

Abstract
Motivation: Whole-genome high-coverage sequencing has been widely used for personal and cancer genomics as well as in various research areas. However, in the lack of an unbiased whole-genome truth set, the global error rate of variant calls and the leading causal artifacts still remain unclear even given the great efforts in the evaluation of variant calling methods.
Results: We made 10 single nucleotide polymorphism and INDEL call sets with two read mappers and five variant callers, both on a haploid human genome and a diploid genome at a similar coverage. By investigating false heterozygous calls in the haploid genome, we identified the erroneous realignment in low-complexity regions and the incomplete reference genome with respect to the sample as the two major sources of errors, which press for continued improvements in these two areas. We estimated that the error rate of raw genotype calls is as high as 1 in 10–15 kb, but the error rate of post-filtered calls is reduced to 1 in 100–200 kb without significant compromise on the sensitivity.
Availability and implementation: BWA-MEM alignment and raw variant calls are available at http://bit.ly/1g8XqRt scripts and miscellaneous data at https://github.com/lh3/varcmp.
Contact: hengli@broadinstitute.org
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Since the sequencing of the first personal genome (Levy et al., 2007), and in particular the first genomes sequenced with the Illumina technologies (Bentley et al., 2008; Wang et al., 2008), resequencing has been widely used for personal and cancer genomics (Watson et al., 2013), for the discovery of de novo mutations associated with Mendelian diseases (Bamshad et al., 2011), for the reconstruction of human population history (Li and Durbin, 2011) and for the understanding of mutation processes (Campbell and Eichler, 2013; Veltman and Brunner, 2012). In most of these studies, mapping-based single nucleotide polymorphism (SNP)/insertion-deletion (INDEL) calling plays a central role. The accuracy of the calls has a fundamental impact on the biological interpretation. In this context, various research groups have attempted to evaluate the performance of variant calling.
The simplest approach to the evaluation of variant calling is to simulate variants and reads from a reference genome (Li et al., 2008). However, we are unable to simulate various artifacts such as the non-random distribution of variants, dependent errors, incomplete reference genome and copy number variations. An improved version is to incorporate real variants instead of using simulated variants (Talwalkar et al., 2013), but it does not address the artifacts caused by large-scale effects either. A better simulation is to take the reads sequenced from one sample with a finished genome, map them to another finished genome, call variants and then compare the calls to the differences found by genome-to-genome alignment (Li et al., 2008). However, this approach is limited to small haploid genomes. There are attempts to apply a similar idea to mammalian genomes (Bolosky et al., Unpublished data; Li et al., 2013), but as the mammalian reference genomes are frequently incomplete and the whole-genome alignment is imperfect, such a simulation is still different from realistic scenarios.
The difficulties in simulation have motivated us to focus more on real data. One simple approach is to thoroughly sequence a small target region with mature technologies, such as the Sanger sequencing technology, and take the resultant sequence as the ground truth (Harismendy et al., 2009). It does not capture large-scale artifacts, though. Another more commonly used method is to measure accuracy either by comparing variant calls from different pipelines, or by comparing calls to variants ascertained with array genotyping or in another study (Boland et al., 2013; Cheng et al., 2014; Clark et al., 2011; Goode et al., 2013; Lam et al., 2012a,b; Li, 2012; Liu et al., 2013; O’Rawe et al., 2013; Zook et al., 2014). However, array genotyping is biased to easier portions of the genome and may have a higher error rate per assayed site than the variant calling error rate (Bentley et al., 2008); simply comparing call sets would only give us an estimate of the relative accuracy––if two pipelines are affected by the same artifact that a third pipeline does not have, then the third pipeline will appear worse even though it is in fact better. In addition, comparative studies usually measure the accuracy with summary statistics such as the fraction of calls present in dbSNP or the transition-to-transversion ratio. They do not tell us the wrong sites.
Many studies also experimentally validated typically up to a few hundred variants with MiSeq or Sanger sequencing or Sequenom genotyping. Nonetheless, such experiments are biased toward easier regions and may also be subjected to other artifacts such as on-primer variants and non-specific amplification (the 1000 Genomes Project analysis subgroup, personal communication). Calling heterozygotes from Sanger sequence data are also challenging by itself.
In the author’s view, it is better to evaluate variant calling by comparing samples from a pedigree (Zook et al., 2014), or from the same individual (Nickles et al., 2012), including cancer samples (Löwer et al., 2012). Because we expect to see only tens to hundreds of somatic mutations or Mendelian errors per genome (Conrad et al., 2011), most other inconsistencies are likely to be errors. However, this method is insensitive to systematic errors. If at a locus, a caller finds erroneous heterozygotes in all samples, the errors would not be identified.
After these efforts, we are still not clear about a basic question: the error rate of SNP and INDEL calling. Although a few papers give an estimate of one error per 100–200 kb, it is either estimated on easy sites (Bentley et al., 2008) or not sufficiently backed with published data (Nickles et al., 2012). In addition, only a few works (Kim and Speed, 2013; Larson et al., 2012; Roberts et al., 2013) have attempted to identify the sources of errors. Analyzing systematic errors is even rarer, as most existing evaluation methods hide them.
