

Abstract
Exposure of zebrafish embryos to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) activates the zebrafish aryl hydrocarbon receptor 2 (AHR) to produce developmental and cardiovascular toxicity. AHR is found in the heart; however, AHR activation by TCDD is not confined to the heart and occurs throughout the organism. In order to understand the cause of cardiotoxicity, we constructed a constitutively active AHR (caAHR) based on the zebrafish AHR2 and expressed it specifically in cardiomyocytes. We show that AHR activation within the cardiomyocytes can account for the heart failure induced by TCDD. Expression of the caAHR within the heart produced cardiac malformations, loss of circulation, and pericardial edema. The heart-specific activation of AHR reproduced several other well-characterized endpoints of TCDD toxicity outside of the cardiovascular system, including defects in swim bladder and craniofacial development. This work identifies a single cellular site of TCDD action, the myocardial cell, that can account for the severe cardiovascular collapse observed following early life stage exposure to TCDD, and contributes to other forms of toxicity.
The prototype dioxin-like compound (DLC) is 2,3,7,8 tetrachlorodibenzo-p-dioxin (TCDD). This group includes some members of the halogenated aromatic hydrocarbons and polycyclic aromatic hydrocarbons (Firestone, 1973; Henry et al., 1997; Incardona et al., 2004; Thackaberry et al., 2005). In 1957, contamination of feed with DLCs caused a syndrome known as chick edema disease, characterized by pericardial and abdominal edema, killing millions of broiler chickens (Firestone, 1973; Metcalfe, 1972). A similar malady in fish, known as blue sac syndrome, characterized by pericardial and yolk sac edema, hemorrhage, reduced blood flow, with heart and craniofacial malformations had been observed in salmonid hatcheries and wild fish populations (Guiney et al., 1997, 2000; Spitsbergen et al., 1991; Walker et al., 1991; Wolf, 1969). Lake trout larvae are particularly sensitive to DLC toxicity, and the decline of lake trout populations in Lake Ontario has been correlated with elevated DLC levels that are sufficient to induce fatal blue sac syndrome (Cook et al., 2003; Tillitt et al., 2008).
The toxicity of compounds such as TCDD is mediated by the aryl hydrocarbon receptor (AHR) reviewed in Beischlag et al. (2008). In its un-activated state, AHR is primarily localized in the cytosol in a complex with a dimer of heat shock protein 90 (HSP90), and a single molecule each of p23 and XAP2 (also known as ARA9 or AIP). These chaperones stabilize the receptor, enhancing its ability to bind ligand, while protecting it from degradation. When bound by ligands such as TCDD, AHR undergoes a structural transformation that causes import into the nucleus and dimerization with the aryl hydrocarbon receptor nuclear translocator (ARNT). The AHR/ARNT heterodimer is a DNA-binding transcription factor. AHR has been associated with the regulation of many biological processes in vertebrates, including hematopoiesis, vascular development, and reproduction (Baba et al., 2005; Gasiewicz et al., 2010; Lahvis et al., 2000); however, it is best known for its role in xenobiotic metabolism. Cytochrome p450 1a (cyp1a) is strongly induced by TCDD-activated AHR and is widely accepted as a biomarker of AHR activation (Safe, 1990).
Zebrafish (Danio rerio) embryos exposed to TCDD immediately after fertilization exhibit a phenotype similar to blue sac syndrome, including pericardial and yolk sac edema, reduced peripheral blood flow and cardiomyocyte proliferation, failed regression of the common cardinal vein (CCV), with impaired heart valve and outflow tract development (Antkiewicz et al., 2005; Bello et al., 2004; Carney et al., 2006; Henry et al., 1997; Mehta et al., 2008). Recently, it has been shown that formation of the epicardial layer of the heart is also disrupted by embryonic exposure to TCDD in zebrafish (Plavicki et al., 2013).
