

Abstract
Aims
Whereas endogenous carbon monoxide (CO) is cytoprotective at physiologic levels, excess CO concentrations are associated with cardiometabolic risk and may represent an important marker of progression from subclinical to clinical cardiovascular disease (CVD).
Methods and results
In 1926 participants of the Framingham Offspring Study (aged 57 ± 10 years, 46% women), we investigated the relationship of exhaled CO, a surrogate of blood CO concentration, with both prevalent subclinical CVD and incident clinical CVD events. Presence of subclinical CVD was determined using a comprehensive panel of diagnostic tests used to assess cardiac and vascular structure and function. Individuals with the highest (>5 p.p.m.) compared with lowest (≤4 p.p.m.) CO exposure were more likely to have subclinical CVD [odds ratios (OR): 1.67, 95% CI: 1.32–2.12; P < 0.001]. During the follow-up period (mean 5 ± 3 years), 193 individuals developed overt CVD. Individuals with both high CO levels and any baseline subclinical CVD developed overt CVD at an almost four-fold higher rate compared with those with low CO levels and no subclinical disease (22.1 vs. 6.3%). Notably, elevated CO was associated with incident CVD in the presence [hazards ration (HR): 1.83, 95% CI: 1.08–3.11; P = 0.026] but not in the absence (HR: 0.80, 95% CI: 0.42–1.53; P = 0.51) of subclinical CVD (Pinteraction = 0.019). Similarly, subclinical CVD was associated with incident CVD in the presence of high but not low CO exposure.
Conclusion
Our findings in a community-based sample suggest that elevated CO is a marker of greater subclinical CVD burden and, furthermore, a potential key component in the progression from subclinical to clinical CVD.
Keywords: Carbon monoxide, Subclinical vascular disease, Cardiovascular outcomes
Introduction
The progression from traditional risk factor exposures to subclinical and, eventually, clinical cardiovascular disease (CVD) remains incompletely understood. Substantial efforts have been made to identify common biological pathways underlying the development of atherosclerotic CVD, and to investigate these pathways across model experimental systems and in humans. A growing body of evidence suggests that carbon monoxide (CO) is a potentially important modulator of CVD risk,1–3 motivating attempts to examine the clinical relevance of endogenous CO.4,5 Carbon monoxide represents a potential mechanistic link between metabolic disease and CVD. Although endogenous CO is known to be cytoprotective at physiologic levels, excess concentrations promote hypertension and endothelial dysfunction in experimental models of obesity and metabolic syndrome.6,7 Although data in humans are limited, we and others have reported that increased exhaled CO (a marker of endogenous levels) is associated with traditional CVD risk factors cross sectionally, and with the development of metabolic syndrome and incident CVD prospectively, even in the absence of smoking.8,9 Thus, variation in endogenous CO may represent an important prognostic as well as pathogenic biomarker.
The mechanisms by which CO can lead to the development of CVD have not been fully investigated. On the one hand, increased CO may represent vascular and metabolic stress that coincides with the presence of subclinical CVD, a known precursor to clinical CVD events.10 Alternately, or in addition, elevated CO may itself promote the development of subclinical CVD per se, and thereby augment the risk for incident CVD in at-risk individuals. To further examine the role of endogenous CO in the progression from risk factor exposure to subclinical and overt clinical CVD, we studied the association between CO exposure, measureable subclinical CVD burden, and risk for incident CVD in a large community-based cohort of ambulatory individuals. We hypothesized that endogenous CO exposure is related positively to the presence of subclinical CVD cross sectionally, and that higher levels of CO are associated with a further increased risk of future CVD events among individuals who have evidence of subclinical disease.
Material and methods
The study design and enrolment criteria of the Offspring Cohort of the Framingham Heart Study have been described previously. Individuals eligible for the present investigation included Offspring Cohort participants who attended the sixth examination cycle (1995–98) and underwent standardized anthropometric measurements, a routine medical history, physical examination, and laboratory assessment of CVD risk factors (n = 3532). Individuals attending this examination cycle also underwent measurement of exhaled CO and testing for the presence of subclinical CVD, as described below. We excluded individuals from the present analysis based on the following criteria: prevalent CVD (n = 412), unavailable exhaled CO measures (n = 11), unavailable electrocardiographic data (n = 6), unavailable urinary albumin measurement (n = 454), unavailable ankle-brachial blood pressure (n = 49), unavailable or inadequate carotid ultrasonography data (n = 69), unavailable or inadequate echocardiographic data for determining left ventricular (LV) mass (n = 588), and unavailable smoking status (n = 2). Of the remaining 1941 individuals, 1926 had at least two serial exhaled CO measures collected over the course of quadrennial Offspring examinations, beginning with the second cycle and leading up to and including the sixth examination cycle (second examination cycle, 1979–83; third examination cycle, 1983–87; fourth examination cycle, 1987–91; fifth examination cycle, 1991–95). These 1926 individuals constituted the study sample for analyses.
All participants provided written informed consent and the study protocol was approved by the Institutional Review Board at Boston University Medical Center.