In this article, we use an exceptional dataset, sequencing data from a haploid human cell line, to evaluate the accuracy of variant calling. As the vast majority of heterozygous calls are supposed to be errors, we almost know the ground truth unbiasedly across the whole genome. We are able to pinpoint errors, investigate their characteristics, experiment filters and get a reasonable estimate of the error rate, not limited to non-systematic errors. In addition to the unique dataset, our study also differs from many previous ones in the use of multiple read mappers, unpublished but well developed variant callers and caller-oblivious genotyping and filtering.
2 DATASETS AND DATA ANALYSIS
2.1 Datasets
In this study, we focused on deep Illumina sequencing data from two cell lines, the CHM1hTERT cell line (Jacobs et al., 1980) and the NA12878 cell line. A crucial and unusual feature of CHM1hTERT, or briefly CHM1, is that this cell line is haploid, which suggests that any heterozygous variant calls are errors. A calling method producing fewer heterozygotes is in theory better. Meanwhile, to avoid overrating a variant calling method with low sensitivity on heterozygotes, we also used NA12878 as a positive control.
Both data sets are available from Sequence Read Archive (SRA). The entire CHM1 dataset (AC:SRP017546) gives over 100-fold coverage. We are only using six SRA runs with the accessions ranging from SRR642636 to SRR642641. The six runs are from the same library, yielding ∼65-fold coverage before the removal of potential duplicates caused by polymerase chain reaction (PCR) during sample preparation.
We acquired the NA12878 dataset (AC:ERR194147) from the Illumina Platinum Genomes project. The library was constructed without PCR amplification. We are only using paired-end data, which yield about 55-fold coverage.
2.2 Alignment and post-alignment processing
We mapped the CHM1 reads with Bowtie2 (Langmead and Salzberg, 2012) and BWA-MEM (Li, 2013), and mapped the NA12878 reads with BWA (Li and Durbin, 2009) in addition to Bowtie2 and BWA-MEM. The detailed command lines can be found in Table 1. Except in Section 3.6, we mapped the reads to hs37d5, the reference genome used by the 1000 Genomes Project in the final phase.
Table 1.
Evaluated mappers and variant callers
| Symbol | Algorithm | Version | Command line |
|---|---|---|---|
| bt2 | Bowtie2 | 2.1.0 | bowtie2 -x ref.fa -1 read1.fq -2 read2.fq -X 500 |
| bwa | BWA-backtrack | 0.7.6 | bwa aln -f read1.sai ref.fa read1.fq; bwa sampe ref.fa read1.sai read2.sai read1.fq read2.fq |
| mem | BWA-MEM | 0.7.6 | bwa mem ref.fa read1.fq read2.fq |
| fb | FreeBayes | 0.9.9 | freebayes -f ref.fa aln.bam |
| st | SAMtools | 0.1.19 | samtools mpileup -Euf ref.fa aln.bam — bcftools view -v - |
| Ug | UnifiedGenotyper | 2.7-4 | java -jar GenomeAnalysisTK.jar -T UnifiedGenotyper -R ref.fa -I aln.bam -stand_call_conf 30 -stand_emit_conf 10 -glm BOTH |
| hc | HaplotypeCaller | 2.7-4 | java -jar GenomeAnalysisTK.jar -T HaplotypeCaller –genotyping_mode DISCOVERY -R ref.fa -I aln.bam -stand_call_conf 30 -stand_emit_conf 10 |
| pt | Platypus | 0.5.2 | Platypus.py callVariants –filterDuplicates=1 –bamFiles=aln.bam –refFile=ref.fa |
After the initial alignment, we run Picard’s MarkDuplicates on both datasets. Picard identified 20% of CHM1 reads as PCR duplicates. For NA12878, Picard reported 1.5% of them as PCR duplicates, which are false positives, as the library was constructed without amplification. We did not apply MarkDuplicates for NA12878 in the subsequent analysis.
For the NA12878 BWA alignment, we also tried GATK’s (Depristo et al., 2011) base quality score recalibration (BQSR) and INDEL realignment around INDEL calls from the 1000 Genomes Project (1000 Genomes Project Consortium, 2012). For both SAMtools (Li, 2011b) and GATK, the number of calls only differs by 0.1%, much smaller than the difference caused by other procedures. We thus did not apply these steps to other alignments because of the additional computational cost. It should be noted that although BQSR and INDEL realignment have little effect on these two high-coverage datasets, it may make difference on low-coverage data or when the base quality is not well calibrated.
2.3 Calling SNPs and short INDELs
We called SNPs and short INDELs with FreeBayes (Garrison and Marth, 2012), GATK UnifiedGenotyper, Platypus, SAMtools and GATK HaplotypeCaller. The command lines can be found in Table 1. Additional details are as follows.
