). However, the role of liver TF and its regulation in liver injury and disease is not fully understood. In mice, the majority of HPC TF is held in a state that lacks procoagulant activity (Sullivan et al., 2013), likely to prevent inappropriate coagulation in the “leaky” sinusoidal vasculature that permits exposure of HPCs to plasma. Defining the mechanisms controlling TF procoagulant activity in the liver could reveal the basis for pathologic coagulation accompanying liver toxicity and disease. Our results here suggest that HPC apoptosis is one cellular process capable of increasing TF procoagulant activity, and that liver TF activates the procoagulant response trigged by Fas-induced apoptosis in vivo.
Stimulation of the Fas receptor induces formation of the death-inducing signaling complex, a complex of several proteins, which ultimately yields cleaved (active) caspase-8. Caspase-8 can directly activate downstream effector caspases, including caspase-3, and can also provoke mitochondrial apoptotic pathways, including the release of cytochrome c (Guicciardi and Gores, 2005; Malhi et al., 2006; Zheng et al., 1998). Fas-directed apoptosis in HPCs is largely considered to be caspase-3-dependent and is associated with other hallmarks of apoptosis, including PS externalization. We demonstrate here that Fas activation of HPCs in vitro is associated with caspase-3 activation, PS externalization, and increased TF-dependent factor Xa generation (Figs. 1, 3, and 4). In this in vitro model, the transcriptional inhibitor ActD prevents up-regulation of anti-apoptotic factors and sensitizes HPCs to Jo2-induced apoptosis (Ni et al., 1994). Notably, this also precludes the possibility that increased TF procoagulant activity is driven by increased TF mRNA expression. TF deficiency did not directly affect caspase-3 activation (Fig. 1D) or cell surface PS levels (Fig. 4A), indicating that the lower level of factor Xa generation by cells from HPC TF(−) mice (Fig. 1C) was a function of TF deficiency. Inhibition of caspase-3 proteolytic activity attenuated Jo2 induction of TF procoagulant activity in HPCs (Figs. 3A and 3B). Consistent with an essential role for anionic phospholipids in TF procoagulant activity (Butenas and Krudysz-Amblo, 2012; Rao et al., 2012), cell surface PS expression increased in Jo2-treated HPCs (Fig. 4A). Utilizing a previously published approach to impair PS interaction with the TF/factor VIIa complex (Shi and Gilbert, 2003), we found that the high-affinity PS-binding protein lactadherin inhibited TF-dependent factor Xa generation by Jo2-treated HPCs (Fig. 4B). Moreover, HPCs have been shown to express protein disulfide isomerase (PDI) on the cell surface (Terada et al., 1995). PDI has been shown to control TF procoagulant activity through Cys186-Cys208 disulfide bond isomerization on the TF molecule (Langer and Ruf, 2014; Rao et al., 2012; Versteeg and Ruf, 2007, 2011). Furthermore, other studies have suggested that PDI could control TF procoagulant activity through modulation of PS externalization (Popescu et al., 2010). Thus, it may be possible that PDI also plays a role in regulation of HPC TF activity by apoptosis. Collectively, our results suggest that caspase- and PS-dependent mechanisms contribute, at least in part, to the increase in TF-dependent factor Xa generation by apoptotic HPCs.
MPs are increasingly recognized as contributors to acute hepatic injury and chronic liver disease (reviewed in Lemoinne et al., 2014). MPs have the potential to contribute to the hepatic and systemic procoagulant response accompanying liver injury and disease. Indeed, procoagulant MPs are increased in patients with acute, drug-induced liver failure and in patients with liver cirrhosis (Lemoinne et al., 2014; Rautou et al., 2014; Stravitz et al., 2013). For the most part, the cellular source of procoagulant MPs in liver disease has not yet been identified and is most certainly context dependent. Complementing the increase in cell plasma membrane-associated TF procoagulant activity, we found that Fas-induced HPC apoptosis was associated with increased release of procoagulant MPs into the culture medium (Fig. 2A). Although MP release from Jo2-treated HPCs was TF-independent (Fig. 2B), the procoagulant activity of MPs released from TF-deficient HPCs was barely detectable (Fig. 2C), indicating that apoptotic HPCs release TF-positive MPs in vitro. The release of procoagulant MPs from HPCs was found to be caspase-3-dependent (Fig. 3C), which is consistent with previous studies identifying MP release as a caspase-driven process in apoptotic cells (Mause and Weber, 2010). Moreover, HPC-derived MP procoagulant activity was abolished by lactadherin (Fig. 4C), consistent with PS contributing to MP procoagulant activity (Freyssinet and Toti, 2010). Overall, our studies provide evidence supporting TF-positive MPs as a potential source of procoagulant activity in liver disease accompanied by HPC apoptosis.
