

In this randomized, placebo-controlled trial, rosuvastatin 10 mg daily improved estimated glomerular filtration rate and reduced plasma cystatin C in HIV-infected patients on antiretroviral therapy. Reductions in cystatin C with statin therapy correlated with reductions in inflammatory biomarkers.
Keywords: statin, HIV, cystatin C, kidney, inflammation
Abstract
Background. In chronic human immunodeficiency virus (HIV) infection, plasma cystatin C may be influenced by factors other than glomerular filtration rate such as inflammation. Statins may improve cystatin C by improving glomerular function or by decreasing inflammation.
Methods. The Stopping Atherosclerosis and Treating Unhealthy Bone With Rosuvastatin in HIV (SATURN-HIV) trial randomized 147 patients on stable antiretroviral therapy (ART) with low-density lipoprotein cholesterol ≤130 mg/dL to blinded 10 mg daily rosuvastatin or placebo. We analyzed relationships of baseline and 0- to 24-week changes in plasma cystatin C concentration with measures of vascular disease, inflammation, and immune activation.
Results. Median age was 46 (interquartile range, 40–53) years; 78% were male, 68% African American. Tenofovir and protease inhibitors were used in 88% and 49% of subjects, respectively. Baseline cystatin C was associated with higher carotid intima-media thickness and epicardial adipose tissue independent of age, sex, and race. Biomarkers of endothelial activation and inflammation were associated with cystatin C in a multivariable model independent of creatinine-based estimated glomerular filtration rate (eGFRcr). After 24 weeks, statin use slowed mean eGFRcr decline (1.61 vs −3.08 mL/minute/1.73 m2 for statin vs placebo; P = .033) and decreased mean cystatin C (−0.034 mg/L vs 0.010 mg/L; P = .008). Within the statin group, changes in cystatin C correlated with changes in endothelial activation, inflammation, and T-cell activation.
Conclusions. Rosuvastatin 10 mg daily reduces plasma cystatin C and slows kidney function decline in HIV-infected patients on ART. Reductions in cystatin C with statin therapy correlate with reductions in inflammatory biomarkers. Relationships between cystatin C, kidney function, and cardiovascular risk in HIV may be mediated in part by inflammation.
Clinical Trials Registration. NCT01218802.
(See the Editorial Commentary by Kalayjian on pages 1157–9.)
Chronic kidney disease (CKD) is a common comorbidity of human immunodeficiency virus (HIV) infection that is associated with substantial cardiovascular risk [1]. Traditional risk factors [2] such as race and diabetes are associated with more rapid kidney function decline, as are HIV-specific factors such as active HIV type 1 (HIV-1) viral replication [3] and certain antiretroviral therapy (ART) such as tenofovir and ritonavir-boosted protease inhibitors (PIs) [4]. The accurate estimation of glomerular filtration rate (GFR) using endogenous filtration markers is critical for the clinical care of these patients, but is impaired in chronic HIV infection by significant non-GFR determinants of creatinine and the novel marker cystatin C [5–7].
Both CKD [8] and HIV infection [9] are characterized by high levels of systemic inflammation; however, whether inflammation is a significant non-GFR determinant of cystatin C levels that explains the strong associations of cystatin C with clinical outcomes in HIV infection is unclear [7, 10]. Statins have anti-inflammatory effects in HIV-uninfected patients with CKD [8, 11], but the ability to slow GFR decline appears to be quite modest [12]. In HIV infection, statins have decreased inflammation and immune activation markers in some studies, with varying results depending on the biomarker studied and ART status [13–18]. To our knowledge, no study has evaluated the effect of statin therapy on glomerular function or cystatin C in HIV-infected patients.
The objectives of this study were 2-fold. First, we aimed to examine the baseline cross-sectional associations of cystatin C levels with markers of inflammation and subclinical vascular disease among HIV-infected patients on ART enrolled in a randomized trial of statin therapy. Second, we studied the 24-week effect of rosuvastatin on cystatin C levels and estimated GFR and whether changes in cystatin C were related to changes in inflammation markers.
METHODS
The Stopping Atherosclerosis and Treating Unhealthy Bone With Rosuvastatin in HIV (SATURN-HIV) trial is a randomized, double-blind, placebo-controlled trial that was designed to study the effect of rosuvastatin 10 mg daily on vascular and bone health. Randomization was stratified by use of protease inhibitor at study entry. Secondary prespecified endpoints include biomarkers of inflammation and immune activation, including cystatin C, and changes in renal function. All subjects were ≥18 years of age without known coronary disease or diabetes, and on stable ART for at least 3 months with HIV-1 RNA <1000 copies/mL. In a similar design as the JUPITER (Justification for the Use of Statins in Prevention—an Intervention Trial Evaluating Rosuvastatin) study in HIV-uninfected adults [19], entry criteria included low-density lipoprotein (LDL) cholesterol ≤130 mg/dL and evidence of increased inflammation and/or T-cell activation (high sensitivity C-reactive protein ≥2 mg/L and/or CD8+CD38+HLA-DR+ T cells ≥19%). Participants with creatinine clearance <50 mL/minute by Cockcroft-Gault were excluded. Full inclusion and exclusion are listed at ClinicalTrials.gov (NCT01218802). The study was approved by the institutional review board of University Hospitals Case Medical Center (Cleveland, Ohio), and all participants signed written informed consent.
Study Evaluations
Demographics and medical history were obtained by self-report and review of past medical records. A targeted physical exam and 12-hour fasting blood draw were performed at each visit (screening, 0, and 24 weeks). Peripheral blood mononuclear cells were separated by centrifugation with Ficoll-Hypaque and were cryopreserved until analyzed by flow cytometry in batch. Frozen plasma samples were stored at −80°C and analyzed in batch. Baseline evaluations also included a whole-body dual-energy absorptiometry scan for peripheral and trunk fat [20]. Insulin resistance was calculated from fasting glucose and insulin using the homeostatic model assessment of insulin resistance [21]. Ten-year Framingham risk score was determined using a published risk calculator [22].