2.3.1 Resolving overlapping variants
Platypus and SAMtools may produce many overlapping variants. To avoid overcounting variants, for two overlapping variants, we always keep the one with the higher variant quality. We repeated this procedure until no overlapping variants remained.
2.3.2 Recalling genotypes
Given the same genotype likelihood, different callers may produce different genotypes. For example, SAMtools estimates genotypes assuming the prior of seeing a heterozygote being , but GATK does not apply a prior. GATK is more likely to call a heterozygote than SAMtools. Genotype calling for a single sample is relatively simple. To avoid the subtle difference in this simple step complicating the final results, we recall the genotypes from genotype likelihoods provided by the callers. We multiplied to the likelihood of heterozygotes, and then called the genotype with the maximum likelihood.
Platypus does not give genotype likelihoods for multi-allelic variants. We kept the reported genotypes in the variant call format (VCF).
2.3.3 Decomposing complex variants
Both FreeBayes and Platypus may report a variant composed of multiple SNPs and/or INDELs. We decomposed such variants into individual events such that the results are more comparable. FreeBayes uses a concise idiosyncratic gapped alignment report notations (CIGAR) string to describe how a complex variant is aligned to the reference. We extracted SNPs and INDELs from the CIGAR. Platypus does not report CIGAR. We assumed the variant allele is always left aligned to the reference allele when decomposing a complex variant.
2.4 Variant filtering
All the callers used in this study come with filtering programs or a recommended set of filters. However, applying caller-specific filters may complicate comparison and obscure artifacts. We decided to choose several universal filters applicable to most callers:
Low-complexity (LC) filter: filtering variants overlapping with low-complexity regions (LCRs) identified with the mdust program (http://bit.ly/mdust-LC), which is a stand-alone implementation of the DUST algorithm first used by BLAST. In GRCh37, 2.0% of A/C/G/T bases on autosomes are identified to be LCRs.
Maximum depth (MD) filter: filtering sites covered by excessive number of reads. It should be noted that different callers may define the depth differently. For example, Platypus apparently only counts reads with unambiguous realignment. The read depth reported in the Platypus VCF is noticeably smaller in comparison with other callers.
Allele balance (AB) filter: filtering sites where the fraction of non-reference reads is too low.
Double strand (DS) filter: filtering variants if either the number of non-reference reads on the forward strand or on the reverse strand is below a certain threshold. This filter is not applicable to GATK calls, as GATK does not report these numbers. DS has been identified to be an effective filter on cancer data (Kim and Speed, 2013; Roberts et al., 2013).
Fisher strand filter (FS): filtering sites where the numbers of reference/non-reference reads are highly correlated with the strands of the reads. More precisely, we counted the number of reference reads on the forward strand and on the reverse strand, and the number of non-reference reads on the forward and reverse strand. With these four numbers, we constructed a 2 × 2 contingency table and used the P-value from a Fisher’s exact test to evaluate the correlation.
Quality filter (QU): filtering sites with the reported variant quality below a threshold.
Among these filters, LC is a regional filter and is entirely independent of alignment and variant calling. Although MD is computed from called variants, its effect is usually not greatly dependent on the mapper and the caller, either. The remaining filters may be dependent of the error models used by the callers. For example, SAMtools effectively gives a higher weight to variants supported on both strand; FreeBayes seems to require a variant to be supported by 20% of reads covering the site. The optimal thresholds for the AB, DS and FS filters are caller dependent.
2.5 Measuring accuracy
The CHM1 and the NA12878 datasets share many properties. They are both sequenced with 100 bp Illumina reads to a similar coverage after the removal of PCR duplicates. The number of called variants per haplotype is also close, usually within 1% difference according to multiple call sets. Under this observation, it is reasonable to assume the number of heterozygous errors in NA12878 is also close to the number of heterozygous calls in CHM1. As a result, we may take as an estimate of the false-positive rate (FPR) of heterozygotes, and as a proxy to sensitivity, where Nh is the number of heterozygous calls in CHM1 and Nd the number in NA12878. This might be the first time that we can unbiasedly measure FPR in a whole genome call set.
2.6 Manual review
To understand the major error modes, we have manually reviewed >200 heterozygous INDELs called by different callers from CHM1. For these sites, we displayed the alignment with SAMtools’ tview alignment viewer to get a sense of the alignment quality, obvious positional biases and the complexity of the reference genome. We often extracted reads in regions around the INDELs, extending to flanking regions with high complexity to eyes. We assembled the extracted reads with fermi (Li, 2012) version 1.1 and mapped the assembled contigs back to the reference genome with BWA-MEM. Fermi tries to preserve heterozygotes. If the INDELs are truly heterozygous, we will typically see two contigs covering a site, one for each allele. We used the local assembly as an orthogonal approach to validate heterozygous calls.