Assessment of exhaled carbon monoxide
Exogenous and endogenous CO concentrations equilibrate across the alveolar-capillary barrier such that exhaled CO reflects blood concentrations of carboxyhemoglobin.11 At the second through sixth examination cycles, exhaled CO was measured in resting participants using the Ecolyzer (2000 series) instrument (Energetics Science Inc., Elmsford, NY, USA), which employs an electrochemical sensor to quantify CO in a sample of exhaled breath (with CO levels ranging from 1 to 100 p.p.m.). A canister of CO gas containing exactly 50 p.p.m. was used to calibrate the Ecolyzer to the midpoint of the scale on each day of testing. The average of two sequential Ecolyzer readings obtained from each participant was recorded. The exhaled CO level was then calculated as this average value minus the base rate of the ambient CO level in the testing room at the Heart Study. This method of determining exhaled CO level has been shown to be reproducible12 and predominantly reflective of endogenous CO, with minimal local environmental (ambient) contamination.13 Reproducibility of sequential Ecolyzer 2000 measurements of exhaled CO has been previously reported, with intra-individual correlations ranging from ≥0.89 to 0.94.12,14
Assessment of subclinical cardiovascular disease
The presence of subclinical vascular disease and target organ damage was determined at the sixth examination using a comprehensive series of non-invasive diagnostic tests, as previously detailed (Supplementary material online).15 Presence vs. absence of subclinical abnormalities detected by each these diagnostic tests was defined using validated cutpoints and criteria (Supplementary material online, Table S1), and used to calculate a subclinical CVD score ranging from 0 to 5, where 1 point each was assigned for the presence of any component of the following abnormalities:15 LV hypertrophy (either by electrocardiography or echocardiography), LV systolic dysfunction (by echocardiography), carotid artery disease (based on abnormal intima-media thickness or presence of stenosis on ultrasound), peripheral arterial disease (based on decreased ankle-brachial index), and glomerular endothelial dysfunction (based on urinary albumin excretion rate).
Cardiovascular outcomes
All study participants were under longitudinal surveillance for incident CVD events, as previously described.15 Briefly, all events were adjudicated by an endpoints review committee of three investigators following an appraisal of outcomes data collected from examination visits, health history updates, and hospitalization and physician office medical records. The primary outcome in the present analysis was incidence of a first CVD event, defined as a composite of coronary heart disease, stroke or transient ischaemic attack, heart failure, and intermittent claudication (as defined previously16). The mean follow-up period was 9.2 ± 2.1 years.
Statistical analyses
Exhaled CO values >50 p.p.m. (n = 4) were considered equivalent to 50 p.p.m. for all analyses. For each participant, all available exhaled CO measurements performed at serial examinations leading up to and including the sixth examination were collected and averaged. Each participant was then grouped into a CO category based on the calculated average value of all available CO levels for that participant, including individual values that may have been above or below the final averaged CO category. These averaged CO values, representing antecedent CO exposure, were categorized into three groups (≤4, >4 and ≤5, and >5 p.p.m.) based on their distribution in the study sample. Categorical thresholds of average CO were determined as whole number approximations of tertiles. For all analyses, exhaled CO was analyzed with the lowest category (≤4 p.p.m.) serving as the referent.
In the first stage, we performed analyses focused on determining the extent to which CO is related cross-sectionally with subclinical CVD. In age- and sex-adjusted logistic regression analyses, we examined the relations of exhaled CO with any prevalent subclinical CVD, defined as presence of ≥1 abnormality detected on testing for subclinical disease (i.e. score ≥1). We also evaluated multivariable models adjusting for the following standard risk factors in addition to age and sex: body mass index, systolic blood pressure (SBP), anti-hypertensive medication use, total/HDL cholesterol ratio, and presence of diabetes. To account for the possible effect of active smoking on exhaled CO levels, we also performed analyses adjusting for current smoking status in addition to other traditional risk factors included in the multivariable models. To assess for potential non-linearity of the relation between exhaled CO and subclinical CVD, above or below any particular cut point, we also examined generalized additive models using penalized splines adjusting for multiple variables including age, sex, SBP, diabetes, anti-hypertensive treatment, and total/HDL cholesterol ratio.
In the second stage, we performed analyses focused on determining the extent to which presence vs. absence of subclinical CVD modifies the prognostic significance of exhaled CO on incidence of CVD events. We calculated age- and sex-adjusted incidence rates of CVD for each CO group overall, and then for each group stratified by subclinical disease status. We used Cox proportional hazards regression in a series of hierarchical models constructed to examine the association of exhaled CO, subclinical disease, and risk of incident CVD. We assessed and confirmed the assumption of proportionality of hazards of the models used in analyses. All models adjusted for age and sex in addition to standard CVD risk factors (SBP, anti-hypertensive treatment, diabetes, total/HDL cholesterol ratio) and smoking status. Model A did not include adjustment for subclinical disease; Model B adjusted for subclinical disease modelled as a dichotomous variable (any vs. none); Model C adjusted for subclinical disease modelled as an ordinal variable; Model D stratified individuals according to CO category and by the presence vs. absence of subclinical disease. For Models A, B, and C, we also used multiplicative interaction terms to assess for the presence of effect modification by subclinical disease status on the association between CO and risk for CVD. Additionally, we used penalized splines to assess the relation between CO and incident CVD while adjusting for presence of baseline subclinical disease. In secondary analyses, we assessed for effect modification of sex and smoking status on the association between CO and risk for CVD.
The null hypothesis was rejected for a two-tailed P-value of <0.05, and all analyses were performed using SAS 9.2 (SAS Institute, Cary, NC, USA).
Results
The characteristics of the study sample at examination cycle 6 are shown in Table 1. Individuals with higher levels of CO exposure were more likely to be younger, men, and current smokers.
Table 1.
