

Abstract
Muscular dystrophy (MD) is a disease characterized by skeletal muscle necrosis and the progressive accumulation of fibrotic tissue. While transforming growth factor (TGF)-β has emerged as central effector of MD and fibrotic disease, the cell types in diseased muscle that underlie TGFβ-dependent pathology have not been segregated. Here, we generated transgenic mice with myofiber-specific inhibition of TGFβ signaling owing to expression of a TGFβ type II receptor dominant-negative (dnTGFβRII) truncation mutant. Expression of dnTGFβRII in myofibers mitigated the dystrophic phenotype observed in δ-sarcoglycan-null (Sgcd−/−) mice through a mechanism involving reduced myofiber membrane fragility. The dnTGFβRII transgene also reduced muscle injury and improved muscle regeneration after cardiotoxin injury, as well as increased satellite cell numbers and activity. An unbiased global expression analysis revealed a number of potential mechanisms for dnTGFβRII-mediated protection, one of which was induction of the antioxidant protein metallothionein (Mt). Indeed, TGFβ directly inhibited Mt gene expression in vitro, the dnTGFβRII transgene conferred protection against reactive oxygen species accumulation in dystrophic muscle and treatment with Mt mimetics protected skeletal muscle upon injury in vivo and improved the membrane stability of dystrophic myofibers. Hence, our results show that the myofibers are central mediators of the deleterious effects associated with TGFβ signaling in MD.
INTRODUCTION
Muscular dystrophy (MD) is a disease characterized by skeletal muscle degeneration owing to myofiber necrosis followed by progressive accumulation of fibrotic tissue (1). Necrosis of myofibers in dystrophic skeletal muscle causes an inflammatory response resulting in secretion of multiple cytokines and growth factors, typically produced by macrophages, fibroblasts and surrounding viable myofibers (2). This enhanced cytokine profile is a two-edge sword, as it promotes resident satellite cell activation to repair the damaged fibers, while at the same time inducing fibroblast activation and fibrosis, as well as detrimental signaling in the remaining myofibers that likely leads to cell death (3–5). Transforming growth factor (TGF)-β has emerged as a primary secreted factor underlying the fibrotic response in MD. TGFβ is upregulated in dystrophic muscles from human patients and animal models (6–9), and inhibition of systemic TGFβ with a monoclonal blocking antibody reduces fibrosis and muscle pathology in MD (10,11). These observations suggest that TGFβ is a central effector of MD, although the role that TGFβ plays in the disease process is not characterized at the level of individual cell types within the muscle (i.e. myofibers versus fibroblasts).
Resident fibroblasts directly respond to TGFβ to initiate fibrosis, and their continual activation promotes the progressivity of MD over time (12). TGFβ also negatively affects skeletal muscle regeneration by exerting direct inhibitory effects on the resident stem cell pool (satellite cells) (13,14). TGFβ can inhibit differentiation of myoblasts into myotubes in culture, and attenuation of TGFβ activity improves the physiologic response of satellite cells following injury, leading to better regenerative capacity in vivo (2,15,16). However, TGFβ signaling is also important for myoblast proliferation and formation from satellite cells, indicating important physiologic roles for this cytokine in proper muscle development (17,18). While skeletal muscle myofibers and myoblasts express the TGFβ receptors and are direct targets of TGFβ signaling, the contribution that TGFβ signaling plays at the level of the myofiber to the dystrophic disease phenotype has not been directly analyzed. In the present study, we elucidate for the first time the role of adult myofibers as primary responders to TGFβ signaling in the dystrophic disease process. We show that specific inhibition of TGFβ signaling only in myofibers of adult skeletal muscle is sufficient to mitigate MD and improve skeletal muscle regeneration.
RESULTS
Generation of dominant-negative TGFβ-receptor type II-expressing transgenic mice
TGFβ is produced and activated in skeletal muscle during injury or in MD, where it is known to negatively affect aspects of healing and regeneration as well as promote fibrosis and disease (9,19). Here, we generated transgenic (TG) mice expressing the well-characterized TGFβ type II receptor dominant-negative (dnTGFβRII) truncation mutant to block TGFβ signaling specifically at the level of the myofiber using the skeletal α-actin muscle-specific promoter (Fig. 1A). This dominant-negative receptor lacks the intracellular kinase domain, yet it still effectively dimerizes with TGFβ type I receptors, thereby generating inactive complexes (20–22). Two independent TG lines were generated with high (TG line 2) and medium (TG line 1) expression (Fig. 1B). In line 1, which was selected for more in-depth analysis, we observed dnTGFβRII expression in every skeletal muscle analyzed but not the heart (Fig. 1C). H&E and Masson's trichrome-stained histological sections showed no obvious phenotype in the muscles of dnTGFβRII TG mice compared with wild-type (WT) littermates (Fig. 1D). Also, we did not observe changes in muscle weights normalized to tibia length at 6 months of age or in fiber size owing to the transgene (data not shown). Analysis of myofiber central nucleation showed a small increase in the quadriceps and tibialis anterior (TA) from TG mice at 6 months of age, although the other muscles analyzed showed no significant change (Fig. 1E). These subtle alterations in myofiber central nucleation are likely non-pathologic and may be the result of greater satellite cell activity, especially because dnTGFβRII TG mice had slightly but significantly better grip strength compared with WT littermates at both 2 and 6 months of age (Fig. 1F). Thus, expression of the dnTGFβRII on skeletal muscle fibers/myoblasts did not induce overt pathology although it did appear to slightly alter the physiology of the muscles in a more adaptive profile.
Figure 1.
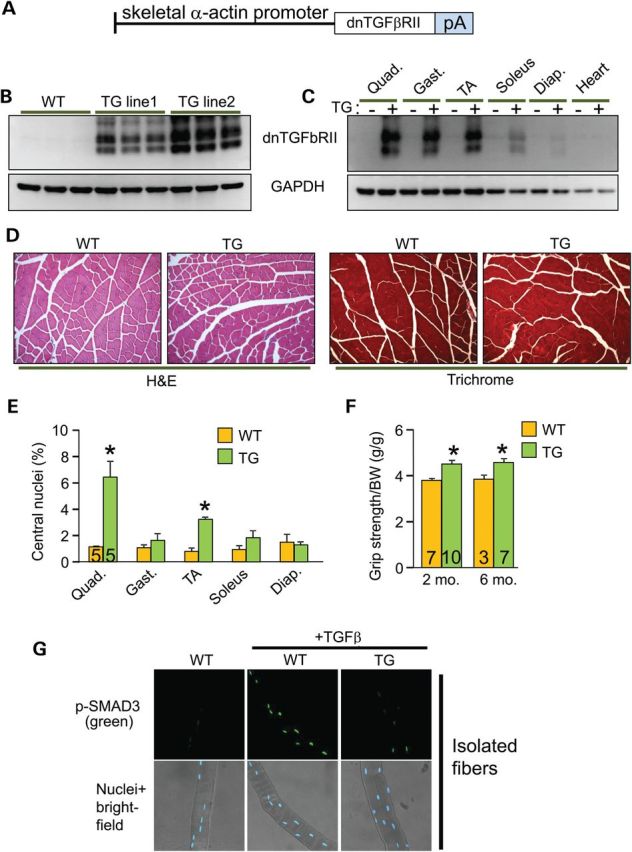
Generation of skeletal muscle-specific TG mice expressing a dominant-negative TGFβ-receptor. (A) Schematic representation of the transgene construct. pA is the abbreviation for polyadenylation sequence. (B) Western blot analysis for dnTGFβRII protein expression in quadriceps isolated from WT and two different TG mouse lines at 2 months of age. (C) Western blot analysis for dnTGFβRII protein expression in different muscle groups isolated from WT and TG mice at 2 months of age. Quad, quadriceps; Gast, gastrocnemius; TA, tibialis anterior; Diap, diaphragm. (D) H&E and Masson's trichrome-stained histological sections of quadriceps from 6-month-old mice of the indicated genotypes. Original magnification is ×200. (E) Percentage of myofibers from histological sections of the indicated muscles with centrally located nuclei from WT and TG mice at 6 months of age. (F) Measurement of grip strength in the indicated genotypes of mice at 2 and 6 months of age. *P < 0.05 versus WT controls. The number of mice used is shown in each of the bars in each panel. (G) Immunocytochemistry for phospho-SMAD3 (top row) in WT or TG myofibers isolated from the EDL and left untreated or stimulated with TGFβ for 20 min. The bottom row shows bright field images of the same fibers and nuclei in blue.