3 RESULTS
When studying the effect of filters on variant calling, we initially applied the filters independently on each call set. However, when presenting the results in the following, we applied the filters in an order, with a filter applied later depending on the filters applied before it. We did this for clarity and to highlight filters having major effects. Figure 1 overviews the breakdown of various filters across multiple call sets. If we consider that there might be true heterozygotes in CHM1 potentially because of somatic mutations, call sets generally have an error rate ∼1 in 100–200 kb (i.e. 15 000–30 000 false heterozygotes per genome) after filtering.
Fig. 1.

Effect of filters. LC filter: not overlapping LCRs identified by the DUST algorithm. MD filter: read depth below , where d is the average read depth. Miscellaneous filter (misc) includes three filters: allele balance above 30%, variants supported by non-reference reads on both strands and Fisher strand P-value is >0.01. Filters are applied in the order of LC, MD and misc, with MD applied to variants passing LC, and misc applied to variants passing both LC and MD. For each call set, the total height of the bar gives the number of raw variant calls with the reported quality in VCF no <30. Note that the Y-axes are scaled differently
3.1 Checking the ploidy of CHM1
Although the CHM1hTERT cell line is supposed to be haploid, we may still see heterozygous variant calls potentially because: (i) the cell line is not truly haploid; (ii) there are somatic mutations in the cell line; (iii) there are library construction and sequencing errors (Robasky et al., 2014), which ought to be considered by the calling algorithms; and (iv) mapping or variant calling algorithms have flaws. In this study, we focus on (iii) and (iv), but first we should make sure heterozygotes resulted from (i) and (ii) occur at a much lower rate.
We note that if the sample submitted for sequencing is not haploid either because of biological artifacts or massive somatic mutations, a large number of heterozygotes should be evident from the sequencing data and get called by all callers. In contrast, if heterozygotes are mostly caused by sequencing errors or algorithm artifacts, owing to the differences in algorithm and error modeling, callers will call a subset of errors with different characteristics, which will result in low consistency between call sets. The small call set intersection in Figure 2 suggests the latter is the case.
Fig. 2.
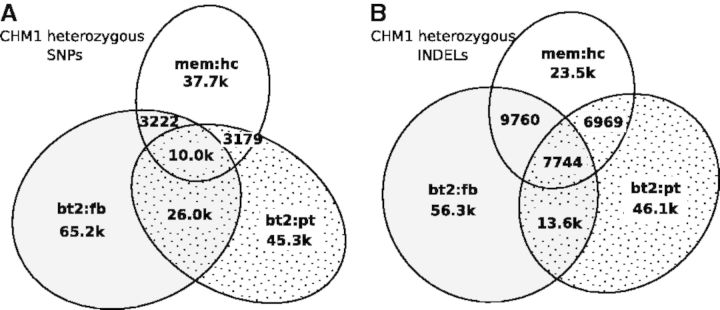
Relationship between CHM1 heterozygous call sets. Raw variant calls were filtered with variant quality no <30, allele balance >20%, Fisher strand P-value >0.001 and maximum read depth below , where d is the average read depth. (A) Relationship between heterozygous SNP call sets. Two SNPs are considered the same if they are at the same position. (B) Relationship between heterozygous INDEL call sets. Two filtered INDELs are said to be linked if the 3′ end of an INDEL is within 20 bp from the 5′ end of the other INDEL, or vice versa. An INDEL cluster is a connected component (not a clique) of linked INDELs. It is possible that in a cluster two INDELs are distant from each other but both overlap a third INDEL. Venn’s diagram shows the number of INDEL clusters falling in each category based on the sources of INDELs in each cluster. In total, 15% of SNPs and 91% of INDELs in the 3-way intersections overlap LCRs
We also manually reviewed tens of heterozygotes called by multiple callers, both on the data in this study and on Illumina data generated from other libraries (AC:SRR642626–SRR642635 and AC:SRR642750), which were mapped with the original BWA algorithm (Li and Durbin, 2009) by the 1000 Genomes Project analysis group. Reviewing the read evidence using an alignment viewer, it appears that more than half of the SNPs are real. Most of these SNPs have averaged read depth, non-overlap with known segmental duplications (http://bit.ly/eelabdb) and are not associated with known error-prone motifs in Illumina sequencing (Nakamura et al., 2011). On the other hand, many INDELs in LCRs look like systematic errors called by all callers (see also Section 3.2). We speculate there may be 5–20 k heterozygotes in CHM1 with strong alignment support from multiple Illumina libraries. It is hard to get a more accurate estimate or to further tell the sources of these heterozygotes with the data we are using. As we were writing up this work, Pacific Biosciences released deep resequencing data for the CHM1 cell line. It could be used to isolate errors caused by the Illumina sample preparation and sequencing. However, mapping and variant calling from PacBio human data is still in the early phase. We decided to omit the comparison with the PacBio data for now.
Anyway, even if we assume the variant calls in the intersection are all present in the CHM1hTERT cell line, we should still be able to measure an error rate up to 1 error per 170 kb (=3 Gbp/17.7 k). Given that there are 10 times more raw heterozygous calls in NA12878 than CHM1 (Fig. 1), it seems likely that CHM1 heterozygotes are likely errors from major sequencing/calling artifacts.