Sample Characteristics
| Characteristic | CO category |
|||
|---|---|---|---|---|
| ≤4 (n = 713) | >4 and ≤5 (n = 568) | >5 (n = 645) | P-value | |
| Clinical features | ||||
| Age (years) | 58.6 ± 9.5 | 57.6 ± 9.7 | 56.0 ± 9.0 | <0.0001 |
| Women (%) | 76 | 50 | 47 | <0.0001 |
| Body mass index (kg/m2) | 26.5 ± 4.5 | 27.8 ± 4.7 | 27.4 ± 4.5 | <0.0001 |
| Systolic blood pressure (mmHg) | 127 ± 20 | 127 ± 19 | 125 ± 18 | 0.096 |
| Diastolic blood pressure (mmHg) | 75 ± 9 | 76 ± 9 | 75 ± 9 | 0.493 |
| Hypertension (%) | 35 | 36 | 34 | 0.706 |
| Diabetes (%) | 5 | 6 | 7 | 0.137 |
| Current smokers (%) | 0 | 1 | 41 | <0.0001 |
| Subclinical disease measures | ||||
| LV hypertrophy by ECG or echocardiography | ||||
| LV hypertrophy by Cornell criteria (%) | 1 | 3 | 1 | 0.269 |
| LV mass-to-height ratio (g/m) | ||||
| In men | 107 ± 24 | 107 ± 22 | 108 ± 23 | 0.856 |
| In women | 84 ± 18 | 87 ± 19 | 86 ± 18 | 0.067 |
| LV hypertrophy by echocardiography (%) | 18 | 20 | 21 | 0.002 |
| LV systolic dysfunction by echocardiography | ||||
| Fractional shortening | 0.38 ± 0.05 | 0.38 ± 0.05 | 0.37 ± 0.05 | <0.0001 |
| LV systolic dysfunction (%) | 2 | 2 | 5 | 0.022 |
| Carotid ultrasound abnormality | ||||
| Increased carotid artery IMT (%) | 17 | 18 | 26 | <0.0001 |
| Extreme increase of common carotid artery IMT (%) | 3 | 5 | 5 | 0.164 |
| Carotid artery stenosis ≥25% (%) | 11 | 13 | 18 | 0.0002 |
| Peripheral arterial disease | ||||
| Ankle-brachial index ≤0.9 (%) | 1 | 1 | 3 | 0.009 |
| Glomerular endothelial dysfunction | ||||
| Microalbuminuria (%) | 9 | 7 | 10 | 0.212 |
| Composite of subclinical disease measures | ||||
| At least one (%) | 40 | 41 | 47 | 0.023 |
| At least two (%) | 10 | 10 | 17 | 0.0001 |
| Three or more (%) | 2 | 1 | 4 | 0.002 |
| Mean score | 0.52 ± 0.73 | 0.53 ± 0.72 | 0.69 ± 0.89 | <0.0001 |
Data are means ± SD (for continuous variables) or percentages (for categorical variables).
Exhaled carbon monoxide and subclinical vascular disease
Individuals with greater CO exposure had a higher frequency of prevalent subclinical disease, as represented by the overall subclinical disease score as well as by abnormal values for many of the component subclinical disease measurements (Table 1). Compared with individuals in the lowest category of CO exposure, individuals with average exhaled CO levels >5 p.p.m. had significantly greater odds of having prevalent subclinical disease (Table 2) in age- and sex-adjusted logistic regression analyses; these results remained unchanged in analyses adjusted for standard cardiovascular risk factors including active smoking (Figure 1).
Table 2.
Relation of carbon monoxide with any evidence of subclinical vascular disease
| Age- and sex- adjusted |
aMultivariable-adjusted |
bMultivariable-adjusted including smoking |
||||
|---|---|---|---|---|---|---|
| OR (95%CI) | P-value | OR (95%CI) | P-value | OR (95%CI) | P-value | |
| Average CO ≤4 | Referent | – | Referent | – | Referent | – |
| Average CO >4 and ≤5 | 1.12 (0.88–1.43) | 0.37 | 1.11 (0.86–1.43) | 0.42 | 1.10 (0.85–1.42) | 0.48 |
| Average CO >5 | 1.67 (1.32–2.12) | <0.0001 | 1.69 (1.32–2.17) | <0.0001 | 1.37 (1.03–1.83) | 0.033 |
| Trend | – | <0.0001 | – | <0.0001 | – | 0.038 |
Values are odds ratios (95% CI).
aAdjusted for age, sex, SBP, anti-hypertensive therapy, diabetes, and total/HDL cholesterol.
bAdjusted for age, sex, SBP, anti-hypertensive therapy, diabetes, total/HDL cholesterol, and smoking.
Figure 1.
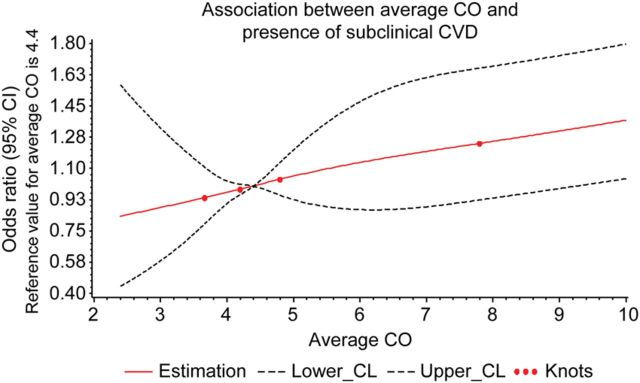
Multivariable-adjusted spline graph displaying the relation of carbon monoxide exposure with prevalent subclinical cardiovascular disease. Bold lines show the association between average exhaled carbon monoxide and presence of subclinical disease; upper and lower 95% confidence limits are plotted as dashed lines, with knots corresponding to carbon monoxide categories (where knots are the points connecting adjoining pieces of the polynomial spline function).