Most, if not all, of the toxicity of DLCs is mediated by the activated AHR/ARNT heterodimer. In zebrafish, AHR2 and Arnt1c mediate the developmental toxicity of TCDD: loss of either protein protects the developing fish from TCDD-induced cardiotoxicity (Antkiewicz et al., 2006; Prasch et al., 2003, 2006). Loss of the AHR2 transcriptional activation domain (TAD) also prevents toxicity (Goodale et al., 2012; Lanham et al., 2011).
Global AHR and ARNT knockdown experiments with morpholino oligonucleotides (MOs) have shown that AHR activation somewhere within the organism is responsible for the observed cardiotoxicity (Antkiewicz et al., 2006). Although AHR and ARNT are found in the zebrafish heart, they are also expressed in essentially all other organs of the body (Andreasen et al., 2002b). This broad expression makes it difficult to determine in which organ and cell type TCDD acts to produce cardiotoxicity. Exposure at 72 hpf produces almost immediate changes in gene expression in the heart, and decreases in cardiac output are observed within 8 h of exposure (Carney et al., 2006). Although this suggests a direct effect on the heart, the loss of cardiac filling might also be explained by changes in central venous pressure and a reduction in venous return. TCDD exposure is known to increase vascular permeability (Dong et al., 2004; Guiney et al., 1997, 2000). Efforts have focused on identifying the earliest visible toxic response; however, the interdependent relationship between heart and vessel function has made it impossible to use simple observation to define the site at which TCDD activation of AHR induces cardiotoxicity.
We have approached this problem by taking advantage of previous work, in which a mouse Ahb1 mutant receptor with constitutive activity was described (McGuire et al., 2001). This approach was previously used to make a mouse that could model the effects of chronic, low-level AHR activation (Andersson et al., 2002; Brunnberg et al., 2011; Moennikes et al., 2004). We used this approach to make a constitutively active zebrafish AHR (caAHR). By using the cardiomyocyte-specific cmlc2 promoter, we have expressed our zebrafish caAHR specifically in myocardial cells, thereby activating the AHR/ARNT pathway only in cells of the zebrafish heart. Our results indicate that AHR activation in cardiomyocytes recapitulates not only TCDD developmental cardiotoxicity, but also many other endpoints of blue sac syndrome produced by TCDD in zebrafish larvae.
MATERIALS AND METHODS
Zebrafish lines, husbandry, and maintenance
Experiments were performed using wild-type AB zebrafish (Danio rerio), Tg(pard3:EGFP) (Poon et al., 2010) (generously supplied by Dr. Vladamir Korzh), Tg(flk1:EGFP), or Tg(cmlc2:caAHR-2AtRFP). Eggs were obtained from spawning adult zebrafish maintained according to procedures in The Zebrafish Book (Akimenko et al., 1995). Zebrafish eggs were collected within 4 h of spawning.
Zebrafish eggs were statically exposed in water to either TCDD (1 ng/ml) or vehicle (DMSO) in 5 ml glass scintillation vials for 1 h with rocking (Belair et al., 2001). After exposure, fish were rinsed in TCDD-free water and placed in housing aquaria until analysis.
All work was done in an ALAC-accredited facility with an IACUC-approved animal care and use protocol no. M489 approved by the University of Wisconsin School of Medicine and Public Health Animal Care and Use Committee.
Plasmid construction
All PCR reactions were performed using PfuUltraII Fusion HS DNA Polymerase (Stratagene), and all primers (Supplementary table 1) were synthesized by Integrated DNA Technologies.
pBK-CMV AHR2-FLAG was constructed by Pfu amplifying AHR2 with the HindIII-AHR2 fwd and AHR2-FLAG XmaI rev primers using the previously described pBK-CMV AHR2 plasmid as a template (Tanguay et al., 1999). pBK-CMV AHR2dbd-FLAG was constructed using a synthetic ahr2 DNA segment encoding a portion of the DNA-binding domain (DBD) with the two mutations (AHR2dbd gBlock; Supplementary table 1) digested at HindIII and AgeI and ligated into the similarly digested pBK-CMV AHR2 plasmid.