To address the in vivo effectiveness of the dnTGFβRII transgene, we isolated primary myofibers from the extensor digitorum longus (EDL) muscle of TG versus WT mice and stimulated them with TGFβ for analysis of phospho-SMAD3 levels and nuclear accumulation. The data show robust activation in WT myofibers, but almost no activation in myofibers from the TG mice (Fig. 1G). Importantly, muscle interstitial cells (mostly fibroblasts) and isolated myoblasts from these same two groups of mice showed equal activation of SMAD3 phosphorylation in response to TGFβ, indicating that the dnTGFβRII transgene had no residual inhibitory effect on these cell types from muscle (Supplementary Material, Fig. S1A and B).
Myofiber-specific inhibition of TGFβ signaling mitigates MD histopathology
To assess how the myofibers contribute to TGFβ-dependent MD, we crossed the dnTGFβRII transgene into the δ-sarcoglycan-null (Sgcd−/−) genetic background, the latter of which is a mouse model with fulminant dystrophic disease (23,24). We first confirmed activation of TGFβ in the Sgcd−/− model of MD by ELISA (Supplementary Material, Fig. S2) as well as showed that the dominant-negative receptor blocked SMAD3 phosphorylation in myofibers from these dystrophic mice, which is normally highly activated with MD (Fig. 2A and B) (9,25). Interstitial cells showed equal SMAD3 phosphorylation levels from muscle of WT versus TG mice, again suggesting specificity of the transgene for TGFβ signaling within the myofibers (Fig. 2B). Inhibition of TGFβ signaling with this transgene reduced histopathology in Sgcd−/− mice (Fig. 2C) with a normalization of the pseudohypertrophy typically observed in the quadriceps, gastrocnemius and TA (Fig. 2D), as well as a significant reduction in central nucleation in every muscle analyzed except the diaphragm (Fig. 2E). The slightly less-potent effect in diaphragm and soleus is likely due to the lower expression of the transgene in those muscles owing to the greater content of slow fibers, where the skeletal α-actin promoter is less active (Fig. 1C). Despite these protective histologic features associated with inhibition of TGFβ signaling within the myofibers, quantification of total muscle fibrosis by analysis of hydroxyproline content revealed no significant changes in the Sgcd−/− mice with or without the transgene (Fig. 1F). Analysis of collagen expression by quantitative PCR confirmed equal collagen induction in Sgcd−/− and Sgcd−/− TG muscles (Supplementary Material, Fig. S3). These observations suggest that the primary role of TGFβ signaling in the myofibers is independent from the well-established pro-fibrotic effect of TGFβ, which is likely more dependent on fibroblasts that do not express the dominant-negative transgene.
Figure 2.
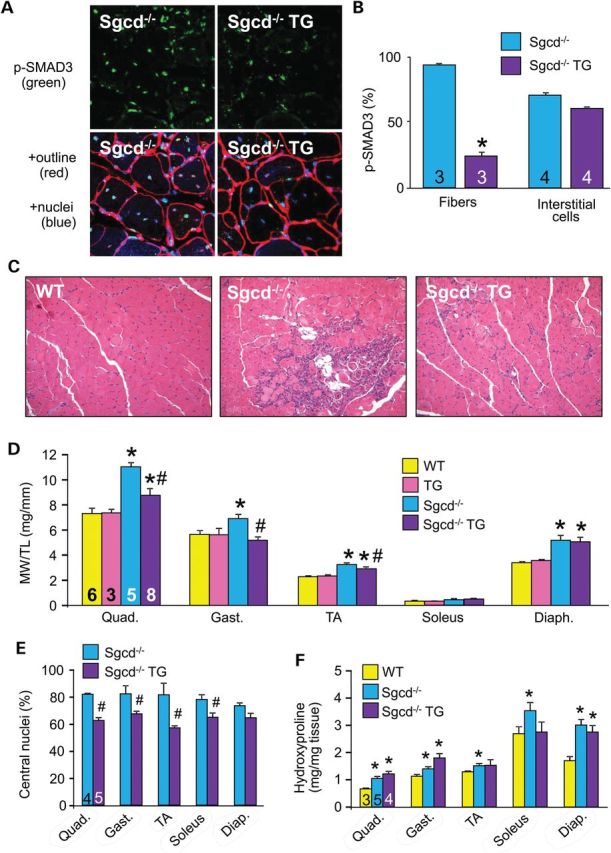
Myofiber-specific inhibition of TGFβ signaling reduces MD histopathology. (A) Representative immunohistochemical images for phospho-SMAD3 in quadriceps (green, top panels) sections from 2-month-old Sgcd−/− mice versus Sgcd−/− mice crossed with the dnTGFβRII transgene (Sgcd−/− TG). The lower panels show blue staining of nuclei (DAPI) and red staining of the myofiber outlines with WGA-TRITC. Original magnification is ×400. (B) Quantification of phospho-SMAD3-positive fibers and interstitial cells from histological sections stained as shown in A, from the indicated genotypes. (C) Representative H&E-stained histological sections from quadriceps of 2-month-old mice of the indicated genotypes. Original magnification is ×200. (D) MW/TL from the indicated muscle groups of mice at 2 months of age. (E) Percentage of myofibers with centrally located nuclei from mice at 2 months of age from the indicated muscles in Sgcd−/− versus Sgcd−/− TG mice. (F) Measurement of hydroxyproline content for fibrosis from the indicated muscle groups of mice at 2 months of age of the indicated genotypes. *P < 0.05 versus WT controls; #P < 0.05 versus Sgcd−/− mice. The number of mice used is shown in the bars within each panel.