Exhaled carbon monoxide, subclinical vascular disease, and cardiovascular outcomes
Of the 1926 individuals in the study, 193 (46% women) developed incident CVD during the follow-up period. The age- and sex-adjusted incidence of CVD increased with rising CO exposure (Figure 2), particularly among individuals with subclinical disease present at baseline (Table 3). Accordingly, individuals with baseline subclinical disease had the highest rates of incident CVD overall. The CVD risk conferred by CO, as reflected by the cumulative incidence of events over the follow-up period, was observed to be stable over time (Figure 3).
Figure 2.
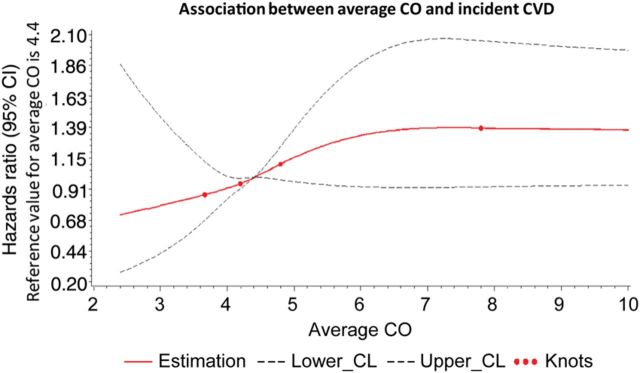
Spline graph displaying the relation of carbon monoxide exposure with incident cardiovascular events, adjusted for baseline presence of subclinical cardiovascular disease. Bold lines show the association between average exhaled carbon monoxide and risk for cardiovascular events; upper and lower 95% confidence limits are plotted as dashed lines, with knots corresponding to carbon monoxide categories.
Table 3.
Incidence of CVD
| Characteristic | No. events/No. at risk | Person-years at risk | Age- and sex-adjusted incidence ratea (95% CI) |
|---|---|---|---|
| Average CO ≤4 | |||
| All | 52/713 | 6675 | 7.64 (5.13–10.03) |
| No subclinical disease | 25/431 | 4099 | 6.32 (2.98–9.50) |
| Any subclinical disease | 27/282 | 2576 | 9.86 (5.58–13.88) |
| Average CO >4 and ≤5 | |||
| All | 62/568 | 5225 | 10.80 (7.54–13.88) |
| No subclinical disease | 20/337 | 3203 | 5.67 (2.51–8.68) |
| Any subclinical disease | 42/231 | 2022 | 18.07 (11.57–23.93) |
| Average CO >5 | |||
| All | 79/645 | 5755 | 13.70 (9.90–17.24) |
| No subclinical disease | 19/345 | 3221 | 5.99 (2.57–9.23) |
| Any subclinical disease | 60/300 | 2534 | 22.13 (15.07–28.37) |
aData shown are number per 100 person years.
Figure 3.

The cumulative incidence of cardiovascular events is shown over the follow-up period by carbon monoxide category.
In age- and sex-adjusted and in multivariable-adjusted Cox proportional hazards analyses, higher average CO was associated with an increased risk for CVD in models without adjustment for subclinical disease (Table 4, Model A). In models adjusting for subclinical disease, as either a dichotomous or ordinal variable, the association of CO with CVD risk was attenuated but remained statistically significant in the multivariable models not including smoking as a covariate (Table 4, Models B and C). In the multivariable models additionally adjusting for current smoking, the association of CO with CVD risk was attenuated and became borderline significant. Results were similar in analyses adjusting for each component measure of subclinical CVD separately, either as dichotomous or continuous variables (Supplementary material online, Tables S2 and S3). Notably, there was significant evidence of effect modification by subclinical disease on CO exposure and the risk for incident CVD in the main multivariable model (P = 0.018) and in the model additionally adjusting for current smoking (P = 0.019). Accordingly, in models stratifying CO exposure groups by the presence vs. absence of subclinical disease at baseline (Table 4, Model D), individuals with both elevated CO (average CO >5 p.p.m.) and baseline subclinical disease experienced an approximately two-fold risk for CVD compared with individuals in the lowest CO group without subclinical disease. In addition, presence of subclinical disease was associated with incident CVD among individuals with moderate CO exposure (average CO >4 and ≤5 p.p.m.) but not among individuals in the lowest CO exposure group. Results were similar in analyses adjusting for number of cigarettes smoked per day, instead of current vs. non-current smoker (Supplementary material online, Table S4). In Models A, B, and C, there was no significant effect modification by smoking status on the association between CO and CVD (data not shown). In all models, there was also no significant effect modification by sex on the association between CO and CVD (data not shown). In analyses of the association between CO and the specific outcome of incident coronary heart disease (n = 109 events), with and without adjustment for subclinical CVD, results were similar (Supplementary material online, Table S5).
Table 4.