pBK-CMV:caAHR-FLAG was constructed by overlap extension. The N-terminal region was synthesized using the HindIII-AHR2 fwd primer and the ZF/M Fusion rev primer. The C-terminal region was synthesized using the ZF/M Fusion fwd primer and the mAHR FLAG XmaI rev primer. These amplicons were gel extracted and used in another PCR reaction without the addition of primers for five cycles. A 1 μl sample from that reaction was used in the final PCR reaction together with the HindIII-AHR2 fwd primer and the mAHR FLAG XmaI rev primer to synthesize the caAHR construct which was then digested and ligated into pBK-CMV at HindIII and XmaI.
pBK-CMV:caAHR−dbd-FLAG was made by digesting the sequenced pBK-CMV AHR2dbd-FLAG construct with HindIII and AgeI, gel extracting the digest and ligating the HindIII/AgeI fragment into the HindIII/AgeI digested pBK-CMV:caAHR-FLAG plasmid.
pME constructs were made using their respective pBK-CMV constructs as a template. Reactions were primed with attB1 AHR2 fwd in conjunction with either the attB2 mAHR (no stop) rev or attB2 AHR2 (no stop) rev primers as appropriate. Amplicons were used in a Gateway BP reaction (Invitrogen) to produce each pME entry plasmid.
Destination vectors were made with Gateway cloning and the Tol2kit (Kwan et al., 2007) using p5E: cmlc2 or p5E: CMV/SP6 as 5’ entry plasmids together with the appropriate pME expression vector and p3E: 2AtRFP (provided by Dr. Mary Halloran) in an LR reaction (Invitrogen). The p3E: 2AtRFP contains the coding sequence for the 18-amino acid porcine teschovirus 2A peptide (Provost et al., 2007) fused to the start of the monomeric red fluorescent protein TagRFP (tRFP) from Evrogen.
Luciferase assays
COS7 cells were seeded into 24-well plates at a density of 5 × 104 cells/well 1 day before transfection. Transfections were performed using Lipofectamine LTX (Invitrogen). On the day of transfection, media was aspirated and replaced with 400 μl of fresh serum-containing media + 100 μl of media containing serum plus pBK-CMV expression constructs in Lipofectamine LTX with PLUS Reagent. The pBK-CMV Arnt1c (200 ng) plasmid was added to each with the following expression constructs as indicated: pBK-CMV AHR2-FLAG (200 ng), pBK-CMV AHR2dbd-FLAG (200 ng), pBK-CMV caAHR-FLAG (200 ng), pBK-CMV caAHR−dbd-FLAG (200 ng), and pBK-CMV EGFP (200 ng). All wells also included the pGudluc 1.1 luciferase reporter vector (100 ng) (Garrison et al., 1996), a generous gift from Dr. Michael Denison (University of California, Davis, CA). To control for transfection efficiency, pBK-CMV β-galactosidase (100 ng) was also included in all transfections. Transfections were incubated for 4 h at 37°C then the transfection media was removed by aspiration and replaced with fresh serum-containing media. The following day, TCDD (10 nM) or vehicle (DMSO) was added and the cells were incubated overnight.
Luciferase assays were performed using The Luciferase Assay System (Promega) according to the manufacturer's instructions. Following addition of Cell Lysis solution and orbital shaking at room temperature for 20 min, cells were further triturated using a micropipette. Lysis of cells was confirmed microscopically and aliquots of 10 μl were transferred to a 96-well luminometer plate. Assays were completed using a Dynatech Laboratories ML-2250 luminometer (Chantilly, VA). Settings were adjusted to inject 50 μl of luciferase assay buffer II into each well, incubate 2 s, and then integrate luminescence over the next 10 s. The luciferase activity was normalized against β-galactosidase activity for each sample (Andreasen et al., 2002c). Significance was determined using a two-tailed Student's t-test assuming unequal variances.