Myofiber-specific inhibition of TGFβ improves MD by enhancing membrane integrity
Here, we subjected mice to forced treadmill running to assess whether the observed improvement in histopathology conferred by the muscle-specific dnTGFβRII transgene correlated with functional benefits. Indeed, Sgcd−/− mice with the dnTGFβRII transgene showed a restoration of their ability to run on a treadmill comparable with WT control mice at both 2 and 6 months of age, whereas Sgcd−/− mice alone were severely compromised (Fig. 3A). In addition, Sgcd−/− TG mice showed a significant reduction in total serum creatine kinase (CK) levels at 2 and 6 months of ages compared with Sgcd−/− mice, suggesting less myofiber membrane ruptures and ongoing myofiber necrosis (Fig. 3B). To further investigate membrane integrity, we employed the Evans blue dye (EBD) uptake assay after forced treadmill running. Using this assay, Sgcd−/− mice showed as much as 30% fiber positivity, whereas WT mice showed no signal (Fig. 3C and D and data not shown). However, the presence of the dnTGFβRII transgene dramatically reduced EBD uptake in the muscles of Sgcd−/− mice, suggesting enhanced membrane stability (Fig. 3C and D). To more directly and acutely evaluate membrane integrity, we employed an assay in which FM1-43 fluorescence dye entry was assessed in isolated myofibers from the flexor digitorium brevis (FDB) after laser-induced damage. Compared with WT control, fibers from Sgcd−/− mice showed significantly greater fluorescence dye entry, which is a typical feature of dystrophic fibers and their greater membrane fragility (Fig. 3E and F). However, the presence of the dnTGFβRII transgene rescued the membrane fragility in Sgcd−/− mice, bringing the levels back to WT control (Fig. 3E and F). These results collectively suggest that inhibiting TGFβ signaling specifically in myofibers increased cell membrane stability in the context of MD.
Figure 3.
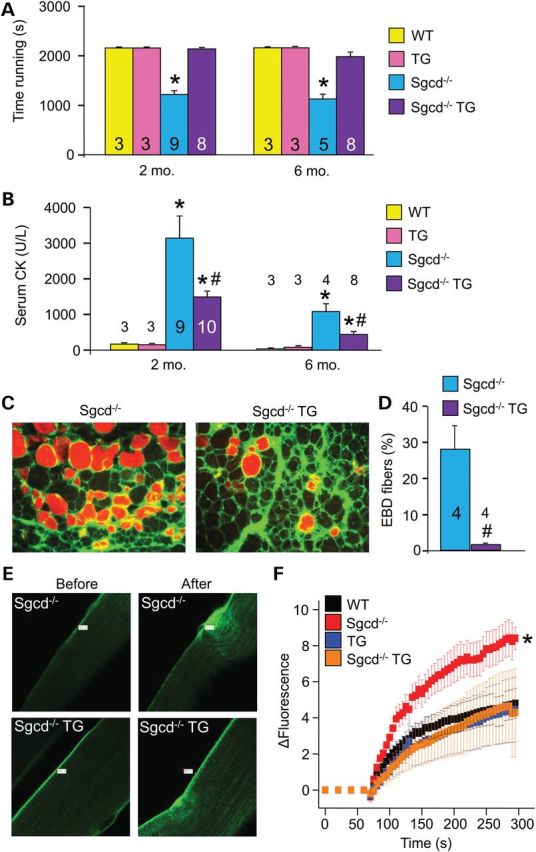
Myofiber-specific inhibition of TGFβ signaling improves muscle performance and protects the sarcolemmal membrane in MD. (A) Time to fatigue in seconds with forced downhill treadmill running in the indicated groups of mice at 2 and 6 months of age. (B) Quantification of CK levels in the blood of the indicated groups of mice at 2 and 6 months of age. (C and D) Representative immunofluorescence images and quantitation of EBD (red) uptake in histological sections from quadriceps of 2-month-old mice subjected to running. Membranes are stained with wheat germ agglutinin (WGA-FITC, green). Original magnification is ×400. (E and F) Representative images and quantitation of FM1-43 dye entry (green fluorescence) in isolated FDB myofibers from the indicated genotypes of mice before and after laser-induced injury. Eight or more fibers were analyzed from each genotype. *P < 0.05 versus WT controls; #P < 0.05 versus Sgcd−/− mice. The number of mice used is shown in the bars of each panel.
Myofiber-specific inhibition of TGFβ protects skeletal muscle from acute injury
To more directly assess myofiber integrity and regenerative potential associated with the dnTGFβRII transgene, we used cardiotoxin (CTX) to acutely injure and cause focal necrosis of the TA muscle. H&E-stained histological sections showed improved muscle architecture with less myofiber death and more newly forming nascent myofibers in TG muscles compared with WT controls 5 days after CTX injury, which also protected against loss in muscle weights (Fig. 4A and B). By Day 14, WT muscle had fully recovered from the injury event, similar to TG muscle (Fig. 4A). New myofiber generation after CTX injury is driven by resident satellite cells that ultimately differentiate into mature myofibers (26). Both newly formed and repaired myofibers re-express developmental proteins such as embryonic myosin heavy chain (eMHC), which is typically used as a marker for this process. Evaluation of eMHC re-expression upon CTX injury revealed higher levels in dnTGFβRII TG muscles compared with WT controls (Fig. 4C and D). The enhanced profile was associated with increased satellite cell numbers in TG fibers from the EDL (Supplementary Material, Fig. S4A), which was confirmed by increased expression of the satellite cell marker Pax7 from total muscle extracts (Supplementary Material, Fig. S4B). These results confirmed that inhibiting TGFβ signaling in myofibers not only protects some fibers from degeneration in the first place (greater membrane stability) but it also enhances satellite cell numbers and activity, leading to an improved regenerative profile after acute injury.
Figure 4.
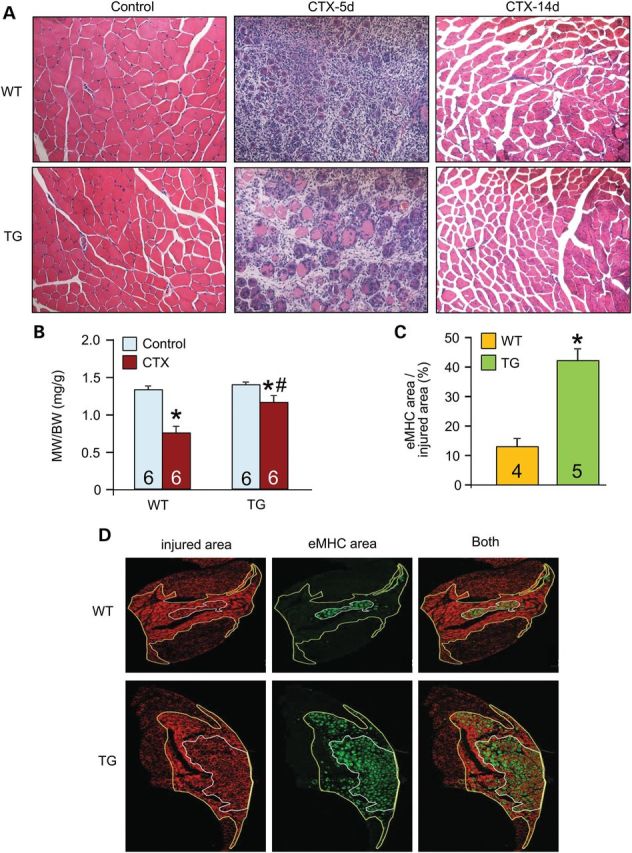
Myofiber-specific inhibition of TGFβ signaling protects skeletal muscle upon CTX injury. (A) Representative H&E-stained histological sections from CTX injury to the TA muscle in WT or TG mice 5 and 14 days afterward. Original magnification is ×200. (B) TA muscle weights (MW) normalized to body weight (BW) from the indicated genotypes of mice with vehicle control or CTX injection. (C and D) Histological sections and quantification of eMHC-positive area (green) within the greater injury area (dark red) of the TA muscle 5 days after CTX injury. The white outline shows the regenerating area whereas the yellow outline is the entire area of injury. Original magnification is ×40. *P < 0.05 versus WT control; #P < 0.05 versus CTX-injured WT mice. The number of mice used is shown in the bars of each panel.