Cystatin C and Kidney Function
Plasma cystatin C concentration was measured by particle-enhanced immunonephelometric assay on a BNII nephelometer (Siemens, Munich, Germany) at the University of Vermont Laboratory for Clinical Biochemistry Research. Interassay variability ranged from 2.0% to 2.8%. Serum creatinine was measured by the enzymatic method in the University Hospitals clinical chemistry laboratory on a VISTA 1500 integrated system (Siemens). Glomerular filtration rate (eGFRcr) was estimated using the creatinine-based 2009 Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation [23].
Subclinical Vascular Disease
Common carotid artery intima-media thickness (CCA-IMT) and brachial artery endothelial function (flow-mediated dilation [FMD] and hyperemic velocity-time integral [VTI]) were measured by ultrasound using semiautomated edge detection software (Medical Imaging Applications LLC, Coralville, Iowa) as previously described [24]. Coronary calcium score and epicardial fat were quantified by gated noncontrast computed tomography (CT) of the chest [25].
Carotid distensibility was measured with semiautomated edge detection software from 10-beat ultrasound cine loops. The diameter of the distal 1 cm of the right carotid artery was measured in systole (Ds) and diastole (Dd). Blood pressure was obtained at the time of carotid ultrasound to determine the pulse pressure (PP). Carotid distensibility was calculated using the same formula [(2*(Ds – Dd) / Dd) / PP] used in the Women's Interagency Health Study and the Multicenter AIDS Cohort Study [26, 27] and is reported in units of 10−6 × N−1m2.
Eight categories of subclinical vascular disease phenotype [24] were prespecified according to the presence of 1 or more of the following: (1) coronary atherosclerosis defined as the presence of any detectable coronary artery calcium (CAC) by CT, (2) carotid atherosclerosis defined as the presence of plaque or a mean-maximum CCA-IMT >1.0 mm, or (3) abnormal endothelial function defined as having below-median values of both FMD (<4%) and hyperemic VTI (<77 cm).
Markers of Inflammation and Immune Activation
Soluble biomarkers of inflammation and monocyte activation were measured by enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, Minnesota). For soluble markers of monocyte activation (soluble CD14 and CD163), the interassay variability ranged from 0.4% to 8.6% and 0.7% to 18.3%, respectively. For markers of systemic inflammation (interleukin 6 [IL-6] and tumor necrosis factor α receptors 1 and 2 [sTNF-RI and -RII]), the interassay variability ranged from 2.02% to 15.36%, 3.66% to 5.77%, and 2.13% to 3.79%, respectively. For markers of endothelial activation (soluble vascular cell adhesion molecule-1 [sVCAM] and soluble intercellular adhesion molecule 1 [sICAM]), the interassay variability ranged from 4.76% to 8.77% and 3.43% to 7.37%, respectively.
Monocytes and T cells were phenotyped by flow cytometry as previously described [28, 29]. Three monocyte subsets were each quantified as a percentage of the overall monocyte population: (1) CD14+CD16+, (2) CD14dimCD16+, and (3) CD14+CD16−. T-cell activation was quantified as the percentage of CD4+ or CD8+ cells that expressed both CD38 and HLA-DR.
Statistical Analysis
This study is a prespecified cross-sectional analysis of the relationship of cystatin C levels to markers of cardiovascular risk and inflammation and a longitudinal analysis of 0- to 24-week changes in cystatin C and eGFR.
Demographics, HIV parameters, and cardiac and metabolic factors are described overall and by group using median and interquartile range (IQR) for continuous variables and frequency and percentage for categorical variables. All baseline variables were compared between groups using unpaired t tests or Wilcoxon rank-sum tests as distributionally appropriate for continuous variables and by χ2 tests, Fisher exact tests, or Pearson exact χ2 tests as appropriate for categorical variables.
Spearman correlation coefficients were determined to test associations between baseline cystatin C concentration and markers of cardiovascular risk. Each cardiovascular risk measure was then dichotomized by median value, with the exception of CAC score, which was categorized as presence of CAC or not. Subsequently, logistic regression was used to explore the relationships between each of these markers and cystatin C adjusted for age, sex, and race. Analysis of variance (ANOVA) was used to compare mean cystatin C levels between the 8 vascular phenotypes described above. To avoid excessive comparisons, phenotypes 0–6 were each individually compared with the highest risk group (phenotype 7) in a prespecified analysis. Additionally, linear regression was used to test for trend across the 8 ordered groups.
Next, univariable followed by multivariable linear regression was used to explore associations between baseline cystatin C and variables of interest, including eGFRcr, demographics, metabolic and traditional kidney risk factors, HIV-related factors, markers of inflammation, and immune activation. Stepwise selection was used to generate final models considering all variables with P ≤ .25 in univariable analyses.
Between and within-group changes in cystatin C and eGFRcr were tested using paired t tests or Wilcoxon signed-rank tests as distributionally appropriate. Analysis of covariance (ANCOVA) was used to adjust mean percentage of change in cystatin C over 24 weeks for age, sex, PI use, and baseline eGFRcr with and without change in eGFRcr. Adjusted means were compared using unpaired t tests.
For ANOVA, linear regression and ANCOVA analyses, all nonnormally distributed dependent variables were log-transformed as well as all markers of inflammation and immune activation. All statistical tests were 2-sided and considered significant with P < .05. Analyses were performed using SAS version 9.2 (SAS Institute, Cary, North Carolina).