A recent study demonstrated that hepatic injury triggered by Jo2 administration to mice was accompanied by intrahepatic coagulation (Weerasinghe et al., 2011). Building on this observation, we found evidence of robust coagulation in mice treated with a sublethal dose of Jo2, as indicated by increased plasma TAT levels and hepatic fibrin deposition (Figs. 5A and 5B). We demonstrate here that liver TF is an essential trigger of the intrahepatic procoagulant response accompanying Fas-induced liver apoptosis induced by both sublethal and lethal doses of Jo2 (Figs. 5A and 5B and Supplementary fig. 1A). Complementing our in vitro studies, the mechanism of TF-dependent coagulation likely involves decryption (activation of procoagulant activity) of hepatic TF/factor VIIa, as hepatic TF mRNA expression was not increased in Jo2-treated mice (Fig. 5C). These studies indicate that liver TF plays an essential role in the procoagulant response triggered by Fas-induced apoptosis in mice. Although multiple studies have examined the role of global TF deficiency in models of chronic liver disease (Kopec and Luyendyk, 2014), to date the role of HPC TF procoagulant activity in chronic liver injury models has not been investigated. Nevertheless, results from this study extend our previous finding showing that coagulation cascade activation in mice given a hepatotoxic dose of acetaminophen was reduced in HPC TF(−) mice (Sullivan et al., 2013). Of importance, in contrast to Jo2-induced apoptosis, HPC necrosis is the predominant lesion produced by acetaminophen (Gujral et al., 2002). Together, these studies demonstrate that liver TF contributes to coagulation triggered by both apoptotic and necrotic cell death pathways.
A number of studies have found that anticoagulant administration inhibits hepatotoxic responses in rodent models (Copple et al., 2002; Fujiwara et al., 1988; Ganey et al., 2007; Pearson et al., 1996). Indeed, heparin administration significantly attenuated Jo2-induced liver injury in mice (Weerasinghe et al., 2011). Complementing this observation, we found that mice with genetically-induced liver TF deficiency had significantly reduced serum ALT activity (Fig. 5E) and a reduction in liver pathology (e.g., hemorrhage) after treatment with Jo2 (Fig. 5F). Heparin pretreatment was previously shown to significantly reduce caspase-3 activation in Jo2-treated mice (Weerasinghe et al., 2011), although in our study caspase-3 levels were not significantly lower in Jo2-treated HPC TF(−) mice (Fig. 5D). Notably, heparin treatment in mice given a lethal dose of Jo2 delayed liver pathology and lethality, but did not completely prevent toxicity (Weerasinghe et al., 2011). This is consistent with our observation that liver TF deficiency caused a diminution in liver pathology at a sublethal (Fig. 5F), but not a higher, lethal dose of Jo2 (Supplementary fig. 1C). Moreover, despite the lack of effect of TF deficiency on liver histopathology at the lethal dose of Jo2, there was a notable decrease in hepatic fibrin deposition in Jo2-treated HPC TF(−) mice (Supplementary fig. 1A). Overall, utilizing a genetic approach we define liver TF as an essential trigger of intrahepatic coagulation accompanying Fas-induced apoptosis. Moreover, our results are consistent with the concept that intrahepatic coagulation amplifies liver injury associated with HPC apoptosis.
In summary, our studies reveal Fas-induced apoptosis as a mechanism capable of increasing HPC TF procoagulant activity through caspase- and PS-dependent mechanisms. Moreover, we demonstrate that liver TF activity is essential for the procoagulant response accompanying Jo2-induced apoptosis in mice. Collectively, the results suggest that HPC apoptosis and associated decryption (activation) of TF/factor VIIa procoagulant activity may be a key feature of procoagulant responses in liver toxicity and disease.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes of Health (NIH) National Institute of Environmental Health Sciences (NIEHS) [R01 ES017537] and [R25 HL103156–03]; National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) [R01 DK087886].
Supplementary Material
Disclaimer:The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIEHS, NIDDK, or NIH.