Altered metallothionein expression underlies TGFβ effects on myofibers
To examine the mechanism whereby muscle-specific inhibition of TGFβ signaling is protective, we performed a genome-wide transcriptome analysis of dnTGFβRII TG and WT control muscles by RNA-seq (Supplementary Material, Table S1). This analysis revealed metallothionein (Mt) family members (Mt1, Mt2 and Mt3) as the most highly upregulated genes in muscle from dnTGFβRII TG mice, which was confirmed by quantitative PCR (Fig. 5A–C). Mts are pro-regenerative and antioxidant proteins involved in protection against free radicals and heavy metals (27–29). Interleukin-6 (IL6) and zinc are known to induce Mt expression (29), a feature that we utilized to better evaluate the mechanistic linkage with TGFβ signaling. C2C12 myotubes were treated with recombinant TGFβ, with or without zinc or IL6. TGFβ was sufficient to inhibit expression of Mt1, Mt2 and Mt3 at baseline and following exposure to the inducers zinc and IL6 (Fig. 5D–I).
Figure 5.

Altered metallothionein mRNA expression underlies TGFβ effects on myofibers. (A–C) Real-time PCR from quadriceps muscle of WT and TG mice for metallothionein 1, 2, 3 (Mt1, Mt2, Mt3) mRNA expression normalized to Rpl7 control mRNA. *P < 0.05 versus WT. (D–I) Real-time PCR from C2C12-derived myotubes with vehicle control (con) or treated with recombinant TGFβ (TGF), recombinant interleukin-6 (IL6) or zinc (Zn) for Mt1, Mt2 and Mt3 mRNA expression normalized to Rpl7 control mRNA. *P < 0.05 versus control; #P < 0.05 versus Zn or IL6 treatment. The number of mice or biological replicates used is shown in each of the graphs.
Inhibition of both canonical and non-canonical TGFβ pathways by adenoviral-mediated overexpression of the inhibitory SMAD6/7 protein or dominant-negative MKK3/6 adenoviruses [inhibit p38 mitogen-activated protein kinase (MAPK)] produced an enhanced profile of Mt1-3 gene expression following Zn and Zn + TGFβ treatment (Fig. 6A–C). However, inhibition of extracellular signal-regulated kinases 1/2 (ERK1/2) with a dominant-negative MEK1 adenovirus was ineffective, whereas inhibition of c-Jun N-terminal kinases 1/2 (JNK1/2) with adenovirus expressing dominant-negative mutant proteins was only partially effective (Fig. 6A–C). The dnMKK3/6 adenoviruses also induced Mt1 and Mt2 expression at baseline (Fig. 6A–C). Finally, considering the antioxidant properties of Mts, we examined whether TGFβ affects reactive oxygen species (ROS) levels in C2C12 cells. To this end, equal numbers of myotubes were treated with TGFβ or vehicle control and ROS were measured by dihydroethidium (DHE) staining, which showed a significant increase with TGFβ treatment (Fig. 6D and E). Collectively, these results suggest that TGFβ signaling utilizes both canonical and non-canonical pathways to regulate Mt expression in muscle cells, with an associated ROS component.
Figure 6.
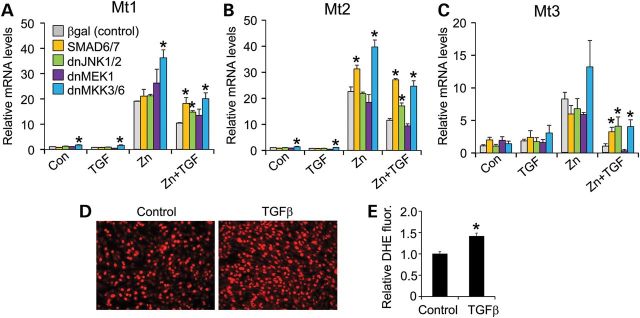
Signals involved in TGFβ-mediated inhibition of Mt expression. (A–C) Real-time PCR for Mt1, 2, 3 mRNA expression from C2C12 cells with no treatment (control) or treated with recombinant TGFβ (TGF) or zinc (Zn) with Adβgal control or AdSMAD6/7, Ad-dnJNK1/2, Ad-dnMEK1, Ad-dnMKK3/6 adenoviral infection. *P < 0.05 versus Adβgal. Data were normalized to Rpl7 control mRNA. (D and E) Representative images and quantitation of the ROS marker DHE (red) from cultured C2C12 myotubes treated with vehicle (control) or recombinant TGFβ. The same numbers of cells are present in each image. Original magnification is ×200.
Treatment with Mt mimetics protects skeletal muscle upon injury in vivo
Increased ROS is a signature of MD that can contribute to membrane fragility and subsequent myofiber necrosis (1,30,31). Here, we quantified ROS by DHE staining and observed that skeletal muscle from Sgcd−/− mice had an increased ROS signature that was significantly reduced by the dnTGFβRII transgene (Fig. 7A and B). Given the prominent induction of Mts associated with the dnTGFβRII transgene, we treated mice with an Mt mimetic, EMTIN B after CTX injury (Fig. 7C) (32). Treatment with EMTIN B protected the injured muscles and improved their regenerative capacity (Fig. 7D–G), reproducing many of the protective features of the dnTGFβRII transgene. We then evaluated the efficacy of an EMTIN B-enriched antioxidant mixture (AO) to protect isolated dystrophic fibers upon laser injury. The EMTIN/AO mix dramatically protected the Sgcd−/− fibers from rupturing after laser injury suggesting increased membrane stability (Fig. 7H and I). Taken together, these results reveal a mechanism whereby TGFβ-mediated pathology at the level of the myofiber regulates membrane integrity through a ROS-dependent mechanism.
Figure 7.
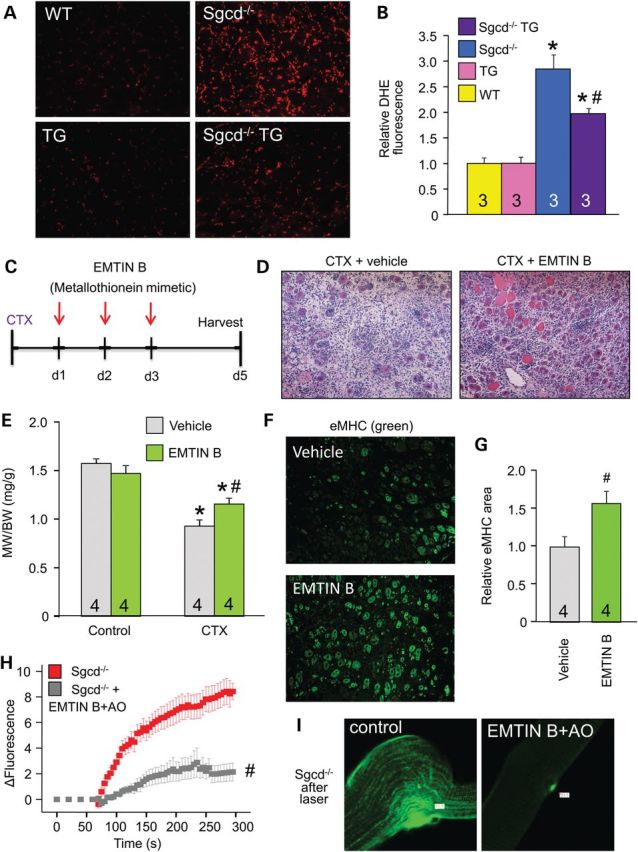
Treatment with Mt mimetics protects skeletal muscle upon injury. (A and B) Representative histological images and quantitation of the ROS marker DHE (red) from quadriceps sections of the indicated genotypes of mice. Original magnification is ×200. (C) Schematic of the protocol used for CTX injury to the TA along with EMTIN B treatment times. (D) Representative H&E-stained histological sections from the TA injured with CTX and treated with vehicle or EMTIN B, 5 days post-injury. Original magnification is ×200. (E) MW/BW from the indicated mouse treatment groups. (F and G) Representative immunohistochemical images and quantitation for eMHC expression (green) from CTX-injured TAs treated with vehicle or EMTIN B. (H and I) Time course and representative images of FM1-43 dye entry into EDL myofibers from Sgcd−/− mice with or without EMTIM B-enriched antioxidant mix (AO) following laser-induced injury. Eight or more fibers were analyzed each. *P < 0.05 versus WT or vehicle control; #P < 0.05 versus Sgcd−/− or CTX-injured vehicle-treated mice. The number of mice used is shown in each of the graphs.