RESULTS
Of 202 participants who were screened, 147 participants were randomized to receive rosuvastatin 10 mg daily (n = 72) or matching placebo (n = 75). Reasons for screen failure and loss to follow-up (n = 11) have been described previously [13, 15] (Supplementary Figure 1). Over the first 24 weeks of the trial, 1 subject switched ART from didanosine to abacavir and 1 switched from lamivudine/zidovudine to emtricitabine/tenofovir plus maraviroc.
The baseline characteristics of the randomized participants are displayed in Table 1 and were similar between treatment groups (all P > .05). Overall, median age was 46 (IQR, 40–53) years; 78% were male, 68% were African American, and 14% had hepatitis C. Tenofovir and PIs were used in 88% and 49% of subjects, respectively. Median eGFRcr was 100 (IQR, 87–118) mL/minute/1.73 m2, and median plasma cystatin C concentration was 0.83 (IQR, 0.73–0.95) mg/L.
Table 1.
Baseline Characteristics of Study Participants by Assigned Treatment Group
| Demographics | Rosuvastatin (n = 72) | Placebo (n = 75) |
|---|---|---|
| Age, y | 45 (41–51) | 47 (39–53) |
| Male sex | 81% | 76% |
| African American race | 69% | 67% |
| HIV parameters | ||
| HIV duration, y | 11 (6–17) | 12 (6–19) |
| Current CD4+ count, cells/µL | 608 (440–848) | 627 (398–853) |
| Nadir CD4+ count, cells/µL | 173 (84–312) | 190 (89–281) |
| Undetectable viral load, <50 copies/mL | 78% | 77% |
| ART duration, y | 5.2 (3.1–9.9) | 5.9 (3.3–9.6) |
| Current protease inhibitor use | 50% | 48% |
| Current TDF use | 89% | 88% |
| Metabolic and cardiovascular risk factors | ||
| Body mass index, kg/m2 | 27 (23–30) | 27 (23–30) |
| Total limb fat, kg | 9.0 (5.5–13) | 8.2 (4.8–14) |
| Total trunk fat, kg | 13 (8.1–18) | 12 (7.0–18) |
| Epicardial fat, cm3 | 70 (43–91) | 63 (50–92) |
| Active hepatitis B | 4% | 5% |
| Active hepatitis C | 7% | 9% |
| Systolic blood pressure, mm Hg | 122 (112–136) | 120 (110–132) |
| Anti–hypertensive medication use | 28% | 24% |
| HDL cholesterol, mg/dL | 47 (38–58) | 46 (37–57) |
| LDL cholesterol, mg/dL | 96 (76–07) | 97 (77–121) |
| HOMA-IR ≥2.5 | 31% | 41% |
| Smoking | ||
| Current | 60% | 67% |
| Past | 15% | 16% |
| Never | 25% | 17% |
| Framingham risk score, % 10-year risk | 3 (1–7) | 4 (1–7) |
| Measures of subclinical vascular disease | ||
| Mean-max common carotid artery IMT, µm | 843 (774–960) | 843 (768–959) |
| Carotid distensibility, 10−6 × Newtons−1 × m2 | 24 (19–32) | 23 (19–30) |
| FMD, % | 3.9 (2.1–6.2) | 4.0 (2.0–6.0) |
| Hyperemic VTI, cm | 80 (60–94) | 76 (67–95) |
| Coronary artery calcium | ||
| 0 | 67% | 60% |
| 1–100 | 25% | 29% |
| >100 | 8% | 11% |
| Kidney function | ||
| eGFRcr, CKD-EPI | 99 (85–117) | 100 (87–118) |
| Cystatin C, mg/L | 0.84 (0.75–0.99) | 0.83 (0.71–0.95) |
| Urine ACR >30 mg/g | 17% | 17% |
Data are presented as median (interquartile range) or percentage. All P > .05 for differences between groups using t test or Wilcoxon rank-sum test for continuous variables and χ2 or Fisher exact tests for categorical variables as appropriate.
Abbreviations: ACR, albumin to creatinine ratio; ART, antiretroviral therapy; CKD-EPI, 2009 Chronic Kidney Disease Epidemiology Collaboration equation; eGFRcr, creatinine-based estimated glomerular filtration rate; FMD, flow-mediated dilation; HDL, high-density lipoprotein; HIV, human immunodeficiency virus; HOMA-IR, homeostatic model assessment of insulin resistance; IMT, intima-media thickness; LDL, low-density lipoprotein; TDF, tenofovir disoproxil fumarate; VTI, velocity-time integral.
At baseline (Table 2), cystatin C concentrations were correlated with carotid artery disease (positively with CCA-IMT and negatively with carotid distensibility), but not with measures of endothelial function (FMD and hyperemic VTI). Cystatin C was also correlated with epicardial adipose tissue—a visceral fat depot that is associated with cardiometabolic risk factors and higher levels of generalized inflammation and immune activation in this population [25]. After adjustment for age, sex, and race, higher cystatin C was independently associated with thicker CCA-IMT (P = .029) and larger epicardial adipose tissue volume (P = .02).
Table 2.
Relationship of Baseline Plasma Cystatin C Concentration With Measures of Subclinical Vascular Disease and Cardiovascular Risk
| Measure | Spearman r | P Value |
|---|---|---|
| Brachial FMD, % | 0.037 | .66 |
| Brachial hyperemic VTI, cm | −0.130 | .12 |
| Mean-max CCA-IMT, µm | 0.339 | <.0001 |
| Carotid distensibility, 10−6*Newtons−1*m2 | −0.193 | .02 |
| Coronary calcium score | 0.170 | .04 |
| Epicardial adipose tissue, cm3 | 0.314 | .0001 |
Bolded text represents statistically significant correlations with P value <.05.
Abbreviations: CCA-IMT, common carotid artery intima-media thickness; FMD, flow-mediated dilation; VTI, velocity-time integral.