REFERENCES
- Bach R. R. Tissue factor encryption. Arterioscler. Thromb. Vasc. Biol. 2006;26:456–461. doi: 10.1161/01.ATV.0000202656.53964.04. [DOI] [PubMed] [Google Scholar]
- Brouckaert G., Kalai M., Krysko D. V., Saelens X., Vercammen D., Ndlovu M. N., Haegeman G., D'Herde K., Vandenabeele P. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol. Biol. Cell. 2004;15:1089–1100. doi: 10.1091/mbc.E03-09-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butenas S., Krudysz-Amblo J. Decryption of tissue factor. Thromb. Res. 2012;129(Suppl. 2):S18–S20. doi: 10.1016/j.thromres.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheang K., Munger M., Terry C., Natisuwan S., Zebrack J., Gilbert E., Callahan K. Enhanced monocyte tissue factor procoagulant activity in heart failure is associated with apoptosis. J. Am. Coll. Cardiol. 2002;39:128A–128A. [Google Scholar]
- Copple B. L., Woolley B., Banes A., Ganey P. E., Roth R. A. Anticoagulants prevent monocrotaline-induced hepatic parenchymal cell injury but not endothelial cell injury in the rat. Toxicol. Appl. Pharmacol. 2002;180:186–196. doi: 10.1006/taap.2002.9394. [DOI] [PubMed] [Google Scholar]
- Donthamsetty S., Mars W. M., Orr A., Wu C., Michalopoulos G. K. Protection against Fas-induced fulminant hepatic failure in liver specific integrin linked kinase knockout mice. Comp. Hepatol. 2011;10:11. doi: 10.1186/1476-5926-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok V. A., Bratton D. L., Frasch S. C., Warner M. L., Henson P. M. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 1998;5:551–562. doi: 10.1038/sj.cdd.4400404. [DOI] [PubMed] [Google Scholar]
- Feng G., Kaplowitz N. Colchicine protects mice from the lethal effect of an agonistic anti-Fas antibody. J. Clin. Invest. 2000;105:329–339. doi: 10.1172/JCI7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyssinet J. M., Toti F. Formation of procoagulant microparticles and properties. Thromb. Res. 2010;125(Suppl. 1):S46–S48. doi: 10.1016/j.thromres.2010.01.036. [DOI] [PubMed] [Google Scholar]
- Fujiwara K., Ogata I., Ohta Y., Hirata K., Oka Y., Yamada S., Sato Y., Masaki N., Oka H. Intravascular coagulation in acute liver failure in rats and its treatment with antithrombin III. Gut. 1988;29:1103–1108. doi: 10.1136/gut.29.8.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganey P. E., Luyendyk J. P., Newport S. W., Eagle T. M., Maddox J. F., Mackman N., Roth R. A. Role of the coagulation system in acetaminophen-induced hepatotoxicity in mice. Hepatology. 2007;46:1177–1186. doi: 10.1002/hep.21779. [DOI] [PubMed] [Google Scholar]
- Greeno E. W., Bach R. R., Moldow C. F. Apoptosis is associated with increased cell surface tissue factor procoagulant activity. Lab. Invest. 1996;75:281–289. [PubMed] [Google Scholar]
- Guicciardi M. E., Gores G. J. Apoptosis: A mechanism of acute and chronic liver injury. Gut. 2005;54:1024–1033. doi: 10.1136/gut.2004.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujral J. S., Knight T. R., Farhood A., Bajt M. L., Jaeschke H. Mode of cell death after acetaminophen overdose in mice: Apoptosis or oncotic necrosis. Toxicol. Sci. 2002;67:322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- Hammad M. A., Abdel-Bakky M. S., Walker L. A., Ashfaq M. K. Tissue factor antisense deoxyoligonucleotide prevents monocrotaline/LPS hepatotoxicity in mice. J. Appl. Toxicol. 2013;33:774–783. doi: 10.1002/jat.2728. [DOI] [PubMed] [Google Scholar]
- Kakinuma C., Takagaki K., Yatomi T., Nakamura N., Nagata S., Uemura A., Shibutani Y. Acute toxicity of an anti-Fas antibody in mice. Toxicol. Pathol. 1999;27:412–420. doi: 10.1177/019262339902700404. [DOI] [PubMed] [Google Scholar]
- Kassel K. M., Owens A. P., Rockwell C. E., Sullivan B. P., Wang R., Tawfik O., Li G., Guo G. L., Mackman N., Luyendyk J. P. Protease-activated receptor 1 and hematopoietic cell tissue factor are required for hepatic steatosis in mice fed a Western diet. Am. J. Pathol. 2011;179:2278–2289. doi: 10.1016/j.ajpath.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J., Okamoto T., Motoyama H., Uchiyama R., Kirchhofer D., Van Rooijen N., Enomoto H., Nishiguchi S., Kawada N., Fujimoto J., Tsutsui H. Interferon-gamma-mediated tissue factor expression contributes to T-cell-mediated hepatitis through induction of hypercoagulation in mice. Hepatology. 2013;57:362–372. doi: 10.1002/hep.26027. [DOI] [PubMed] [Google Scholar]