As a side technical note, we applied milder filters in Figure 2 in comparison with Figure 1. We found the intersection between call sets often becomes smaller with more stringent thresholds because stringent thresholds reduce the sensitivity in different aspects of call sets and amplify the subtle differences between calling algorithms. In addition, in Figure 2B, we were clustering INDELs within 20 bp from each other. Increasing the distance threshold to 100 bp only changed the numbers slightly.
3.2 The LC
On CHM1, low-complexity regions (LCRs), 2% of the human genome, harbor 80–90% of heterozygous INDEL calls and up to 60% of heterozygous SNPs (Fig. 1). Recall that if we let be the number of CHM1 heterozygous INDELs in LCRs and the number of NA12878 heterozygous INDELs in LCRs, estimates the FPR of heterozygotes. The FPR in LCRs is ranged from 10% to as high as 40% depending on call sets. With a similar estimator, the FPR of heterozygous INDELs outside LCRs is much lower, ∼1–8% depending on call sets. We have also tried lobSTR (Gymrek et al., 2012). It called 65 k heterozygous INDELs from microsatellites, still yielding a high FPR. To understand why errors are enriched in LCRs, we reviewed >100 sites and identified two major sources of INDEL genotyping errors: potential PCR errors and realignment errors.
3.2.1 Potential PCR amplification errors
PCR errors are known to be responsible for many INDEL errors in long homopolymer runs (1000 Genomes Project Consortium, 2012). On CHM1, we have observed many apparent 1 bp heterozygous INDELs (Fig. 1) inserted to or deleted from long poly-A or poly-T runs, which may be because of PCR errors. Although most callers deploy advanced models for homopolymer INDELs, they are calling vastly different number of 1 bp heterozygous INDELs. It is still not clear to us that we can model PCR errors well. Maybe the most effective solution is to avoid PCR in sample preparation.
Potential PCR errors are not the only error source. On the PCR-free NA12878 data, the call set intersection in LCRs is noticeably smaller than in high-complexity regions (Fig. 3), which suggests the presence of other error sources in LCRs. In addition, PCR errors introduced during sample preparation are believed to affect SNPs to a lesser extent. The small intersections between CHM1 heterozygous SNP call sets (Fig. 2) and PCR-free SNP call sets in LCRs (Fig. 3) should be caused by other types of errors.
Fig. 3.

Relationship between NA12878 heterozygous call sets
3.2.2 Realignment errors
When mapping a read to the reference genome, a read mapper chooses the optimal pairwise alignment for each read independent of others. For reads mapped to the same region, the combination of optimal pairwise alignments does not always yield the optimal multi-alignment of reads. If a variant caller simply trusts the suboptimal multi-alignment, it may produce false variants or genotypes (Fig. 4). Therefore, more recent variant callers, including HaplotypeCaller, Platypus and FreeBayes in this study, heavily rely on realignment for both SNP and INDEL calling.
Fig. 4.

Example of misalignment around chr1:26608841 in CHM1. The truth allele is derived from local assembly. Three erroneous read alignments and their correct alignments are shown below it. Each of the three reads is an exact substring of the truth allele, but their alignments are different. The first read ‘errRead1’ is aligned without gaps, as the 3′ end of the read is a substring of the 18 bp deletion. Read ‘errRead2’ is aligned with a 6 bp insertion, as this alignment is better than having two long deletions. Read ‘errRead3’ is also aligned without gaps but with seven mismatches. It is possible for an aligner to find its correct alignment given a small gap extension penalty. On this example, Bowtie2 did not align any reads with gaps. BWA-MEM aligned four reads correctly. Except HaplotypeCaller which locally assembled reads, other callers all called multiple heterozygotes around this region
However, with our manual review, we found that variant callers often failed to produce the optimal realignment in LCRs. About 50–70% of the reviewed >1 bp heterozygous INDELs from CHM1 can be corrected away with better realignment. Without the thorough understanding of the very details of the realignment process, we are unable to explain why the callers fail even on some obvious cases. Nonetheless, as we can often manually derive a better multi-alignment, it is possible that a good realignment algorithm may replace our manual work and achieve higher accuracy than all the tools in our evaluation.
In the process of manual review, we found local assembly with fermi is frequently more effective than the INDEL callers, which may be because of the independence of the reference sequence, the requirement of long-range consistency and the more powerful topology-based error cleaning (Zerbino and Birney, 2008). Some difficult errors such as Figure 4 are trivial to resolve with local assembly.
3.3 The maximum read depth filter
3.3.1 The effectiveness of the MD filter
Other filters require a threshold on a single value. To study which filter, in addition to LC, is more effective, we used a receiver operating characteristic (ROC)-like plot, as shown in Figure 5. In this figure, the X-axis indicates the number of heterozygous SNPs in CHM1, which is proportional to the FPR; the Y-axis indicates the difference of the number of heterozygous SNPs between NA12878 and CHM1, which serves as a proxy to the sensitivity. Similar to a standard ROC plot, a curve closer to the top left corner implies a better classifier of errors.