As reported previously, more than two-thirds of this population had endothelial dysfunction or subclinical atherosclerosis in at least 1 vascular bed [24]. Figure 1A describes the number of participants that fall into the various combinations of vascular disease (ie, “vascular phenotype”), and Figure 1B shows the median cystatin C concentration across each of these 8 vascular phenotypes. In general, more severe vascular phenotypes were associated with higher cystatin C concentration (P for trend = .004). Median cystatin C was 26% higher among participants with the worst subclinical disease compared with normal subjects (median, 1.02 [IQR, 0.86–1.07] vs 0.81 [IQR, 0.73–0.92] mg/dL for group 7 vs group 0; P = .003).
Figure 1.
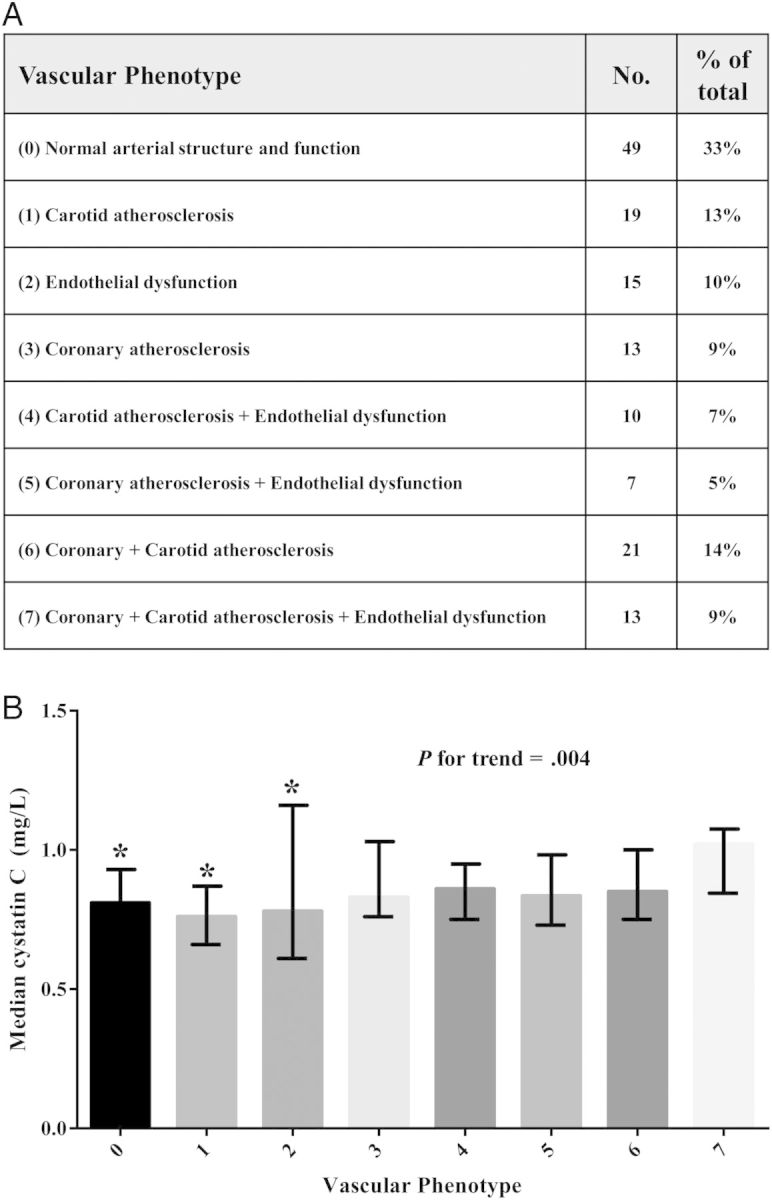
The association of baseline cystatin C with extent of subclinical vascular disease. A, Number (%) of participants who were categorized into 8 vascular phenotypes representing the extent of subclinical vascular disease in multiple vascular beds. B, Median cystatin C levels of participants in each of the 8 vascular phenotypes. Error bars represent interquartile range. *P < .05 compared with group 7.
Baseline plasma cystatin C concentrations were associated with circulating markers of generalized inflammation (IL-6 and soluble TNF-α receptors), endothelial activation (sVCAM-1), monocyte activation (soluble CD163), and T-cell activation (CD4+CD38+HLA-DR+ T cells) in univariable analyses (Table 3). Importantly, cystatin C levels remained independently associated with soluble TNF-α receptors and sICAM-1 in a model that included estimated GFR (Table 3). Other independent associations were observed with eGFRcr, systolic blood pressure, and PI use. Tenofovir use was not associated with baseline cystatin C in univariable analyses (P = .99), nor when it was added to the final multivariable model (P = .77).
Table 3.