Carbon monoxide, subclinical vascular disease, and risk of incident cardiovascular disease
| Age- and sex- adjusted |
aMultivariable-adjusted |
bMultivariable-adjusted including smoking |
||||
|---|---|---|---|---|---|---|
| HR (95% CI) | P-value | HR (95% CI) | P-value | HR (95% CI) | P-value | |
| Model A: no adjustment for subclinical disease | ||||||
| Average CO ≤4 | Referent | – | Referent | – | Referent | – |
| Average CO >4 and ≤5 | 1.45 (0.99–2.11) | 0.054 | 1.47 (1.01–2.14) | 0.045 | 1.45 (1.00–2.12) | 0.053 |
| Average CO >5 | 1.91 (1.33–2.75) | 0.0005 | 1.95 (1.35–2.82) | 0.0004 | 1.55 (1.02–2.36) | 0.042 |
| Trend | – | 0.0005 | – | 0.0004 | – | 0.034 |
| Model B: Adjustment for subclinical disease as dichotomous variable (any vs. none) | ||||||
| Average CO ≤4 | Referent | – | Referent | – | Referent | – |
| Average CO >4 and ≤5 | 1.43 (0.99–2.09) | 0.059 | 1.46 (1.00–2.13) | 0.049 | 1.44 (0.99–2.10) | 0.057 |
| Average CO >5 | 1.77 (1.23–2.55) | 0.002 | 1.84 (1.27–2.66) | 0.001 | 1.48 (0.97–2.25) | 0.070 |
| Trend | – | 0.002 | – | 0.001 | – | 0.056 |
| Model C: Adjustment for subclinical disease as ordinal variable (sum score) | ||||||
| Average CO ≤4 | Referent | – | Referent | – | Referent | – |
| Average CO >4 and ≤5 | 1.47 (1.01–2.14) | 0.042 | 1.51 (1.04–2.21) | 0.031 | 1.50 (1.03–2.18) | 0.036 |
| Average CO >5 | 1.67 (1.16–2.40) | 0.006 | 1.79 (1.23–2.59) | 0.002 | 1.48 (0.97–2.26) | 0.068 |
| Trend | – | 0.007 | – | 0.002 | – | 0.052 |
| Model D: Risks by presence vs. absence of any subclinical disease | ||||||
| Average CO ≤4 with no subclinical CVD | Referent | – | Referent | – | Referent | – |
| Average CO ≤4 with any subclinical CVD | 1.16 (0.67–2.02) | 0.60 | 0.91 (0.52–1.60) | 0.75 | 0.89 (0.50–1.56) | 0.68 |
| Average CO >4 and ≤5 with no subclinical CVD | 0.95 (0.53–1.73) | 0.87 | 0.95 (0.52–1.72) | 0.86 | 0.93 (0.51–1.69) | 0.81 |
| Average CO >4 and ≤5 with any subclinical CVD | 2.18 (1.31–3.62) | 0.003 | 1.76 (1.05–2.95) | 0.033 | 1.70 (1.01–2.85) | 0.046 |
| Average CO >5 with no subclinical CVD | 1.03 (0.56–1.90) | 0.92 | 1.00 (0.54–1.85) | 0.99 | 0.80 (0.42–1.53) | 0.51 |
| Average CO >5 with any subclinical CVD | 2.77 (1.71–4.47) | <0.0001 | 2.33 (1.44–3.79) | 0.0006 | 1.83 (1.08–3.11) | 0.026 |
Values are Cox proportional hazards ratios (95% CI).
aAdjusted for age, sex, SBP, anti-hypertensive therapy, diabetes, and total/HDL cholesterol.
bAdjusted for age, sex, SBP, anti-hypertensive therapy, diabetes, total/HDL cholesterol, and smoking.
Discussion
The main findings of this study were four-fold. First, higher mean levels of CO exposure were associated cross-sectionally with a higher prevalence of subclinical CVD detected using a comprehensive panel of diagnostic tests. Second, on prospective follow-up, individuals with both high CO levels and evidence of subclinical disease developed new-onset CVD at rates that were nearly four times those of individuals with low CO levels and no subclinical disease. Third, when adjusting for traditional risk factors, CO conferred increased risk for incident CVD in the presence of subclinical disease but not in its absence (consistent with the observed statistical interaction between CO and subclinical disease for incident CVD). Finally, participants with subclinical CVD were more likely to develop clinical CVD in the setting of high but not low CO levels. Taken together, these findings indicate that higher CO levels could represent an aggregate marker of greater subclinical disease burden affecting different end organs and, furthermore, a critical component in the progression from subclinical to clinical CVD.
Endogenous CO has garnered attention as a gaseous second messenger that is biologically similar to nitric oxide. Like nitric oxide, CO is also a diatomic small molecule implicated in multiple vasoactive, inflammatory, and oxidative pathways.1,17–19 Compared with nitric oxide, however, endogenous CO may exhibit even more involvement in metabolic pathways.20 In experimental models, CO has been shown to stimulate insulin and glucagon release from islet cells21 as well as modulate insulin sensitivity and glucose tolerance in obesity and diabetes.22 Thus, CO represents a plausible mechanistic link between metabolic and vascular disease. Indeed, we and others have previously shown that elevation of CO levels in humans is associated with metabolic risk factors, diabetes, and incident CVD.8,9 Findings from the present study now further demonstrate an association between CO and multiple measures of subclinical CVD, including carotid atherosclerosis, peripheral arterial disease, and LV hypertrophy.