RNA synthesis
Capped mRNA was synthesized using the mMessage mMachine SP6 Kit (Ambion) according to manufacturer's instructions. Synthesis reactions were allowed to go for 2 h as recommended by the manufacturer. RNA was purified by LiCl precipitation, washed in 70% ethanol, and resuspended in sterile Danieau's solution.
RNA synthesis was performed using the pDestTol2pA2-CMV/SP6:caAHR-2AtRFP or pDestTol2pA2-CMV/SP6:caAHR−dbd-2AtRFP plasmids linearized with AflII. Tol2 transposase was synthesized using pCS2FA-Tol2 transposase plasmid linearized at NotI.
Injections and transient expression analysis
Fertilized eggs were obtained from adult AB strain zebrafish bred in our laboratory as described by Westerfield (2000). Newly fertilized eggs were injected with 50 pg of either caAHR, caAHR−dbd, AHR2, or AHR2dbd expression plasmids, together with 50–100 pg of Tol2 transposase RNA in Danieau's solution at the 1–2 cell stage. For RNA injections of caAHR-2AtRFP and caAHR−dbd-2AtRFP, RNA was prepared as described and 300 pg of RNA was injected into embryos at the 1-cell stage. Embryos were screened at 24 hpf for expression of the construct using an epifluorescence microscope.
Assessment of blood flow
Embryos were injected with the relevant constructs or exposed to TCDD or vehicle as described above. At 96 hpf, larvae were placed in 4% methylcellulose and the intersegmental arteries of the posterior trunk were imaged with video microscopy. The number of red blood cells passing through a point within the vessel in 10 s was recorded for each animal. Significance was determined using a two-tailed Student's t-test assuming unequal variances.
Immunohistochemistry
Antibody staining was performed as previously described (Dong et al., 2007). The antibody against activated leukocyte cell adhesion molecule (ALCAM) was used at a 1:50 dilution in phosphate buffered saline with 4% bovine albumin serum and 0.3% Triton (PBT). Cyp1a staining used either mouse Mab 1–12–3 (0.3 μg/ml) or mouse α fish Cyp1a monoclonal (1:500 dilution, Biosense laboratories). Rabbit anti-tRFP was obtained from Axxora, LLC and used at 1:8000 dilution. Secondary anti-mouse antibodies and anti-rabbit antibodies (Alexa 488, Alexa 568; Invitrogen) were used at 1:200 dilution in PBT. Embryos were mounted in Vectashield with DAPI (Vector Laboratories). Confocal images were collected on an Olympus Fluoview FV1000 microscope. Brightest point projections were made using Olympus Fluoview software and images were processed using Adobe Photoshop.
RESULTS
Construction of a Constitutively Active Zebrafish AHR
Our initial attempts to construct a caAHR for zebrafish were based on reports that deletion of the PAS B domain of the mouse Ahb-1 allele made a mouse caAHR (McGuire et al., 2001). However, the comparable change in the zebrafish AHR2 did not produce constitutive activation as measured in an in vitro transient transfection assay. We do not know why deletion of the ligand-binding domain did not have the same effect on the zebrafish receptor as it did on the mouse Ahb1 receptor. Sequence conservation with the mouse AHR declines substantially beyond the PAS B domain of the zebrafish AHR2 and although we tried to closely match the deletion it is possible that retention or loss of a few key residues could account for differences in transactivation potential. We note that in unrelated experiments using a yeast reporter assay, the AHR2 TAD had lower activity when coupled to the Gal4 DBD than that from either ARNT or mouse AHR (Warren H. unpublished). Further deletions up to and including the acidic TAD of AHR2 did not produce high levels of constitutive activity, although we note that low levels of constitutive activity comparable to DMSO exposed controls remained. Because our goal was to obtain a caAHR, rather than explore the protein itself, we ultimately chose to make a chimeric receptor. To accomplish this we fused the activation domain from the mouse caAHR (422–805) with the amino terminal domains of the zebrafish AHR2 (1–298). In this construct, the DNA binding and dimerization domains were contributed by the zebrafish, whereas the activation domain was from the mouse (Fig. 1). We considered other well-characterized TADs such as that from VP16, but judged this to be the most conservative and well-established choice.