Univariable and Multivariable Analyses of the Relationship Between Baseline Cystatin C Concentration and Markers of Inflammation and Immune Activation
| Marker | Univariable Analysisa |
Multivariable Analysisb |
||
|---|---|---|---|---|
| Estimate | P Value | Estimate | P Value | |
| Kidney function | ||||
| eGFRcr (per 10 mL/min/1.73 m3) | −0.06 | <.0001 | −0.022 | .005 |
| Demographics and traditional kidney risk factors | ||||
| Age (per decade) | 0.077 | <.0001 | ||
| Caucasian race (vs all other) | 0.089 | .028 | ||
| Male sex (vs female) | 0.069 | .123 | ||
| SBP (per 10 mm Hg) | 0.016 | .152 | 0.021 | .018 |
| DBP (per 10 mm Hg) | 0.022 | .245 | ||
| HIV-specific factors and coinfections | ||||
| Nadir CD4 (per 100 cells/m3) | −0.02 | .042 | ||
| HIV duration (per year) | 0.006 | .024 | ||
| ART duration (per year) | 0.006 | .093 | ||
| Current PI use (vs no PI use) | 0.079 | .034 | 0.072 | .009 |
| Hepatitis B coinfection | 0.132 | .129 | ||
| Hepatitis C coinfection | 0.079 | .246 | ||
| Markers of inflammation and immune activation | ||||
| CD14+CD16+ monocytes | 0.094 | .018 | ||
| Soluble CD163 | 0.174 | <.0001 | ||
| CD4+DR+38+ | 0.091 | .031 | ||
| CD8+DR+38+ | 0.052 | .13 | ||
| IL-6 | 0.120 | <.0001 | ||
| sICAM-1 | 0.049 | .156 | −0.058 | .041 |
| sVCAM-1 | 0.216 | <.0001 | ||
| sTNF-RI | 0.306 | <.0001 | 0.299 | <.0001 |
| sTNF-RII | 0.246 | <.0001 | 0.272 | <.0001 |
Cystatin C and all markers of inflammation and immune activation were log-transformed prior to analyses.
Bolded text represents statistically significant estimates with P value <.05.
Abbreviations: ART, antiretroviral therapy; DBP, diastolic blood pressure; eGFRcr, creatinine-based estimated glomerular filtration rate; HIV, human immunodeficiency virus; IL-6, interleukin 6; PI, protease inhibitor; SBP, systolic blood pressure; sICAM-1, soluble intercellular adhesion molecule 1; sTNF-RI, soluble tumor necrosis factor α receptor 1; sTNF-RII, soluble tumor necrosis factor α receptor 2; sVCAM, soluble vascular cell adhesion molecule 1.
a Univariable associations at a level of P < .25 are shown. Variables tested but not shown in table include smoking status, body mass index, current tenofovir use, HIV RNA >50 copies/mL, current CD4, CD14dimCD16+ monocytes, and soluble CD14.
b Multivariable analysis was performed using stepwise selection of variables with univariable associations at a level of P < .25. Adjusted R2 = 0.58. All variance inflation factors <1.5.
Cystatin C concentration decreased significantly in those taking daily rosuvastatin compared with placebo over the first 24 weeks of this trial (Figure 2). Additionally, there was a decrease in eGFRcr in the placebo group but not in the rosuvastatin group, with a significant between group difference (Figure 2). In ANCOVA analyses of cystatin C changes, adjustment for PI use and baseline eGFRcr did not change the results; however, after adjustment for 0–24 week change in eGFRcr, the difference between statin and placebo arms was no longer statistically significant (P = .099). Changes in cystatin C concentration were correlated with changes in sTNF-RII receptors in both arms of the study, whereas significant correlations with changes in CD4+ T-cell activation and sICAM-1 were only observed in the statin arm (Table 4). Within the statin arm, changes in cystatin C did not correlate with changes in LDL cholesterol (r = −0.1274, P = .3056).
Figure 2.

Comparison of cystatin C and creatinine-based estimated glomerular filtration rate (eGFRcr) changes from 0 to 24 weeks by treatment group. The graph represents mean percentage change with 95% confidence bands. The tables display mean (standard deviation) absolute and percentage changes for cystatin C and eGFRcr, respectively. Abbreviation: eGFRcr, creatinine-based estimated glomerular filtration rate.
Table 4.
Correlations of Cystatin C Changes With Inflammation and Immune Activation Marker Changes by Treatment Assignment
| Marker | Rosuvastatin |
Placebo |
||
|---|---|---|---|---|
| Spearman r | P Value | Spearman r | P Value | |
| CD14+CD16+ monocytes | −0.178 | .163 | 0.008 | .951 |
| CD14dimCD16+ monocytes | 0.004 | .973 | −0.118 | .343 |
| Soluble CD163 | −0.003 | .978 | −0.064 | .607 |
| Soluble CD14 | 0.118 | .348 | −0.018 | .885 |
| CD4+CD38+HLA-DR+ T cells | 0.281 | .025 | 0.022 | .862 |
| CD8+CD38+HLA-DR+ T cells | 0.159 | .210 | 0.049 | .691 |
| IL-6 | 0.010 | .933 | 0.058 | .634 |
| sICAM-1 | 0.460 | <.0001 | 0.186 | .126 |
| sVCAM-1 | 0.180 | .144 | 0.109 | .375 |
| sTNF-RI | 0.184 | .136 | 0.111 | .362 |
| sTNF-RII | 0.372 | .002 | 0.314 | .009 |
Bolded text represents statistically significant correlations with P value <.05.
Abbreviations: IL-6, interleukin 6; sICAM-1, soluble intercellular adhesion molecule 1; sTNF-RI, soluble tumor necrosis factor α receptor 1; sTNF-RII, soluble tumor necrosis factor α receptor 2; sVCAM, soluble vascular cell adhesion molecule 1.
DISCUSSION
In this randomized clinical trial of HIV-infected patients on ART, 10 mg of daily rosuvastatin decreased plasma cystatin C levels and preserved eGFRcr. Interestingly, plasma cystatin C was associated with inflammation and endothelial activation at baseline independent of eGFRcr, and changes in cystatin C correlated with changes in inflammation, endothelial activation, and T-cell activation. This trial provides the first evidence in patients with HIV infection that statin therapy may have salutary effects on kidney function, and also suggests that some of the effect on plasma cystatin C levels may be related to changes in systemic inflammation and immune activation.