Fig. 5.

Effect of filters after removing variants in LCRs. Each filter is associated with one value. For each filter, the number of heterozygous SNPs called from CHM1 and NA12878 are counted accumulatively from the most stringent threshold on the filter value to the most relax threshold. Thresholds are chosen such that they approximately evenly divide variants into 100 bins. Each chosen threshold yields a point in the plot. An arrow points to a point on the MD curve when the corresponding read depth is right above , where d is the mean read depth across called variants
Figure 5 implies that the MD filter is the most effective against false heterozygotes, especially those found from the BWA-MEM alignment. On our data with depth , a maximum depth threshold between and removes many false positives with little effect on the sensitivity. These false positives are mostly caused by copy number variations (CNVs) or paralogous sequences not present in the human reference genome.
3.3.2 The difference between Bowtie2 and BWA-MEM alignment
It is clear that Bowtie2 is less affected by the presence of CNVs and an incomplete genome (Figs 1 and 5). With manual review, it seems to us that in comparison with BWA-MEM, Bowtie2 tends to give the same alignment a lower mapping quality when the read has other suboptimal hits. At the same time, missing paralogous sequences from the reference genome is often associated with existing segmental duplications in the reference genome. Therefore, Bowtie2 is more likely to correctly give a low mapping quality to a read from these paralogous sequences. As variant callers usually distrust mismatches on alignments with low mapping quality, their calls from the Bowtie2 alignment are less susceptible to CNVs or an incomplete reference genome.
However, being conservative on the mapping quality estimate may lead to more false negatives. For example, we found a read pair having one mismatch around 13.7 Mb in chr1 but two mismatches around 13.5 Mb. Both Bowtie2 and BWA-MEM mapped the ends of the pair at the same positions. Bowtie2 gives the pair a mapping quality 6, whereas BWA-MEM gives a mapping quality 27. The similar scenario happens to the other reads mapped to this region. As a result, a SNP is called from the BWA-MEM alignment but not from the Bowtie2 alignment. Variant callers usually call more variants from the BWA-MEM alignment (Fig. 1), many of which are located in segmental duplications.
Another difference, not relevant to the mapping quality, comes from the alignment around long INDELs. HaplotypeCaller always called ≥15 bp INDELs from the BWA-MEM alignment (data not shown). Other callers made three times as many ≥15 bp deletion calls from the BWA-MEM alignment, either in LCRs or not, and called 40% more insertions outside LCRs. Interestingly, except HaplotypeCaller, others called more ≥15 bp insertions from the Bowtie2 alignment in LCRs instead. We have not found a good explanation to the apparently conflictive observations.
3.3.3 An alternative to the MD filter
While the MD filter is effective against false heterozygotes, it is only applicable to high-coverage data with uniform read depth. It does not work with exome sequencing data, or is not powerful on data with shallow coverage.
To overcome the limitation, we derived an alternative filter. We obtained unfiltered SAMtools SNP calls from the 1000 Genomes Project and computed the inbreeding coefficient and the Hardy–Weinberg P-value using genotype likelihoods (Li, 2011b). We extracted sites satisfying: (i) the reported read depth above 25 000; (ii) the inbreeding coefficient <0; (iii) the P-value <. We then clustered the sites within 10 kb into regions. These regions are susceptible to common CNVs or artifacts in the reference genome. We call this filter as the Hardy–Weinberg filter or HW in brief.
On CHM1, the HW filter is almost as effective as the MD filter. It could be a valid alternative when the MD filter cannot be applied. However, the derivation of the HW filter requires multiple thresholds and depends on populations, the mapper (BWA) and the caller (SAMtools). Therefore, we decided to use the much simpler MD filter here.
3.4 Other filters
The remaining filters, including AB, DS, FS and QU (Section 2.4), can filter additional false heterozygous called from CHM1, but their effectiveness varies with call sets. It is also difficult to find the optimal thresholds on these filters as they affect both the false-negative rate and the FPR. In the end, we arbitrarily chose reasonable thresholds based on the ROC-like curves (Fig. 5), which may not be optimal for all call sets.
3.5 Effect of PCR duplicates
Twenty percent of CHM1 data are discarded in our analysis because of PCR duplicates. We have also tried variant calling with mem:hc without the MarkDuplicates step. Before filtering, this approach yields 3% more heterozygous SNPs and 12% more heterozygous INDELs, suggesting INDELs are more susceptible to PCR artifacts than SNPs. After filtering, the total numbers of SNPs and INDELs are about the same with or without duplicates.
3.6 Effect of the reference genome
In this work, we mapped reads to hs37d5, the reference genome used by the 1000 Genomes Project. This reference genome contains extra 35.4 Mb sequences present in several de novo assemblies but likely to be missing from the primary assembly of GRCh37. These sequences are supposed to attract many mismapped reads, so are called as decoy sequences.