- Kopec A. K., Luyendyk J. P. Coagulation in liver toxicity and disease: Role of hepatocyte tissue factor. Thromb. Res. 2014;133(Suppl. 1):S57–S59. doi: 10.1016/j.thromres.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer F., Ruf W. Synergies of phosphatidylserine and protein disulfide isomerase in tissue factor activation. Thromb. Haemost. 2014;111:565–597. doi: 10.1160/TH13-09-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoinne S., Thabut D., Housset C., Moreau R., Valla D., Boulanger C. M., Rautou P. E. The emerging roles of microvesicles in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2014;11:350–361. doi: 10.1038/nrgastro.2014.7. [DOI] [PubMed] [Google Scholar]
- Lisman T., Caldwell S. H., Burroughs A. K., Northup P. G., Senzolo M., Stravitz R. T., Tripodi A., Trotter J. F., Valla D. C., Porte R. J., et al. Hemostasis and thrombosis in patients with liver disease: The ups and downs. J. Hepatol. 2010;53:362–371. doi: 10.1016/j.jhep.2010.01.042. [DOI] [PubMed] [Google Scholar]
- Luyendyk J. P., Flanagan K. C., Williams C. D., Jaeschke H., Slusser J. G., Mackman N., Cantor G. H. Tissue factor contributes to neutrophil CD11b expression in alpha-naphthylisothiocyanate-treated mice. Toxicol. Appl. Pharmacol. 2011;250:256–262. doi: 10.1016/j.taap.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luyendyk J. P., Sullivan B. P., Guo G. L., Wang R. Tissue factor-deficiency and protease activated receptor-1-deficiency reduce inflammation elicited by diet-induced steatohepatitis in mice. Am. J. Pathol. 2010;176:177–186. doi: 10.2353/ajpath.2010.090672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhi H., Gores G. J., Lemasters J. J. Apoptosis and necrosis in the liver: A tale of two deaths. Hepatology. 2006;43:S31–44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- Mause S. F., Weber C. Microparticles: Protagonists of a novel communication network for intercellular information exchange. Circ. Res. 2010;107:1047–1057. doi: 10.1161/CIRCRESAHA.110.226456. [DOI] [PubMed] [Google Scholar]
- Müschen M., Warskulat U., Douillard P., Gilbert E., Häussinger D. Regulation of CD95 (APO-1/Fas) receptor and ligand expression by lipopolysaccharide and dexamethasone in parenchymal and nonparenchymal rat liver cells. Hepatology. 1998;27:200–208. doi: 10.1002/hep.510270131. [DOI] [PubMed] [Google Scholar]
- Ni R., Tomita Y., Matsuda K., Ichihara A., Ishimura K., Ogasawara J., Nagata S. Fas-mediated apoptosis in primary cultured mouse hepatocytes. Exp. Cell Res. 1994;215:332–337. doi: 10.1006/excr.1994.1349. [DOI] [PubMed] [Google Scholar]
- Ogasawara J., Watanabe-Fukunaga R., Adachi M., Matsuzawa A., Kasugai T., Kitamura Y., Itoh N., Suda T., Nagata S. Lethal effect of the anti-Fas antibody in mice. Nature. 1993;364:806–809. doi: 10.1038/364806a0. [DOI] [PubMed] [Google Scholar]
- Pawlinski R., Tencati M., Holscher T., Pedersen B., Voet T., Tilley R. E., Marynen P., Mackman N. Role of cardiac myocyte tissue factor in heart hemostasis. J. Thromb. Haemost. 2007;5:1693–1700. doi: 10.1111/j.1538-7836.2007.02649.x. [DOI] [PubMed] [Google Scholar]
- Pearson J. M., Schultze A. E., Schwartz K. A., Scott M. A., Davis J. M., Roth R. A. The thrombin inhibitor, hirudin, attenuates lipopolysaccharide-induced liver injury in the rat. J. Pharmacol. Exp. Ther. 1996;278:378–383. [PubMed] [Google Scholar]
- Pinkoski M. J., Brunner T., Green D. R., Lin T. Fas and Fas ligand in gut and liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2000;278:G354–366. doi: 10.1152/ajpgi.2000.278.3.G354. [DOI] [PubMed] [Google Scholar]