There are several mechanisms by which CO may be related to the development of subclinical CVD. Endogenous CO is primarily produced from heme catabolism via the enzymatic activity of heme-oxygenases (HO), which have wide tissue distribution.19 On the one hand, excess levels of HO activity and circulating CO may directly promote subclinical atherosclerosis, given that CO is involved in the regulation of vascular smooth muscle cell growth23 as well as the inhibition of nitric oxide-induced vascular relaxation.24 On the other hand, elevated CO levels could represent an endogenous compensatory response to worsening subclinical atherosclerosis, given that physiologic concentrations of CO have been shown to promote large vessel and myocardial angiogenesis25,26 as well as exhibit anti-oxidant,27 anti-inflammatory,28,29 anti-thrombotic,30 and anti-apoptotic31,32 properties. Accordingly, investigations of human vascular tissue have observed increased levels of HO expression in relation to features of vulnerable atheromatous plaque, including increased thrombogenecity and MMP-9 levels.33 Notably, HO-1 dependent production of CO may be closely interrelated with NO synthase-dependent production of NO in pathways related to endothelial cell protection or angiogenesis. Experimental studies indicate that NO upregulates HO-1 activity; in turn, high levels of CO can inhibit NO synthase activity while low levels of CO can induce NO release.34
To investigate the extent to which CO may be a marker or promoter of CVD, we examined the association of CO with incident CVD among individuals with and without evidence of subclinical disease. We observed that in the absence of subclinical disease, CO exposure was not significantly associated with risk for CVD in multivariable analyses. Thus, elevated CO may be regarded, at the very least, as a marker of prevalent subclinical disease. However, we also observed that subclinical disease was not associated with incident CVD in the setting of very low CO levels. Together, these findings suggest that CO could be involved not only in the development of subclinical CVD but, importantly, also in the progression from subclinical to clinical CVD. Alternately, or in addition, elevated CO could indicate the presence of more severe rather than less severe subclinical disease. In this regard, the activity of HO enzymes and their by-products, which are known to play a central role in endogenous CO cycling,18 may be closely related to the pathogenesis of clinical CVD. Thus, the clinical importance of the specific factors involved in endogenous CO cycling (including HO and its other metabolites) remains the focus of ongoing cardiovascular investigations.35
Several limitations of the present investigation merit comment. The reference standard for measuring circulating concentrations of CO in humans is gas chromatography; however, exhaled CO has been validated as a surrogate measure that can reliably capture variation in CO levels.13,36 Furthermore, the exhaled CO measurements in this study were performed in the controlled clinic setting of the Framingham Heart Study and using instrumentation that was routinely calibrated with ambient indoor CO levels, serving to reduce excess intra- and inter-individual variability in measurements. Measured levels of exhaled CO can be influenced by micro- and macro-environmental exposure to products of combustion.37 In our study, reliable measures were not available for second-hand smoke, occupational smoke exposure, or environmental sources such as areas with a high density of automobile traffic. In addition, neither individual nor regional level data on atmospheric CO levels were available at time points contemporaneous with our study.38 Residual confounding from additional unmeasured covariates is also possible. Individuals in our study sample also did not have concurrent measures of exhaled or endogenous nitric oxide, a theoretical potential confounder in the present analyses.18 Because our results are based on observational analyses, causal relationships cannot be inferred. Furthermore, our study sample was limited to individuals with available data for all subclinical CVD measures and, thus, excluded individuals missing any single component of the subclinical CVD score. Because participants who were able to complete separate diagnostic tests for multiple subclinical CVD measure likely represent a healthier sample at baseline, with less variation in both CO levels and subclinical CVD measures, sampling in our study likely biased our results to the null. Finally, our study was conducted in a cohort that included predominantly middle-aged white individuals of European ancestry; thus, the extent to which our findings are generalizable to other ethnicities is not known.
In summary, we observed that measurably elevated levels of CO exposure (assessed in exhaled breath) are associated both with the presence of subclinical CVD cross-sectionally, and with a greater risk of incident clinical CVD prospectively. The presence of subclinical CVD is conventionally defined as anatomic or functional evidence of end-organ damage, which is known to predate the incidence of clinical CVD events in many but not all affected individuals. In this context, our data suggest that CO could be a marker of pathways that are active in both the development and progression of CVD. Further research is needed to determine the possible mechanisms underlying our results and the extent to which measures of CO may be useful for prognostication among individuals both with and without subclinical CVD.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
From the Framingham Heart Study of the National Heart, Lung, and Blood Institute of the National Institutes of Health and Boston University School of Medicine. This work was supported by the National Heart, Lung and Blood Institute's Framingham Heart Study (Contract No. N01-HC-25195). This work was supported in part by the National Heart, Lung and Blood Institute's Framingham Heart Study (contract no. N01-HC-25195) and grants R01-NS017950 and K99HL107642 and the American College of Cardiology/Merck Research Fellowship in Cardiovascular Disease and Cardiometabolic Disorders (S.C.).
Conflict of interest: none declared.