We have also mapped the CHM1 reads to the GRCh37 and GRCh38 primary assemblies and called variants. The number of homozygous non-LC SNPs called from each reference is close: 2.408, 2.405 and 2.412 million from GRCh37, hs37d5 and GRCh38, respectively. However, the numbers of heterozygous SNPs/INDELs are distinct (Fig. 6). We called twice as many heterozygotes from GRCh37 in comparison with hs37d5. This indicates that the 35.4 Mb decoy sequences attracted many mismapped reads and consequently improved the variant calls in chromosomal regions. GRCh38 further resolves 39.8 k (=36.9 k + 2909) heterozygotes called from hs37d5. However, it also retains 36.8 k heterozygotes called from GRCh37 but not from hs37d5. Intriguingly, GRCh38 further adds 24.6 k autosomal heterozygotes not called from GRCh37 or hs37d5. We are unclear of the source of these false heterozygous SNPs. In general, we conclude that hs37d5 and GRCh38 are more complete than GRCh37.
Fig. 6.

Relationship of CHM1 heterozygous SNPs called from mappings to different reference genomes. CHM1 reads were mapped with BWA-MEM. Autosomal SNPs were called with GATK HaplotypeCaller and passed the LC filter. Heterozygous calls from GRCh38 were lifted to GRCh37 with the liftOver tool from UCSC under the default setting
4 DISCUSSIONS AND CONCLUSIONS
A distinct feature of our work is the use of a haploid human sample, CHM1, from which heterozygous calls are supposed to be errors. This allows us to unbiasedly investigate the causal artifacts and to experiment effective filters with the diploid NA12878 dataset as a positive control.
When we called SNPs and INDELs from CHM1, we were surprised to find 10% of raw variant calls were heterozygotes. Honestly, our immediate reaction was that CHM1 was not truly haploid. However, after careful analysis, we have convinced ourselves that the heterozygosity of CHM1 should be of an order of magnitude lower than the raw error rate of variant calling. The vast majority of heterozygotes are calling errors. In the raw call set, we usually see an error per 10–15 kb.
It was also to our surprise that the LC is the most effective against false heterozygotes, especially short INDELs. Although we knew that INDEL errors may be introduced by PCR during sample preparation, we underestimated its substantial effect. We were also unaware that realignment of INDELs in LCRs remains a great challenge even after the many existing efforts in this direction (Albers et al., 2011; Homer and Nelson, 2010; Li, 2011a; Narzisi et al., 2014). Without the suggestion from (P.Sudmant, personal communication), we would not have tried this filter.
Before we understand and resolve the issues in variant calling in LCRs, it might be better to filter out all variants overlapping these regions. Although >50% of single-sample INDEL calls fall in LCRs (Figs 1 and 3), only 1.25% of autosomal INDELs in the ClinVar database (http://clinvar.com) overlap with LCRs––most INDELs in LCRs have unknown clinical functionality. For certain applications, it might be safe to drop or downweigh these difficult calls.
Outside LCRs, different call sets usually agree well with each other if the same set of filters is applied (Fig. 3). Based on Figure 1, we estimate that a caller usually makes a wrong call per 100–200 kb without significant compromise on the sensitivity, similar to the previous estimates (Bentley et al., 2008; Nickles et al., 2012). Many of these errors are likely to be systematic. In the context of somatic or de novo mutation discovery by sample contrast, systematic errors will appear in all samples. They will not lead to false mutation calls, fortunately.
A simple method to improve the variant accuracy is to use two distinct pipelines, take the intersection of the raw calls and then apply caller-oblivious filters to derive the final call set. As callers agree well on post-filtered sites (Fig. 3) but badly on false positives (Fig. 2), we should be able to remove most errors without much hit to the sensitivity. Such a consensus approach has been applied to cancer data with limited success (Goode et al., 2013; Löwer et al., 2012). Without subclonal mutations, it should be much more effective on the variant discovery from normal samples.
Finally, the advances in sequencing technologies lead to the development of algorithms. We are heavily relying on mapping-based variant calling because with short reads or at low coverage, the traditional assembly-and-mapping approach would not work. With increased read lengths and decreased sequencing cost, we might go back to de novo assembly. An assembly does not only encode small variants but also retains large-scale structural variations and is free of the artifacts in the reference genome. Another possible direction which we mentioned 4 years ago (Li et al., 2010) is to map sequence reads to the ensemble of multiple genomes. Recently, there has been significant progress toward this goal (Paten et al., 2014; Sirén et al., 2010), but a practical solution is yet to be concluded.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Richard Wilson and his team who sequenced the CHM1 cell line and granted us the permission to use the data in this study. The authors also thank the 1000 Genomes Project analysis group for the helpful comments and discussions, and Mike Lin and the anonymous reviewers whose comments have helped us to improve the manuscript.
Funding: NHGRI U54HG003037; NIH GM100233.
Conflict of Interest: none declared.