- Popescu N. I., Lupu C., Lupu F. Extracellular protein disulfide isomerase regulates coagulation on endothelial cells through modulation of phosphatidylserine exposure. Blood. 2010;116:993–1001. doi: 10.1182/blood-2009-10-249607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao L. V., Kothari H., Pendurthi U. R. Tissue factor encryption and decryption: Facts and controversies. Thromb. Res. 2012;129(Suppl. 2):S13–S17. doi: 10.1016/j.thromres.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautou P. E., Vion A. C., Luyendyk J. P., Mackman N. Circulating microparticle tissue factor activity is increased in patients with cirrhosis. Hepatology. 2014 doi: 10.1002/hep.27033. doi:10.1002/hep.27033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangwan V., Paliouras G. N., Cheng A., Dubé N., Tremblay M. L., Park M. Protein-tyrosine phosphatase 1B deficiency protects against Fas-induced hepatic failure. J. Biol. Chem. 2006;281:221–228. doi: 10.1074/jbc.M507858200. [DOI] [PubMed] [Google Scholar]
- Shi J., Gilbert G. E. Lactadherin inhibits enzyme complexes of blood coagulation by competing for phospholipid-binding sites. Blood. 2003;101:2628–2636. doi: 10.1182/blood-2002-07-1951. [DOI] [PubMed] [Google Scholar]
- Stéphenne X., Vosters O., Najimi M., Beuneu C., Dung K. N., Wijns W., Goldman M., Sokal E. M. Tissue factor-dependent procoagulant activity of isolated human hepatocytes: Relevance to liver cell transplantation. Liver Transpl. 2007;13:599–606. doi: 10.1002/lt.21128. [DOI] [PubMed] [Google Scholar]
- Stravitz R. T., Bowling R., Bradford R. L., Key N. S., Glover S., Thacker L. R., Gabriel D. A. Role of procoagulant microparticles in mediating complications and outcome of acute liver injury/acute liver failure. Hepatology. 2013;58:304–313. doi: 10.1002/hep.26307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukocheva O. A., Carpenter D. O. Anti-apoptotic effects of 3,5,3’-tri-iodothyronine in mouse hepatocytes. J. Endocrinol. 2006;191:447–458. doi: 10.1677/joe.1.07061. [DOI] [PubMed] [Google Scholar]
- Sullivan B. P., Kopec A. K., Joshi N., Cline H., Brown J. A., Bishop S. C., Kassel K. M., Rockwell C., Mackman N., Luyendyk J. P. Hepatocyte tissue factor activates the coagulation cascade in mice. Blood. 2013;121:1868–1874. doi: 10.1182/blood-2012-09-455436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke F., Schoffski P., Trautwein C., Manns M. P., Ganser A., von Depka M. Tissue factor and thrombomodulin levels are correlated with stage of cirrhosis in patients with liver disease. Blood Coagul. Fibrinolysis. 2001;12:539–545. doi: 10.1097/00001721-200110000-00005. [DOI] [PubMed] [Google Scholar]
- Terada K., Manchikalapudi P., Noiva R., Jauregui H. O., Stockert R. J., Schilsky M. L. Secretion, surface localization, turnover, and steady state expression of protein disulfide isomerase in rat hepatocytes. J. Biol. Chem. 1995;270:20410–20416. doi: 10.1074/jbc.270.35.20410. [DOI] [PubMed] [Google Scholar]
- Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermes I., Haanen C., Steffens-Nakken H., Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- Versteeg H. H., Ruf W. Thiol pathways in the regulation of tissue factor prothrombotic activity. Curr. Opin. Hematol. 2011;18:343–348. doi: 10.1097/MOH.0b013e32834981de. [DOI] [PubMed] [Google Scholar]
- Versteeg H. H., Ruf W. Tissue factor coagulant function is enhanced by protein-disulfide isomerase independent of oxidoreductase activity. J. Biol. Chem. 2007;282:25416–25424. doi: 10.1074/jbc.M702410200. [DOI] [PubMed] [Google Scholar]
- Weerasinghe S. V., Moons D. S., Altshuler P. J., Shah Y. M., Omary M. B. Fibrinogen-γ proteolysis and solubility dynamics during apoptotic mouse liver injury: Heparin prevents and treats liver damage. Hepatology. 2011;53:1323–1332. doi: 10.1002/hep.24203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolberg A. S., Monroe D. M., Roberts H. R., Hoffman M. R. Tissue factor de-encryption: Ionophore treatment induces changes in tissue factor activity by phosphatidylserine-dependent and -independent mechanisms. Blood Coagul. Fibrinolysis. 1999;10:201–210. [PubMed] [Google Scholar]
- Zheng T. S., Schlosser S. F., Dao T., Hingorani R., Crispe I. N., Boyer J. L., Flavell R. A. Caspase-3 controls both cytoplasmic and nuclear events associated with Fas-mediated apoptosis in vivo. Proc. Natl Acad. Sci. U. S. A. 1998;95:13618–13623. doi: 10.1073/pnas.95.23.13618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.