DISCUSSION
TGFβ signaling underlies diverse cellular functions such as proliferation, differentiation, cell death and motility (33). The exact biologic response to TGFβ depends on the cell type and the context in which the signal is received (33). Although enhanced TGFβ activity is associated with MD and inhibition of its signaling has been proposed as a therapeutic strategy in this disease (2,34,35), the known mechanisms of action for this cytokine are highly pleiotropic with a number of beneficial effects; hence, a more cell-type selective inhibitory strategy might be necessary. Our results provide the first evidence that myofibers are central mediators of the deleterious effects associated with TGFβ signaling in skeletal muscle, both during acute injury of skeletal muscle and during MD.
Unlike post-mitotic tissues with little regenerative capacity, such as the heart or the brain, skeletal muscle robustly regenerates after injury owing to the activity of resident satellite cells (26). However, during MD, skeletal muscle regeneration progressively becomes impaired and excessive TGFβ activation and signaling is thought to be a primary mechanism underlying this effect (2). TGFβ can inhibit the activity of myoblasts to differentiate in culture, as well as mediating a progressive fibrotic response that eventually restricts the activity of satellite cells to form nascent myofibers (12,13). However, we did not observe differences in collagen deposition in our dnTGFβRII TG mice crossed to the Sgcd−/− model of MD, in agreement with an unchanged profile of TGFβ signaling in interstitial cells. Excessive TGFβ signaling also appears to enhance the death of skeletal muscle precursor cells and myoblasts, possibly through activation of p38 MAPK and/or JNK1/2 (36–39). Our observation that inhibition of TGFβ signaling at the level of the myofiber enhances membrane stability, thus lessening myofiber degeneration and even death with CTX injury, is consistent with past observations of less cell death or enhanced regeneration when TGFβ signaling is dampened (2,40,41). Preservation of membrane integrity is also crucial for skeletal muscle regeneration. Indeed, interaction between integrins on myofiber membranes and laminins in the matrix is crucial for proper satellite cell adhesion to the myofiber and for fusion of myoblasts with injured fibers (42).
Oxidative stress and ROS formation are important contributors to membrane fragility in the pathology of several of the MDs (30,31). Moreover, TGFβ signaling, which is enhanced in MD, is known to enhance ROS formation (43–46). Our unbiased expression analysis by RNA-seq revealed the Mts as being dramatically upregulated by the dnTGFβRII transgene, although many other candidate genes were also changed in expression suggesting additional mechanisms at play. However, Mts appear to favor regeneration in both brain and heart (47–49), and their antioxidant properties appear to be a main determinant in protecting tissue from injury as well as enhancing regenerative activity by creating a more favorable environment for stem cells (50,51). Indeed, we observed that TGFβ had a direct inhibitory effect on both baseline and induced Mt expression and that inhibition of TGFβ signaling within the myofibers reduced ROS levels and was protective. We also showed that TGFβ treatment was sufficient to elevate ROS in C2C12 cells. Treatment of mice with EMTIM B promoted greater recovery after acute injury with CTX, and an EMTIM B antioxidant cocktail directly protected the degree of muscle membrane injury after acute laser damage. These results suggest that a properly formulated antioxidant therapy might protect MD patients during the course of their disease. However, clinical trials in humans have thus far failed to conclusively show efficacy of an antioxidant approach (52,53). These negative findings may be due to, in part, a lack of understanding of the precise mechanism responsible for ROS elevation and the ensuing cellular damage in skeletal muscle or it is possibly due to an ineffective therapeutic regimen to fully extinguish ROS in the proper locations where oxidative damage is most central to the disease.
Our data show that the myofibers are central mediators of the deleterious aspects of TGFβ signaling in muscle. TGFβ appears to enhance the ROS status within skeletal muscle fibers, which increases membrane fragility, leading to greater myofiber necrosis, and also reduced levels of effective satellite cell activity with regeneration. Hence, while global TGFβ inhibition may not be a desirable therapeutic strategy for treating human MD, in part owing to adverse systemic side effects as well as owing to inhibiting some beneficial aspects of TGFβ signaling within muscle, our results suggest that targeted inhibition of TGFβ at the level of the myofibers would be beneficial and protective against dystrophic disease.
MATERIAL AND METHODS
Ethics statement
All animal procedures and usage were approved by the Institutional Animal Care and Use Committee of the Cincinnati Children's Hospital Medical Center, protocol 2E11104. No human subjects were used or human tissue or cells.
Animals
A modified human skeletal α-actin promoter (54) was used to create TG mice overexpressing a truncated TGFβ type II receptor mutant (dnTGFβRII) (20). Two TG lines were generated and analyzed for experiments. Sgcd–/– mice were described previously (24).
Western blotting, ELISA and hydroxyl-proline assay
Western blot analysis of skeletal muscles homogenates was performed using standard procedures. TGFβRII was detected using an antibody purchased from Santa Cruz Biotechnology (sc-33929). ELISA assays were performed on muscle homogenates using an ELISA kit for TGF-β1 purchased from R&D Systems (SMB100B). Hydroxyproline content was performed as previously described (55).
Fiber isolation and cell culture
Muscle fibers were isolated by incubation of the EDL muscle in 0.2% collagenase (Type I, Sigma–Aldrich, C-0130) for 1 h after careful removal of the fascia. Following collagenase treatment, muscles were transferred to DMEM (Gibco Invitrogen Life Technologies; 11995065) containing 10% horse serum and triturated to release individual fibers. Interstitial cells were obtained from the EDL fiber isolation procedure by plating the supernatant derived after trituration and seeding of the fibers. Myoblasts were obtained by culturing isolated fibers in ECL Cell Attachment Matrix (Millipore, 08-110)-coated dishes for 3 days in F12 medium containing 10% bovine growth serum. Fibers, interstitial cells and myoblasts were treated with 10 ng/ml of recombinant TGFβ (R&D Systems; 101-b1-010) for 20 min prior to being processed for immunostaining. C2C12 cells were differentiated into myotubes in DMEM containing 2% horse serum. Forty-eight hours post-induction of differentiation, cells were treated with 10 ng/ml of recombinant TGFβ or 40 ng/ml recombinant IL6 (PeproTech; 216-16) for 15 h, and 100 μm ZnSO4 for 6 h. Overexpression of SMAD6, SMAD7, dnJNK1, dnJNK2, dnMKK3, dnMKK6, dnMEK1 or β-galactosidase was performed by recombinant adenoviral infection in differentiated C2C12 myotubes for 4 h the day before applying specific treatments. For ROS quantification, cells were incubated with DHE for 10 min at 37°C, and red fluorescence was visualized immediately afterward.