REFERENCES
- 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers CA, et al. Dindel: accurate indel calls from short-read data. Genome Res. 2011;21:961–973. doi: 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland JF, et al. The new sequencer on the block: comparison of Life Technology’s Proton sequencer to an Illumina HiSeq for whole-exome sequencing. Hum. Genet. 2013;132:1153–1163. doi: 10.1007/s00439-013-1321-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CD, Eichler EE. Properties and rates of germline mutations in humans. Trends Genet. 2013;29:575–584. doi: 10.1016/j.tig.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AY, et al. Assessing single nucleotide variant detection and genotype calling on whole-genome sequenced individuals. Bioinformatics. 2014;30:1707–1713. doi: 10.1093/bioinformatics/btu067. [DOI] [PubMed] [Google Scholar]
- Clark MJ, et al. Performance comparison of exome DNA sequencing technologies. Nat. Biotechnol. 2011;29:908–914. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, et al. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. 2012 arXiv:1207.3907. [Google Scholar]
- Goode DL, et al. A simple consensus approach improves somatic mutation prediction accuracy. Genome Med. 2013;5:90. doi: 10.1186/gm494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gymrek M, et al. lobSTR: a short tandem repeat profiler for personal genomes. Genome Res. 2012;22:1154–1162. doi: 10.1101/gr.135780.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harismendy O, et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homer N, Nelson SF. Improved variant discovery through local re-alignment of short-read next-generation sequencing data using SRMA. Genome Biol. 2010;11:R99. doi: 10.1186/gb-2010-11-10-r99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs PA, et al. Mechanism of origin of complete hydatidiform moles. Nature. 1980;286:714–716. doi: 10.1038/286714a0. [DOI] [PubMed] [Google Scholar]
- Kim SY, Speed TP. Comparing somatic mutation-callers: beyond Venn diagrams. BMC Bioinformatics. 2013;14:189. doi: 10.1186/1471-2105-14-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam HYK, et al. Detecting and annotating genetic variations using the HugeSeq pipeline. Nat. Biotechnol. 2012a;30:226–229. doi: 10.1038/nbt.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam HY, et al. Performance comparison of whole-genome sequencing platforms. Nat. Biotechnol. 2012b;30:78–82. doi: 10.1038/nbt.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DE, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–317. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Improving SNP discovery by base alignment quality. Bioinformatics. 2011a;27:1157–1158. doi: 10.1093/bioinformatics/btr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011b;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Exploring single-sample SNP and INDEL calling with whole-genome de novo assembly. Bioinformatics. 2012;28:1838–1844. doi: 10.1093/bioinformatics/bts280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013 arXiv:1303.3997. [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. A survey of sequence alignment algorithms for next-generation sequencing. Brief Bioinform. 2010;11:473–483. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, et al. SOAPindel: efficient identification of indels from short paired reads. Genome Res. 2013;23:195–200. doi: 10.1101/gr.132480.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, et al. Variant callers for next-generation sequencing data: a comparison study. PLoS One. 2013;8:e75619. doi: 10.1371/journal.pone.0075619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löwer M, et al. Confidence-based somatic mutation evaluation and prioritization. PLoS Comput. Biol. 2012;8:e1002714. doi: 10.1371/journal.pcbi.1002714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, et al. Sequence-specific error profile of illumina sequencers. Nucleic Acids Res. 2011;39:e90. doi: 10.1093/nar/gkr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narzisi G, et al. Accurate detection of de novo and transmitted indels within exome-capture data using micro-assembly. bioRxiv. 2014 doi: 10.1038/nmeth.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickles D, et al. In depth comparison of an individual’s DNA and its lymphoblastoid cell line using whole genome sequencing. BMC Genomics. 2012;13:477. doi: 10.1186/1471-2164-13-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rawe J, et al. Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med. 2013;5:28. doi: 10.1186/gm432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paten B, et al. Mapping to a reference genome structure. 2014 arXiv:1404.5010. [Google Scholar]
- Robasky K, et al. The role of replicates for error mitigation in next-generation sequencing. Nat. Rev. Genet. 2014;15:56–62. doi: 10.1038/nrg3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts ND, et al. A comparative analysis of algorithms for somatic SNV detection in cancer. Bioinformatics. 2013;29:2223–2230. doi: 10.1093/bioinformatics/btt375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirén J, et al. Indexing finite language representation of population genotypes. CoRR. 2010 abs/1010.2656. [Google Scholar]
- Talwalkar A, et al. SMaSH: a benchmarking toolkit for human genome variant calling. 2013 doi: 10.1093/bioinformatics/btu345. arXiv:1310.8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- Wang J, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson IR, et al. Emerging patterns of somatic mutations in cancer. Nat. Rev. Genet. 2013;14:703–718. doi: 10.1038/nrg3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, et al. Integrating human sequence data sets provides a resource of benchmark snp and indel genotype calls. Nat. Biotechnol. 2014;32:246–251. doi: 10.1038/nbt.2835. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.