Statins substantially reduce the risk of cardiovascular events among patients with mild to moderate CKD in the general population [30, 31], although their effect on eGFR decline is less clear [30–32]. In JUPITER [30], 20 mg of rosuvastatin decreased the risk of the primary endpoint by 45% and all-cause mortality by 44% among subjects with eGFRcr <60 mL/minute/1.73 m2. High-sensitivity CRP reductions were similar in those with and without CKD. In contrast, 12-month median eGFRcr loss was no different for those with CKD (−3 mL/minute/1.73 m2 for both statin and placebo). Furthermore, concerns about possible renal effects of rosuvastatin compared to atorvastatin in patients with known CKD have been raised by the PLANET 1 and 2 trials [33, 34]. Among nondiabetic patients with CKD (mean eGFR of 75 mL/minute) in PLANET 2 (Prospective Evaluation of Proteinuria and Renal Function in Nondiabetic Patients With Progressive Renal Disease), atorvastatin 80 mg daily decreased proteinuria and caused a smaller 52-week mean eGFR decline (−1.74 mL/minute) compared with rosuvastatin 10 mg (−2.71 mL/minute) or rosuvastatin 40 mg (−3.30 mL/minute) [33]. Similar results were seen among diabetic patients in PLANET 1 (Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients With Progressive Renal Disease) [34].
In a combined analysis of pravastatin trials [32], pravastatin 40 mg daily decreased the rate of eGFR loss by 8% overall, and by 34% among those with moderate CKD (eGFR 30–60 mL/minute/1.73 m2). In the CARE (Cholesterol and Recurrent Events) trial [35], the most “inflamed” participants (highest tertile of CRP and sTNF-RI) derived the greatest renal benefit from pravastatin. Consistent with these findings from CARE, our study suggests that HIV-infected subjects with elevated markers of inflammation despite effective ART are a subgroup of patients that may gain a protective renal benefit from statins.
The extent to which cystatin C changes in our study were due to changes in eGFR is not clear. Although adjustment for eGFRcr attenuated the observed changes in cystatin C, there was still a trend toward statistical significance. It is possible that residual cystatin C changes were due to changes in true GFR that were not accurately captured by changes in eGFRcr. Alternatively, changes in cystatin C may have resulted from reductions in non-GFR determinants of cystatin C such as inflammation, as the anti-inflammatory properties of statins have been well described [8]. The fact that changes in cystatin C within the statin arm of our study correlated with changes in T-cell activation and endothelial activation suggests that this may be the case. This interpretation is supported further by evidence that T-cell activation may partially explain the bias and inaccuracy of cystatin C based estimated GFR in HIV infection compared with measured GFR [7]. On the other hand, cystatin C and TNF-α receptor changes correlated similarly in both arms of our study, which cannot be attributed to the anti-inflammatory effect of statins. TNF-α receptors are primarily renally cleared and are more strongly correlated with glomerular function than other biomarkers of inflammation [36]. It is also possible that inflammation itself (particularly endothelial activation) may be causally related to glomerular function [8], an effect that may be more prominent among subjects of black race [37]. Our study population is notably two-thirds African American.
This study furthers our understanding of cystatin C as a predictor of cardiovascular risk in patients with treated HIV infection and the potential of statins to decrease that risk. Cystatin C and cystatin C–based estimates of GFR are stronger predictors of cardiovascular events in HIV when compared to creatinine or creatinine-based GFR [10]. The mechanism of this association between cystatin C levels and clinical events may be multifactorial and partly related to inflammation and/or abnormalities of vascular structure and function. Two studies have examined the relationship of cystatin C to subclinical carotid disease in HIV. Incipient renal disease (defined as eGFRcr-cys <90 mL/minute/1.73 m2 + >3% annual decrease in eGFR + albumin/creatinine ratio >5 mg/g) was independently associated with carotid IMT in a Spanish cohort [38]. In the Study of Fat Redistribution and Metabolic Change (FRAM), cystatin C was a potent independent predictor of mortality [39], but was not related to carotid intima media thickness after adjustment for age, sex, and race [40]. In our study, plasma cystatin C levels were associated with multiple measures of subclinical vascular disease in univariable analyses. Adjustment for age, sex, and race removed the association with some measures, although CCA-IMT and epicardial adipose tissue remained independently associated with cystatin C. Our study adds to these results by including endothelial function testing and coronary calcium scoring to provide a more complete phenotype of vascular structure and function. The highest cystatin C levels were seen in participants with the combination of brachial endothelial dysfunction, detectable coronary calcium, and carotid disease.
The major strength of our study was the double-blind, placebo-controlled randomized trial design. In addition, the extent of inflammation, immune activation, and subclinical vascular disease in the study subjects were very well characterized. The study was limited by the lack of a gold-standard direct measure of GFR. The study may have lacked power to detect an effect of statin therapy on cystatin C that is independent of eGFRcr. We chose to use the creatinine-based CKD-EPI equation to estimate GFR rather than the cystatin C–based or the combined equation, because we were testing the hypothesis that cystatin C is influenced by inflammation. We recognize that eGFRcr is biased by other non-GFR determinants of creatinine such as muscle mass, which may have influenced our findings. Finally, we did not account for the effect of other potentially nephrotoxic medications besides ART.
In conclusion, rosuvastatin 10 mg daily reduces plasma cystatin C and slows eGFR decline compared to placebo over 24 weeks in HIV-infected patients on ART. HIV-infected subjects with elevated levels of inflammation despite effective ART are a subgroup of patients that may gain a protective renal benefit from statins. Furthermore, reductions in cystatin C with statin therapy correlate with reductions in T-cell activation, endothelial activation, and inflammation. Our study suggests that relationships between cystatin C, kidney function, and cardiovascular risk in HIV are complex but may be mediated in part by inflammation. Future studies should evaluate whether other anti-inflammatory therapies improve cystatin C and/or glomerular function in patients with treated HIV infection.
Supplementary Data
Supplementary materials are available at Clinical Infectious Diseases online (http://cid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. The authors thank the study participants.