Histological analysis and immunohistochemistry
Muscles were paraffin-embedded and 5-μm histological sections were cut at the center of the muscle and stained with H&E or Masson's trichrome. Immunohistochemistry for eMHC was performed on cryosections within optimal cutting temperature compound (OCT)-embedded muscles using the antibody BF-G6 from Developmental Studies Hybridoma Bank. Antibody for laminin was purchased from Sigma–Aldrich (L9393). Antibody for vimentin (Sigma–Aldrich, V5255) was used to label interstitial cells whereas antibody for MyoD (BD Biosciences, 554130) was used to identify myoblasts. For ROS quantification, cryosections were incubated with DHE for 10 min at 37°C. Satellite cells were visualized in isolated EDL fibers using the antibodies against CD34 (rat anti-mouse CD34 monoclonal antibody; eBioscience, RAM34 14-0341-82) and Pax7 (mouse anti-chicken Pax7 monoclonal antibody; Developmental Studies Hybridoma Bank). Quantitation was performed on fibers fixed in 4% paraformaldehyde for 20 min at room temperature, washed three times for 20 min each in PBS, pH 7.4 and processed for immunohistochemistry. Absence of endothelial contamination was confirmed by staining with rat anti-mouse CD31 antibody (BD Biosciences; Clone MEC 13.3). Wheat germ agglutinin-TRITC was also used to show fiber outlines in the Phospho-SMAD3 immunohistochemistry experiments; nuclei are shown in blue with DAPI.
mRNA expression analysis
RNA was extracted from muscles and C2C12 cells using the RNeasy Kit according to manufacturer's instructions (Qiagen). Quadriceps-derived samples from two separate mice per group were submitted for RNAseq (University of Cincinnati sequencing and genome analysis core laboratory). For RNAseq data analysis, P-values were calculated using a negative binomial statistical model as implemented in DESeq [Bioconductor version: Release (2.14)], meanwhile false discover rates were used to obtain the adjusted P-values. For qPCR assays, RNA samples were retrotranscribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Selected gene differences were analyzed by real-time qPCR using SYBR green (Applied Biosystems). Quantified mRNA expression was normalized to Rpl7 and expressed relative to WT or untreated C2C12 controls.
Forced treadmill running and grip strength evaluation
Mice were placed in individual lanes of an electrically driven four-lane treadmill (Omni-Pacer LC4/M; Columbus Instruments International) at the speed of 6 m/min for 3 min. The treadmill measured 19 inches wide by 20 inches in length, with a conveyor belt measuring 3 inches wide by 12 inches in length. A training regimen was first instituted for 10 min to familiarize the mice with the environment and shock grids (adjustable from 0 to 2.0 mA). The speed was increased in increments of 2 m/min every 3 min to a maximum speed of 22 m/min. Exhaustion was assessed as >5 consecutive seconds on the shock grid without attempting to reengage the treadmill. Time spent on the treadmill before exhaustion or time to complete the protocol was recorded as average maximum time spent in exercise. The treadmill regimen was performed downhill. Grip strength test was performed using the Chatillon model DFIS-2 digital force gauge, and each animal was assessed 5 times consecutively on a given day.
CTX injury
CTX derived from Naja mossambica (Sigma) was dissolved in sterile saline to a final concentration of 10 μm, divided into aliquots and stored at −20°C. Mouse legs were shaved and cleaned with alcohol, and then, the TA muscle was injected with 50 μl of this CTX solution through a 26-gauge needle. EMTIN B (peptide SAGSCKCKESKSTS) was synthetized by Selleck Chemicals, dissolved in PBS and used intramuscular (50 ng/TA) once a day for 3 consecutive days post-CTX injury. PBS was used with the same regimen as vehicle control.
Laser-induced membrane injury assay
FDB muscles were excised from the foot and processed for laser-induced membrane injury assay as previously published method (56). The experiment was performed with and without adding EMTIN B at 10 ng/ml and an antioxidant supplement (A1345, from Sigma–Aldrich) on isolated fibers.
EBD uptake
Mice were injected with EBD (10 mg/ml, 0.1 ml/10 g body weight) and subjected to forced treadmill running 12 h later. Mice were then sacrificed, and quadriceps were embedded in OCT and snap-frozen in liquid nitrogen for viewing as previously described (57).
Statistics
All results are presented as mean ± SEM. Statistical analysis was performed with unpaired two-tailed t-test (for two groups) and one-way ANOVA with Bonferroni correction (for groups of three or more). P-values of <0.05 were considered significant.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from the NIH (J.D.M. and E.M.M.). J.D.M. was also supported by the Howard Hughes Medical Institute.
Supplementary Material
Acknowledgments
Conflict of Interest statement. None declared.