References
- 1.Durante W. Carbon monoxide and bile pigments: surprising mediators of vascular function. Vasc Med. 2002;7:195–202. doi: 10.1191/1358863x02vm424ra. [DOI] [PubMed] [Google Scholar]
- 2.Wang X, Wang Y, Kim HP, Nakahira K, Ryter SW, Choi AM. Carbon monoxide protects against hyperoxia-induced endothelial cell apoptosis by inhibiting reactive oxygen species formation. J Biol Chem. 2007;282:1718–1726. doi: 10.1074/jbc.M607610200. [DOI] [PubMed] [Google Scholar]
- 3.Ndisang JF, Tabien HE, Wang R. Carbon monoxide and hypertension. J Hypertens. 2004;22:1057–1074. doi: 10.1097/00004872-200406000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Chan KH, Ng MK, Stocker R. Haem oxygenase-1 and cardiovascular disease: mechanisms and therapeutic potential. Clin Sci (Lond) 2011;120:493–504. doi: 10.1042/CS20100508. [DOI] [PubMed] [Google Scholar]
- 5.Ryter SW, Morse D, Choi AM. Carbon monoxide and bilirubin: potential therapies for pulmonary/vascular injury and disease. Am J Respir Cell Mol Biol. 2007;36:175–182. doi: 10.1165/rcmb.2006-0333TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson FK, Johnson RA, Durante W, Jackson KE, Stevenson BK, Peyton KJ. Metabolic syndrome increases endogenous carbon monoxide production to promote hypertension and endothelial dysfunction in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol. 2006;290:R601–R608. doi: 10.1152/ajpregu.00308.2005. [DOI] [PubMed] [Google Scholar]
- 7.Owens EO. Endogenous carbon monoxide production in disease. Clin Biochem. 2010;43:1183–1188. doi: 10.1016/j.clinbiochem.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Paredi P, Biernacki W, Invernizzi G, Kharitonov SA, Barnes PJ. Exhaled carbon monoxide levels elevated in diabetes and correlated with glucose concentration in blood: a new test for monitoring the disease? Chest. 1999;116:1007–1011. doi: 10.1378/chest.116.4.1007. [DOI] [PubMed] [Google Scholar]
- 9.Cheng S, Lyass A, Massaro JM, O'Connor GT, Keaney JF, Jr, Vasan RS. Exhaled carbon monoxide and risk of metabolic syndrome and cardiovascular disease in the community. Circulation. 2010;122:1470–1477. doi: 10.1161/CIRCULATIONAHA.110.941013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuller LH, Shemanski L, Psaty BM, Borhani NO, Gardin J, Haan MN, O'Leary DH, Savage PJ, Tell GS, Tracy R. Subclinical disease as an independent risk factor for cardiovascular disease. Circulation. 1995;92:720–726. doi: 10.1161/01.cir.92.4.720. [DOI] [PubMed] [Google Scholar]
- 11.Jones RH, Ellicott MF, Cadigan JB, Gaensler EA. The relationship between alveolar and blood carbon monoxide concentrations during breathholding; simple estimation of COHb saturation. J Lab Clin Med. 1958;51:553–564. [PubMed] [Google Scholar]
- 12.Hedblad B, Ogren M, Engstrom G, Wollmer P, Janzon L. Heterogeneity of cardiovascular risk among smokers is related to degree of carbon monoxide exposure. Atherosclerosis. 2005;179:177–183. doi: 10.1016/j.atherosclerosis.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Kharitonov SA, Barnes PJ. Exhaled markers of pulmonary disease. Am J Respir Crit Care Med. 2001;163:1693–1722. doi: 10.1164/ajrccm.163.7.2009041. [DOI] [PubMed] [Google Scholar]
- 14.Woodman G, Wintoniuk DM, Taylor RG, Clarke SW. Time course of end-expired carbon monoxide concentration is important in studies of cigarette smoking. Clin Sci (Lond) 1987;73:553–555. doi: 10.1042/cs0730553. [DOI] [PubMed] [Google Scholar]
- 15.Ingelsson E, Sullivan LM, Murabito JM, Fox CS, Benjamin EJ, Polak JF, Meigs JB, Keyes MJ, O'Donnell CJ, Wang TJ, D'Agostino RB, Sr, Wolf PA, Vasan RS. Prevalence and prognostic impact of subclinical cardiovascular disease in individuals with the metabolic syndrome and diabetes. Diabetes. 2007;56:1718–1726. doi: 10.2337/db07-0078. [DOI] [PubMed] [Google Scholar]
- 16.Kannel WB, Wolf PA, Garrison RJ. The Framingham Study: An Epidemiological Investigation of Cardiovascular Disease. Section 34. Some Risk Factors Related to the Annual Incidence of Cardiovascular Disease and Death Using Pooled Repeated Biennial Measurements: Framingham Heart Study, 30-year follow-up. Bethesda, MD: National Heart, Lung, and Blood Institute; 1987. NIH publication no. 87–2703. [Google Scholar]
- 17.Bauer I, Pannen BH. Bench-to-bedside review: Carbon monoxide – from mitochondrial poisoning to therapeutic use. Crit Care. 2009;13:220. doi: 10.1186/cc7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu L, Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev. 2005;57:585–630. doi: 10.1124/pr.57.4.3. [DOI] [PubMed] [Google Scholar]
- 19.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 20.Joe Y, Zheng M, Kim SK, Kim S, Uddin JM, Min TS, Ryu do G, Chung HT. The role of carbon monoxide in metabolic disease. Ann N Y Acad Sci. 2011;1229:156–161. doi: 10.1111/j.1749-6632.2011.06121.x. [DOI] [PubMed] [Google Scholar]