FIG. 1.
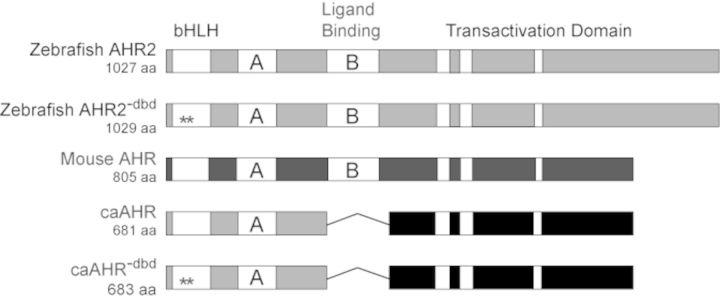
Construction of a zebrafish caAHR. Schematics of zebrafish AHR2 and mouse AHR (Ahb1) are shown with domains depicted. PAS A and PAS B domains indicated as A and B; the white bars within the transactivation domains indicate conserved acidic and Q-rich regions. The asterisks indicate Gly-Ser insertions between residues R38 and D39 in the bHLH region to make the DNA-binding mutant control constructs, AHR2−dbd and caAHR−dbd. The fusion of the caAHR chimera consisting of the amino terminus of zebrafish AHR2 (amino acids 1–298) fused to the transactivation domain of the mouse AHR (amino acids 422–805) is shown, with deletion of the PAS B region indicated by a line.
For use in control experiments, we made DNA-binding deficient AHR2 mutants, by inserting a glycine and a serine in between arginine-38 (R38) and aspartate-39 (D39). This disrupts DNA binding, but leaves other functional aspects of the receptor intact (Bunger et al., 2008). These control constructs were made for both the wild-type AHR2 and caAHR, and are referred to as AHR2−dbd and caAHR−dbd, respectively.
To test the different constructs, we used an AHRE-driven luciferase reporter assay (pGudLuc) in the Cos7 cell line. Each AHR construct was expressed from the CMV promoter, together with ARNT1c, and assayed for induction of the AHRE-driven luciferase reporter on the plasmid pGudLuc. The transfected cells were exposed to either TCDD or vehicle for 24 h prior to assay.
As has been repeatedly observed, TCDD produced an approximately twofold luciferase induction with wild-type AHR2 (Fig. 2). We also found that simply expressing AHR2 in the Cos7 cells produced luciferase activity without TCDD; however, this was dependent on co-expression of ARNT1 and the AHRE-driven reporter construct. Therefore, this TCDD-independent activity in cell culture has normal specificity for dimerization partner and DNA target motif.
FIG. 2.

Cos7 Transactivation assay. Plasmids expressing AHR2, AHR2dbd, caAHR, caAHRdbd, or GFP were transfected into Cos7 cells along with an ARNT1c expression vector and the AHRE-driven luciferase reporter pGudLuc as detailed in the Materials and Methods. Cells were treated with either TCDD (black bars) or DMSO (white bars). The average luciferase activity is shown relative to a transfection control. Error bars represent standard error of the mean. Lowercase letters indicate a significant difference between samples with other letters (P < 0.05), n = 3.
In contrast, the caAHR construct produced high luciferase activity that was completely TCDD-independent, and of a higher magnitude than produced by the normal AHR2. This showed the successful production of a constitutively active AHR2 fusion. As expected, the AHR2−dbd and caAHR−dbd constructs did not induce luciferase activity.
caAHR Activity in Zebrafish Embryos
To test the caAHR in vivo, we prepared mRNA with an open reading frame (ORF) encoding the caAHR linked to tRFP by the viral 2A peptide sequence. The 2A sequence induces ribosomal skipping, so that for every caAHR protein made, an independent tRFP protein is also made: the RFP signal indicates expression of both proteins (Provost et al., 2007). A comparable mRNA was made expressing caAHR−dbd as a control. The mRNAs were used to inject newly fertilized eggs at the 1-cell stage.