REFERENCES
- 1.Wallace G.Q., McNally E.M. Mechanisms of muscle degeneration, regeneration, and repair in the muscular dystrophies. Annu. Rev. Physiol. 2009;71:37–57. doi: 10.1146/annurev.physiol.010908.163216. [DOI] [PubMed] [Google Scholar]
- 2.Cohn R.D., van Erp C., Habashi J.P., Soleimani A.A., Klein E.C., Lisi M.T., Gamradt M., ap Rhys C.M., Holm T.M., Loeys B.L., et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 2007;13:204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Merly F., Lescaudron L., Rouaud T., Crossin F., Gardahaut M.F. Macrophages enhance muscle satellite cell proliferation and delay their differentiation. Muscle Nerve. 1999;22:724–732. doi: 10.1002/(sici)1097-4598(199906)22:6<724::aid-mus9>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 4.Lescaudron L., Peltekian E., Fontaine-Perus J., Paulin D., Zampieri M., Garcia L., Parrish E. Blood borne macrophages are essential for the triggering of muscle regeneration following muscle transplant. Neuromuscul. Disord. 1999;9:72–80. doi: 10.1016/s0960-8966(98)00111-4. [DOI] [PubMed] [Google Scholar]
- 5.Brunelli S., Rovere-Querini P. The immune system and the repair of skeletal muscle. Pharmacol. Res. 2008;58:117–121. doi: 10.1016/j.phrs.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 6.Iannaccone S., Quattrini A., Smirne S., Sessa M., de Rino F., Ferini-Strambi L., Nemni R. Connective tissue proliferation and growth factors in animal models of Duchenne muscular dystrophy. J. Neurol. Sci. 1995;128:36–44. doi: 10.1016/0022-510x(94)00219-e. [DOI] [PubMed] [Google Scholar]
- 7.Murakami N., McLennan I.S., Nonaka I., Koishi K., Baker C., Hammond-Tooke G. Transforming growth factor-beta2 is elevated in skeletal muscle disorders. Muscle Nerve. 1999;22:889–898. doi: 10.1002/(sici)1097-4598(199907)22:7<889::aid-mus12>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 8.Sun G., Haginoya K., Dai H., Chiba Y., Uematsu M., Hino-Fukuyo N., Onuma A., Iinuma K., Tsuchiya S. Intramuscular renin-angiotensin system is activated in human muscular dystrophy. J. Neurol. Sci. 2009;280:40–48. doi: 10.1016/j.jns.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 9.Bernasconi P., Torchiana E., Confalonieri P., Brugnoni R., Barresi R., Mora M., Cornelio F., Morandi L., Mantegazza R. Expression of transforming growth factor-beta 1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J. Clin. Invest. 1995;96:1137–1144. doi: 10.1172/JCI118101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brooke B.S., Habashi J.P., Judge D.P., Patel N., Loeys B., Dietz H.C., 3rd Angiotensin II blockade and aortic-root dilation in Marfan's syndrome. N. Engl. J. Med. 2008;358:2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andreetta F., Bernasconi P., Baggi F., Ferro P., Oliva L., Arnoldi E., Cornelio F., Mantegazza R., Confalonieri P. Immunomodulation of TGF-beta 1 in mdx mouse inhibits connective tissue proliferation in diaphragm but increases inflammatory response: implications for antifibrotic therapy. J. Neuroimmunol. 2006;175:77–86. doi: 10.1016/j.jneuroim.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Wynn T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olson E.N., Sternberg E., Hu J.S., Spizz G., Wilcox C. Regulation of myogenic differentiation by type beta transforming growth factor. J. Cell Biol. 1986;103:1799–1805. doi: 10.1083/jcb.103.5.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charge S.B., Rudnicki M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 15.Li Y., Foster W., Deasy B.M., Chan Y., Prisk V., Tang Y., Cummins J., Huard J. Transforming growth factor-beta1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: a key event in muscle fibrogenesis. Am. J. Pathol. 2004;164:1007–1019. doi: 10.1016/s0002-9440(10)63188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen R.E., Boxhorn L.K. Inhibition of skeletal muscle satellite cell differentiation by transforming growth factor-beta. J. Cell Physiol. 1987;133:567–572. doi: 10.1002/jcp.1041330319. [DOI] [PubMed] [Google Scholar]
- 17.Kollias H.D., McDermott J.C. Transforming growth factor-beta and myostatin signaling in skeletal muscle. J. Appl. Physiol. 2008;104:579–587. doi: 10.1152/japplphysiol.01091.2007. [DOI] [PubMed] [Google Scholar]
- 18.Mu X., Li Y. Conditional TGF-beta1 treatment increases stem cell-like cell population in myoblasts. J. Cell Mol. Med. 2011;15:679–690. doi: 10.1111/j.1582-4934.2010.01042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamazaki M., Minota S., Sakurai H., Miyazono K., Yamada A., Kanazawa I., Kawai M. Expression of transforming growth factor-beta 1 and its relation to endomysial fibrosis in progressive muscular dystrophy. Am. J. Pathol. 1994;144:221–226. [PMC free article] [PubMed] [Google Scholar]
- 20.Brand T., MacLellan W.R., Schneider M.D. A dominant-negative receptor for type beta transforming growth factors created by deletion of the kinase domain. J. Biol. Chem. 1993;268:11500–11503. [PubMed] [Google Scholar]
- 21.Yoo B.M., Yeo M., Oh T.Y., Choi J.H., Kim W.W., Kim J.H., Cho S.W., Kim S.J., Hahm K.B. Amelioration of pancreatic fibrosis in mice with defective TGF-beta signaling. Pancreas. 2005;30:e71–e79. doi: 10.1097/01.mpa.0000157388.54016.0a. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y.F., Feng J.A., Li P., Xing D., Zhang Y., Serra R., Ambalavanan N., Majid-Hassan E., Oparil S. Dominant negative mutation of the TGF-beta receptor blocks hypoxia-induced pulmonary vascular remodeling. J. Appl. Physiol. 2006;100:564–571. doi: 10.1152/japplphysiol.00595.2005. [DOI] [PubMed] [Google Scholar]
- 23.Durbeej M., Campbell K.P. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr. Opin. Genet. Dev. 2002;12:349–361. doi: 10.1016/s0959-437x(02)00309-x. [DOI] [PubMed] [Google Scholar]
- 24.Hack A.A., Lam M.Y., Cordier L., Shoturma D.I., Ly C.T., Hadhazy M.A., Hadhazy M.R., Sweeney H.L., McNally E.M. Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J. Cell Sci. 2000;113(Pt 14):2535–2544. doi: 10.1242/jcs.113.14.2535. [DOI] [PubMed] [Google Scholar]
- 25.Burks T.N., Cohn R.D. Role of TGF-beta signaling in inherited and acquired myopathies. Skelet. Muscle. 2011;1:19. doi: 10.1186/2044-5040-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Relaix F., Zammit P.S. Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development. 2012;139:2845–2856. doi: 10.1242/dev.069088. [DOI] [PubMed] [Google Scholar]
- 27.Leung Y.K., Pankhurst M., Dunlop S.A., Ray S., Dittmann J., Eaton E.D., Palumaa P., Sillard R., Chuah M.I., West A.K., et al. Metallothionein induces a regenerative reactive astrocyte phenotype via JAK/STAT and RhoA signalling pathways. Exp. Neurol. 2010;221:98–106. doi: 10.1016/j.expneurol.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 28.Leung J.Y., Bennett W.R., Herbert R.P., West A.K., Lee P.R., Wake H., Fields R.D., Chuah M.I., Chung R.S. Metallothionein promotes regenerative axonal sprouting of dorsal root ganglion neurons after physical axotomy. Cell Mol. Life Sci. 2012;69:809–817. doi: 10.1007/s00018-011-0790-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruttkay-Nedecky B., Nejdl L., Gumulec J., Zitka O., Masarik M., Eckschlager T., Stiborova M., Adam V., Kizek R. The role of metallothionein in oxidative stress. Int. J. Mol. Sci. 2013;14:6044–6066. doi: 10.3390/ijms14036044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terrill J.R., Radley-Crabb H.G., Iwasaki T., Lemckert F.A., Arthur P.G., Grounds M.D. Oxidative stress and pathology in muscular dystrophies: focus on protein thiol oxidation and dysferlinopathies. FEBS J. 2013;280:4149–4164. doi: 10.1111/febs.12142. [DOI] [PubMed] [Google Scholar]