Financial support. This project was supported by the National Institutes of Health (grant numbers NR012642 to G. A. M. and HL116209 to C. O. H.) and by a Virology Fellowship grant from Bristol-Myers Squibb to C. T. L. Technical assistance was provided by the Center for AIDS Research, Case Western Reserve University (P30 AI36219). Study drugs and matching placebo were donated by AstraZeneca.
Potential conflicts of interest. C. T. L. has received grants from Bristol-Myers Squibb and the Medtronic Foundation. G. A. M. has served as a scientific advisor or speaker for Bristol-Myers Squibb, GlaxoSmithKline, Tibotec, Merck, and Gilead Sciences; has received research grants from Bristol-Myers Squibb, GlaxoSmithKline, and Gilead Sciences; and is currently serving as the data and safety monitoring board chair for a Pfizer-sponsored study. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Choi AI, Li Y, Deeks SG, Grunfeld C, Volberding PA, Shlipak MG. Association between kidney function and albuminuria with cardiovascular events in HIV-infected persons. Circulation. 2010;121:651–8. doi: 10.1161/CIRCULATIONAHA.109.898585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi AI, Rodriguez RA, Bacchetti P, Bertenthal D, Volberding PA, O'Hare AM. The impact of HIV on chronic kidney disease outcomes. Kidney Int. 2007;72:1380–7. doi: 10.1038/sj.ki.5002541. [DOI] [PubMed] [Google Scholar]
- 3.Longenecker CT, Scherzer R, Bacchetti P, Lewis CE, Grunfeld C, Shlipak MG. HIV viremia and changes in kidney function. AIDS. 2009;23:1089–96. doi: 10.1097/QAD.0b013e32832a3f24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mocroft A, Kirk O, Reiss P, et al. Estimated glomerular filtration rate, chronic kidney disease and antiretroviral drug use in HIV-positive patients. AIDS. 2010;24:1667–78. doi: 10.1097/QAD.0b013e328339fe53. [DOI] [PubMed] [Google Scholar]
- 5.Gagneux-Brunon A, Delanaye P, Maillard N, et al. Performance of creatinine and cystatin C-based glomerular filtration rate estimating equations in a European HIV-positive cohort. AIDS. 2013;27:1573–81. doi: 10.1097/QAD.0b013e32835fac30. [DOI] [PubMed] [Google Scholar]
- 6.Inker LA, Wyatt C, Creamer R, et al. Performance of creatinine and cystatin C GFR estimating equations in an HIV-positive population on antiretrovirals. J Acquir Immune Defic Syndr. 2012;61:302–9. doi: 10.1097/QAI.0b013e31826a6c4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhasin B, Lau B, Atta MG, et al. HIV viremia and T-cell activation differentially affect the performance of glomerular filtration rate equations based on creatinine and cystatin C. PLoS One. 2013;8:e82028. doi: 10.1371/journal.pone.0082028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krane V, Wanner C. Statins, inflammation and kidney disease. Nat Rev Nephrol. 2011;7:385–97. doi: 10.1038/nrneph.2011.62. [DOI] [PubMed] [Google Scholar]
- 9.Hunt PW. HIV and inflammation: mechanisms and consequences. Curr HIV/AIDS Rep. 2012;9:139–47. doi: 10.1007/s11904-012-0118-8. [DOI] [PubMed] [Google Scholar]
- 10.Lucas G, Cozzi-Lepri A, Wyatt C, et al. Glomerular filtration rate estimated using creatinine, cystatin C or both markers and the risk of clinical events in HIV-infected individuals. HIV Med. 2013;15:116–23. doi: 10.1111/hiv.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goicoechea M, de Vinuesa SG, Lahera V, et al. Effects of atorvastatin on inflammatory and fibrinolytic parameters in patients with chronic kidney disease. J Am Soc Nephrol. 2006;17:S231–5. doi: 10.1681/ASN.2006080938. [DOI] [PubMed] [Google Scholar]
- 12.Savarese G, Musella F, Volpe M, Paneni F, Perrone-Filardi P. Effects of atorvastatin and rosuvastatin on renal function: a meta-analysis. Int J Cardiol. 2013;167:2482–9. doi: 10.1016/j.ijcard.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 13.Eckard AR, Jiang Y, Debanne SM, Funderburg NT, McComsey GA. The effect of 24 weeks of statin therapy on systemic and vascular inflammation in HIV-infected subjects on antiretroviral therapy. J Infect Dis. 2014;209:1156–64. doi: 10.1093/infdis/jiu012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aslangul E, Fellahi S, Assoumou LK, Bastard JP, Capeau J, Costagliola D. High-sensitivity C-reactive protein levels fall during statin therapy in HIV-infected patients receiving ritonavir-boosted protease inhibitors. AIDS. 2011;25:1128–31. doi: 10.1097/QAD.0b013e328346be29. [DOI] [PubMed] [Google Scholar]
- 15.Funderburg NT, Jiang Y, Debanne SM, et al. Rosuvastatin treatment reduces markers of monocyte activation in HIV infected subjects on antiretroviral therapy. Clin Infect Dis. 2014;58:588–95. doi: 10.1093/cid/cit748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fichtenbaum CJ, Yeh TM, Evans SR, Aberg JA. Treatment with pravastatin and fenofibrate improves atherogenic lipid profiles but not inflammatory markers in ACTG 5087. J Clin Lipidol. 2010;4:279–87. doi: 10.1016/j.jacl.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganesan A, Crum-Cianflone N, Higgins J, et al. High dose atorvastatin decreases cellular markers of immune activation without affecting HIV-1 RNA levels: results of a double-blind randomized placebo controlled clinical trial. J Infect Dis. 2011;203:756–64. doi: 10.1093/infdis/jiq115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker JV, Huppler Hullsiek K, Prosser R, et al. Angiotensin converting enzyme inhibitor and HMG-CoA reductase inhibitor as adjunct treatment for persons with HIV infection: a feasibility randomized trial. PLoS One. 2012;7:e46894. doi: 10.1371/journal.pone.0046894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 20.McComsey GA, Kitch D, Sax PE, et al. Peripheral and central fat changes in subjects randomized to abacavir-lamivudine or tenofovir-emtricitabine with atazanavir-ritonavir or efavirenz: ACTG Study A5224s. Clin Infect Dis. 2011;53:185–96. doi: 10.1093/cid/cir324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 22.Wilson PW, D'Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–47. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 23.Inker LA, Schmid CH, Tighiouart H, et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med. 2012;367:20–9. doi: 10.1056/NEJMoa1114248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Longenecker CT, Jiang Y, Orringer CE, et al. Soluble CD14 is independently associated with coronary calcification and extent of subclinical vascular disease in treated HIV infection. AIDS. 2014;28:969–77. doi: 10.1097/QAD.0000000000000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Longenecker CT, Jiang Y, Yun CH, et al. Perivascular fat, inflammation, and cardiovascular risk in HIV-infected patients on antiretroviral therapy. Int J Cardiol. 2013;168:4039–45. doi: 10.1016/j.ijcard.2013.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaplan RC, Sinclair E, Landay AL, et al. T cell activation predicts carotid artery stiffness among HIV-infected women. Atherosclerosis. 2011;217:207–13. doi: 10.1016/j.atherosclerosis.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seaberg EC, Benning L, Sharrett AR, et al. Association between human immunodeficiency virus infection and stiffness of the common carotid artery. Stroke. 2010;41:2163–70. doi: 10.1161/STROKEAHA.110.583856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Longenecker C, Funderburg N, Jiang Y, et al. Markers of inflammation and CD8 T-cell activation, but not monocyte activation, are associated with subclinical carotid artery disease in HIV-infected individuals. HIV Med. 2013;14:385–90. doi: 10.1111/hiv.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Funderburg NT, Zidar DA, Shive C, et al. Shared monocyte subset phenotypes in HIV-1 infection and in uninfected subjects with acute coronary syndromes. Blood. 2012;120:4599–608. doi: 10.1182/blood-2012-05-433946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ridker PM, MacFadyen J, Cressman M, Glynn RJ. Efficacy of rosuvastatin among men and women with moderate chronic kidney disease and elevated high-sensitivity C-reactive protein: a secondary analysis from the JUPITER (Justification for the Use of Statins in Prevention-an Intervention Trial Evaluating Rosuvastatin) trial. J Am Coll Cardiol. 2010;55:1266–73. doi: 10.1016/j.jacc.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 31.Strippoli GF, Navaneethan SD, Johnson DW, et al. Effects of statins in patients with chronic kidney disease: meta-analysis and meta-regression of randomised controlled trials. BMJ. 2008;336:645–51. doi: 10.1136/bmj.39472.580984.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tonelli M, Isles C, Craven T, et al. Effect of pravastatin on rate of kidney function loss in people with or at risk for coronary disease. Circulation. 2005;112:171–8. doi: 10.1161/CIRCULATIONAHA.104.517565. [DOI] [PubMed] [Google Scholar]
- 33.Rutter MK, Prais HR, Charlton-Menys V, et al. Protection against nephropathy in diabetes with atorvastatin (PANDA): a randomized double-blind placebo-controlled trial of high- vs. low-dose atorvastatin(1) Diabet Med. 2011;28:100–8. doi: 10.1111/j.1464-5491.2010.03139.x. [DOI] [PubMed] [Google Scholar]
- 34.Cooper RD, Wiebe N, Smith N, Keiser P, Naicker S, Tonelli M. Systematic review and meta-analysis: renal safety of tenofovir disoproxil fumarate in HIV-infected patients. Clin Infect Dis. 2010;51:496–505. doi: 10.1086/655681. [DOI] [PubMed] [Google Scholar]
- 35.Tonelli M, Sacks F, Pfeffer M, et al. Biomarkers of inflammation and progression of chronic kidney disease. Kidney Int. 2005;68:237–45. doi: 10.1111/j.1523-1755.2005.00398.x. [DOI] [PubMed] [Google Scholar]
- 36.Keller C, Katz R, Cushman M, Fried LF, Shlipak M. Association of kidney function with inflammatory and procoagulant markers in a diverse cohort: a cross-sectional analysis from the Multi-Ethnic Study of Atherosclerosis (MESA) BMC Nephrol. 2008;9:9. doi: 10.1186/1471-2369-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dubin R, Shlipak M, Li Y, et al. Racial differences in the association of pentraxin-3 with kidney dysfunction: the Multi-Ethnic Study of Atherosclerosis. Nephrol Dial Transplant. 2011;26:1903–8. doi: 10.1093/ndt/gfq648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serrano-Villar S, Estrada V, Gomez-Garre D, et al. Incipient renal impairment as a predictor of subclinical atherosclerosis in HIV-infected patients. J Acquir Immune Defic Syndr. 2012;59:141–8. doi: 10.1097/QAI.0b013e3182414366. [DOI] [PubMed] [Google Scholar]
- 39.Choi A, Scherzer R, Bacchetti P, et al. Cystatin C, albuminuria, and 5-year all-cause mortality in HIV-infected persons. Am J Kidney Dis. 2010;56:872–82. doi: 10.1053/j.ajkd.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jotwani V, Scherzer R, Choi A, et al. Reduced kidney function and preclinical atherosclerosis in HIV-infected individuals: the study of fat redistribution and metabolic change in HIV infection (FRAM) Am J Nephrol. 2011;33:453–60. doi: 10.1159/000327606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.