Activated AHR induces cyp1a, so we used the tRFP signal to monitor the AHR expression, and Cyp1a immunohistochemistry (green) to monitor cyp1a induction in the injected fish (Fig. 3). Although similar tRFP intensities were observed for both constructs, we observed a substantial increase in the Cyp1a signal in the caAHR-injected embryos, compared with the controls. We note that in these experiments, no TCDD was added. The induction of cyp1a by caAHR was ligand-independent, as it was in cell culture.
FIG. 3.
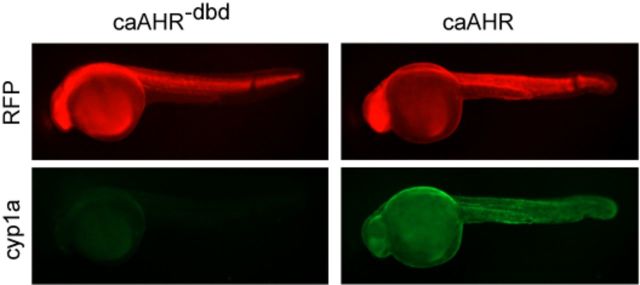
caAHR induces Cyp1a in vivo. Images show 24 hpf embryos injected at the 1-cell stage with RNA encoding the caAHR−dbd control (left column) or the caAHR (right column) as described in the Materials and Methods. RFP shows expression of the construct; Cyp1a immunostaining is shown as green; n = 10 for each condition from two separate experiments.
The caAHR−dbd mutation was made in a highly conserved region of the DBD, and the effects of this mutation have been extensively characterized in the original version. This has been shown to interrupt DNA binding but not dimerization or nuclear transport. The absence of an effect with the caAHR−dbd shows that the activity of the caAHR requires the DNA-binding function. Induction of cyp1a is consistent with this view.
caAHR Expression in Cardiomyocytes Causes Heart Malformation
To determine how AHR activation in cardiomyocytes contributes to TCDD-induced cardiotoxicity, we developed a strategy to express our newly developed caAHR specifically in cardiomyocytes using the cardiomyocyte-specific cmlc2 promoter. Our hypothesis predicts that this should cause embryonically lethal cardiotoxicity, so we focused on transient expression in DNA-injected embryos, rather than trying to generate transgenic lines.
However, a disadvantage of transient DNA injection experiments is that the DNA is incorporated randomly into the chromosomes of dividing cells, causing a mosaic pattern in which some regions of the embryo carry the transgene, and others do not. The cmlc2 promoter is cardiomyocyte-specific, so the construct was expressed only if it was incorporated into cells destined to make the heart: injected DNA remained silent if incorporated into other cell types, producing neither tRFP nor caAHR. We used the tRFP signal to identify those embryos in which the caAHR was expressed in the developing heart.
We found that those embryos expressing the caAHR throughout the myocardial layer of the heart consistently exhibited the hallmark signs of TCDD cardiotoxicity. Figure 4 shows a comparison of TCDD-exposed and caAHR-injected fish, along with their respective DMSO vehicle, and caAHR−dbd controls. The caAHR fish shows clear RFP expression in the heart, along with an un-looped, elongated heart and yolk/pericardial edema. In contrast, whereas the tRFP signal was as strong in the heart of the control injected with the caAHR−dbd (Fig. 4), the fish was normal with no signs of heart malformation.
FIG. 4.