- 31.Jung C., Martins A.S., Niggli E., Shirokova N. Dystrophic cardiomyopathy: amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc. Res. 2008;77:766–773. doi: 10.1093/cvr/cvm089. [DOI] [PubMed] [Google Scholar]
- 32.Ambjorn M., Asmussen J.W., Lindstam M., Gotfryd K., Jacobsen C., Kiselyov V.V., Moestrup S.K., Penkowa M., Bock E., Berezin V. Metallothionein and a peptide modeled after metallothionein, EmtinB, induce neuronal differentiation and survival through binding to receptors of the low-density lipoprotein receptor family. J. Neurochem. 2008;104:21–37. doi: 10.1111/j.1471-4159.2007.05036.x. [DOI] [PubMed] [Google Scholar]
- 33.Ikushima H., Miyazono K. Biology of transforming growth factor-beta signaling. Curr. Pharm. Biotechnol. 2011;12:2099–2107. doi: 10.2174/138920111798808419. [DOI] [PubMed] [Google Scholar]
- 34.Ceco E., McNally E.M. Modifying muscular dystrophy through transforming growth factor-beta. FEBS J. 2013;280:4198–4209. doi: 10.1111/febs.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heydemann A., Ceco E., Lim J.E., Hadhazy M., Ryder P., Moran J.L., Beier D.R., Palmer A.A., McNally E.M. Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice. J. Clin. Invest. 2009;119:3703–3712. doi: 10.1172/JCI39845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cencetti F., Bernacchioni C., Tonelli F., Roberts E., Donati C., Bruni P. TGFbeta1 evokes myoblast apoptotic response via a novel signaling pathway involving S1P4 transactivation upstream of Rho-kinase-2 activation. FASEB J. 2013;27:4532–4546. doi: 10.1096/fj.13-228528. [DOI] [PubMed] [Google Scholar]
- 37.Li X., McFarland D.C., Velleman S.G. Transforming growth factor-beta1-induced satellite cell apoptosis in chickens is associated with beta1 integrin-mediated focal adhesion kinase activation. Poult. Sci. 2009;88:1725–1734. doi: 10.3382/ps.2008-00534. [DOI] [PubMed] [Google Scholar]
- 38.Wissing E.R., Boyer J.G., Kwong J.Q., Sargent M.A., Karch J., McNally E.M., Otsu K., Molkentin J.D. P38alpha MAPK underlies muscular dystrophy and myofiber death through a Bax-dependent mechanism. Hum. Mol. Genet. 2014;23:5452–5463. doi: 10.1093/hmg/ddu270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sinha-Hikim I., Braga M., Shen R., Sinha Hikim A.P. Involvement of c-Jun NH2-terminal kinase and nitric oxide-mediated mitochondria-dependent intrinsic pathway signaling in cardiotoxin-induced muscle cell death: role of testosterone. Apoptosis. 2007;12:1965–1978. doi: 10.1007/s10495-007-0120-6. [DOI] [PubMed] [Google Scholar]
- 40.Zimowska M., Duchesnay A., Dragun P., Oberbek A., Moraczewski J., Martelly I. Immunoneutralization of TGFbeta1 improves skeletal muscle regeneration: effects on myoblast differentiation and glycosaminoglycan content. Int. J. Cell Biol. 2009;2009:659372. doi: 10.1155/2009/659372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fakhfakh R., Lamarre Y., Skuk D., Tremblay J.P. Losartan enhances the success of myoblast transplantation. Cell Transplant. 2012;21:139–152. doi: 10.3727/096368911X576045. [DOI] [PubMed] [Google Scholar]
- 42.Schwander M., Leu M., Stumm M., Dorchies O.M., Ruegg U.T., Schittny J., Muller U. Beta1 integrins regulate myoblast fusion and sarcomere assembly. Dev. Cell. 2003;4:673–685. doi: 10.1016/s1534-5807(03)00118-7. [DOI] [PubMed] [Google Scholar]
- 43.Sharma K., Cook A., Smith M., Valancius C., Inscho E.W. TGF-beta impairs renal autoregulation via generation of ROS. Am. J. Physiol. Renal Physiol. 2005;288:F1069–F1077. doi: 10.1152/ajprenal.00345.2004. [DOI] [PubMed] [Google Scholar]
- 44.Hagler M.A., Hadley T.M., Zhang H., Mehra K., Roos C.M., Schaff H.V., Suri R.M., Miller J.D. TGF-beta signalling and reactive oxygen species drive fibrosis and matrix remodelling in myxomatous mitral valves. Cardiovasc. Res. 2013;99:175–184. doi: 10.1093/cvr/cvt083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang L., Qu M., Wang Y., Duan H., Chen P., Wang Y., Shi W., Danielson P., Zhou Q. Trichostatin A inhibits transforming growth factor-beta-induced reactive oxygen species accumulation and myofibroblast differentiation via enhanced NF-E2-related factor 2-antioxidant response element signaling. Mol. Pharmacol. 2013;83:671–680. doi: 10.1124/mol.112.081059. [DOI] [PubMed] [Google Scholar]
- 46.Hsieh H.L., Wang H.H., Wu W.B., Chu P.J., Yang C.M. Transforming growth factor-beta1 induces matrix metalloproteinase-9 and cell migration in astrocytes: roles of ROS-dependent ERK- and JNK-NF-kappaB pathways. J. Neuroinflammation. 2010;7:88. doi: 10.1186/1742-2094-7-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santos C.R., Martinho A., Quintela T., Goncalves I. Neuroprotective and neuroregenerative properties of metallothioneins. IUBMB Life. 2012;64:126–135. doi: 10.1002/iub.585. [DOI] [PubMed] [Google Scholar]
- 48.Ceylan-Isik A.F., Zhao P., Zhang B., Xiao X., Su G., Ren J. Cardiac overexpression of metallothionein rescues cardiac contractile dysfunction and endoplasmic reticulum stress but not autophagy in sepsis. J. Mol. Cell. Cardiol. 2010;48:367–378. doi: 10.1016/j.yjmcc.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oshima Y., Fujio Y., Nakanishi T., Itoh N., Yamamoto Y., Negoro S., Tanaka K., Kishimoto T., Kawase I., Azuma J. STAT3 mediates cardioprotection against ischemia/reperfusion injury through metallothionein induction in the heart. Cardiovasc. Res. 2005;65:428–435. doi: 10.1016/j.cardiores.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 50.Lim K.S., Cha M.J., Kim J.K., Park E.J., Chae J.W., Rhim T., Hwang K.C., Kim Y.H. Protective effects of protein transduction domain-metallothionein fusion proteins against hypoxia- and oxidative stress-induced apoptosis in an ischemia/reperfusion rat model. J. Control Release. 2013;169:306–312. doi: 10.1016/j.jconrel.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 51.Nielsen A.E., Bohr A., Penkowa M. The balance between life and death of cells: roles of metallothioneins. Biomark. Insights. 2007;1:99–111. [PMC free article] [PubMed] [Google Scholar]
- 52.Malik V., Rodino-Klapac L.R., Mendell J.R. Emerging drugs for Duchenne muscular dystrophy. Expert Opin. Emerg. Drugs. 2012;17:261–277. doi: 10.1517/14728214.2012.691965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Luca A. Pre-clinical drug tests in the mdx mouse as a model of dystrophinopathies: an overview. Acta Myol. 2012;31:40–47. [PMC free article] [PubMed] [Google Scholar]
- 54.Tinsley J.M., Potter A.C., Phelps S.R., Fisher R., Trickett J.I., Davies K.E. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384:349–353. doi: 10.1038/384349a0. [DOI] [PubMed] [Google Scholar]
- 55.Accornero F., van Berlo J.H., Benard M.J., Lorenz J.N., Carmeliet P., Molkentin J.D. Placental growth factor regulates cardiac adaptation and hypertrophy through a paracrine mechanism. Circ. Res. 2011;109:272–280. doi: 10.1161/CIRCRESAHA.111.240820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai C., Masumiya H., Weisleder N., Matsuda N., Nishi M., Hwang M., Ko J.K., Lin P., Thornton A., Zhao X., et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009;11:56–64. doi: 10.1038/ncb1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Millay D.P., Sargent M.A., Osinska H., Baines C.P., Barton E.R., Vuagniaux G., Sweeney H.L., Robbins J., Molkentin J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008;14:442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.