- 21.Henningsson R, Alm P, Ekstrom P, Lundquist I. Heme oxygenase and carbon monoxide: regulatory roles in islet hormone release: a biochemical, immunohistochemical, and confocal microscopic study. Diabetes. 1999;48:66–76. doi: 10.2337/diabetes.48.1.66. [DOI] [PubMed] [Google Scholar]
- 22.Li M, Kim DH, Tsenovoy PL, Peterson SJ, Rezzani R, Rodella LF, Aronow WS, Ikehara S, Abraham NG. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes. 2008;57:1526–1535. doi: 10.2337/db07-1764. [DOI] [PubMed] [Google Scholar]
- 23.Morita T, Mitsialis SA, Koike H, Liu Y, Kourembanas S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J Biol Chem. 1997;272:32804–32809. doi: 10.1074/jbc.272.52.32804. [DOI] [PubMed] [Google Scholar]
- 24.Marazioti A, Bucci M, Coletta C, Vellecco V, Baskaran P, Szabo C, Cirino G, Marques AR, Guerreiro B, Goncalves AM, Seixas JD, Beuve A, Romao CC, Papapetropoulos A. Inhibition of nitric oxide-stimulated vasorelaxation by carbon monoxide-releasing molecules. Arterioscler Thromb Vasc Biol. 2011;31:2570–2576. doi: 10.1161/ATVBAHA.111.229039. [DOI] [PubMed] [Google Scholar]
- 25.Lin HH, Chen YH, Yet SF, Chau LY. After vascular injury, heme oxygenase-1/carbon monoxide enhances re-endothelialization via promoting mobilization of circulating endothelial progenitor cells. J Thromb Haemost. 2009;7:1401–1408. doi: 10.1111/j.1538-7836.2009.03478.x. [DOI] [PubMed] [Google Scholar]
- 26.Lakkisto P, Kyto V, Forsten H, Siren JM, Segersvard H, Voipio-Pulkki LM, Laine M, Pulkki K, Tikkanen I. Heme oxygenase-1 and carbon monoxide promote neovascularization after myocardial infarction by modulating the expression of HIF-1alpha, SDF-1alpha and VEGF-B. Eur J Pharmacol. 2010;635:156–164. doi: 10.1016/j.ejphar.2010.02.050. [DOI] [PubMed] [Google Scholar]
- 27.Wu BJ, Kathir K, Witting PK, Beck K, Choy K, Li C, Croft KD, Mori TA, Tanous D, Adams MR, Lau AK, Stocker R. Antioxidants protect from atherosclerosis by a heme oxygenase-1 pathway that is independent of free radical scavenging. J Exp Med. 2006;203:1117–1127. doi: 10.1084/jem.20052321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orozco LD, Kapturczak MH, Barajas B, Wang X, Weinstein MM, Wong J, Deshane J, Bolisetty S, Shaposhnik Z, Shih DM, Agarwal A, Lusis AJ, Araujo JA. Heme oxygenase-1 expression in macrophages plays a beneficial role in atherosclerosis. Circ Res. 2007;100:1703–1711. doi: 10.1161/CIRCRESAHA.107.151720. [DOI] [PubMed] [Google Scholar]
- 29.Lee TS, Chang CC, Zhu Y, Shyy JY. Simvastatin induces heme oxygenase-1: a novel mechanism of vessel protection. Circulation. 2004;110:1296–1302. doi: 10.1161/01.CIR.0000140694.67251.9C. [DOI] [PubMed] [Google Scholar]
- 30.True AL, Olive M, Boehm M, San H, Westrick RJ, Raghavachari N, Xu X, Lynn EG, Sack MN, Munson PJ, Gladwin MT, Nabel EG. Heme oxygenase-1 deficiency accelerates formation of arterial thrombosis through oxidative damage to the endothelium, which is rescued by inhaled carbon monoxide. Circ Res. 2007;101:893–901. doi: 10.1161/CIRCRESAHA.107.158998. [DOI] [PubMed] [Google Scholar]
- 31.Brunt KR, Fenrich KK, Kiani G, Tse MY, Pang SC, Ward CA, Melo LG. Protection of human vascular smooth muscle cells from H2O2-induced apoptosis through functional codependence between HO-1 and AKT. Arterioscler Thromb Vasc Biol. 2006;26:2027–2034. doi: 10.1161/01.ATV.0000236204.37119.8d. [DOI] [PubMed] [Google Scholar]
- 32.Yet SF, Layne MD, Liu X, Chen YH, Ith B, Sibinga NE, Perrella MA. Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 2003;17:1759–1761. doi: 10.1096/fj.03-0187fje. [DOI] [PubMed] [Google Scholar]
- 33.Cheng C, Noordeloos AM, Jeney V, Soares MP, Moll F, Pasterkamp G, Serruys PW, Duckers HJ. Heme oxygenase 1 determines atherosclerotic lesion progression into a vulnerable plaque. Circulation. 2009;119:3017–3027. doi: 10.1161/CIRCULATIONAHA.108.808618. [DOI] [PubMed] [Google Scholar]
- 34.Thorup C, Jones CL, Gross SS, Moore LC, Goligorsky MS. Carbon monoxide induces vasodilation and nitric oxide release but suppresses endothelial NOS. Am J Physiol. 1999;277:F882–F889. doi: 10.1152/ajprenal.1999.277.6.F882. [DOI] [PubMed] [Google Scholar]
- 35.Stec DE, Ishikawa K, Sacerdoti D, Abraham NG. The emerging role of heme oxygenase and its metabolites in the regulation of cardiovascular function. Int J Hypertens. 2012;2012:593530. doi: 10.1155/2012/593530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevenson DK, Ostrander CE, Johnson JD. Effect of erythrocyte destruction on the pulmonary excretion rate of carbon monoxide in adult male Wistar rats. J Lab Clin Med. 1979;94:649–654. [PubMed] [Google Scholar]
- 37.Chau CK, Tu EY, Chan DW, Burnett J. Estimating the total exposure to air pollutants for different population age groups in Hong Kong. Environ Int. 2002;27:617–630. doi: 10.1016/s0160-4120(01)00120-9. [DOI] [PubMed] [Google Scholar]
- 38.EPA. Regional Trends in CO Levels. http://www.epa.gov/airtrends/carbon.html#coreg (5 November 2013) [Google Scholar]