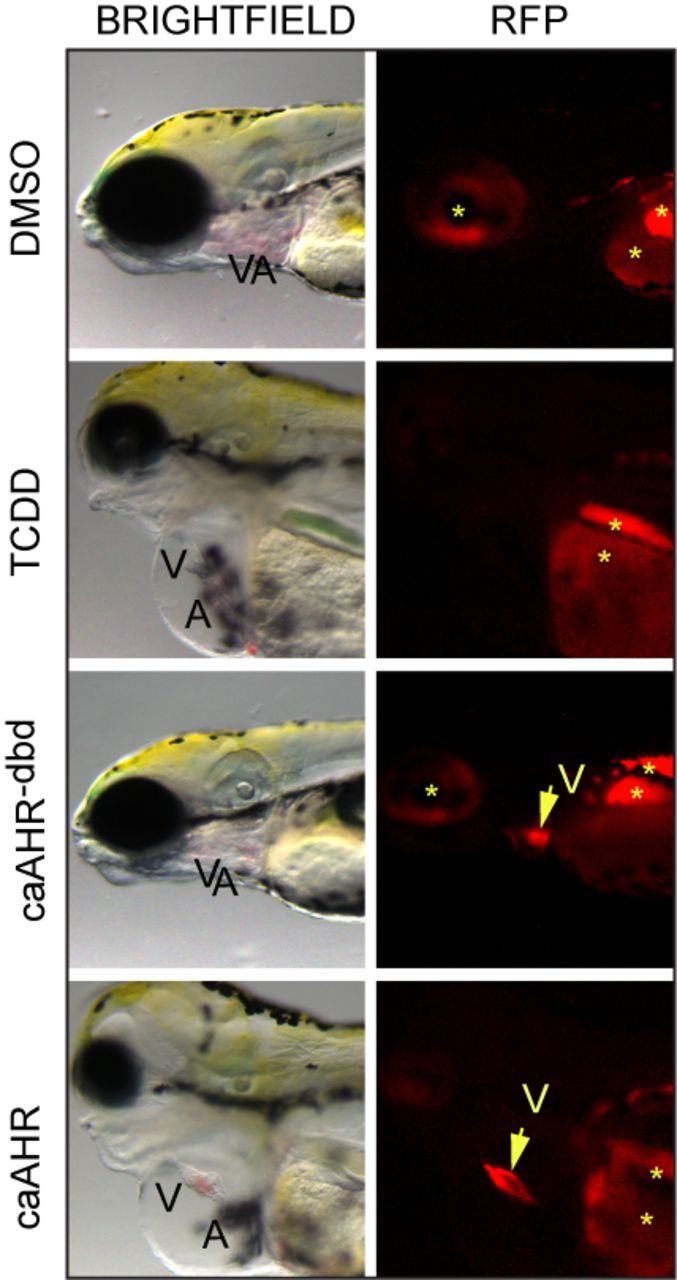
Cardiomyocyte expression of caAHR causes heart malformation. Immediately after fertilization, embryos were treated with DMSO, or TCDD in water, or they were injected with cmlc2:caAHR−dbd-2AtRFP or cmlc2:caAHR-2AtRFP DNA as described in the Materials and Methods. Brightfield (left) and RFP fluorescence (right) images at 96 hpf of representative samples. Specimens are positioned such that the heart is at center with the eye at the left edge and the yolk sac at right. Ventricle (V) and atrium (A) are labeled. The arrow indicates the ventricle, asterisks indicate regions of autofluorescence.
We consistently observed similar signs of cardiotoxicity in the TCDD-treated and caAHR positive fish: the DMSO and caAHR−dbd control fish were consistently healthy. The incidence of heart malformation and circulation collapse scored at 96 hpf was 0% for DMSO (n = 5), 100% for TCDD (n = 5), 0% for caAHR−dbd (n = 20), and 100% for caAHR (n = 21). Fish with hearts visually indistinguishable from controls were scored as negative. Fish with elongated hearts, prominent pericardial and yolk sac edema, and circulation failure were scored as positive. We observed no intermediate phenotypes: all fish matched either one or the other description.
Using normal epifluorescence microscopy, we generally observed non-specific signal in the red channel from the region of the yolk, from melanophores, and from the eye. This can be seen with some variation in all fish examined and cannot be tRFP. The only red signal that we observed that was dependent on the construct DNA was found in the heart.
Furthermore, after fixation and immunostaining with anti-RFP antibodies, only the signal from the heart was retained. This specificity is reinforced by the images in Figure 5. We compared a cmlc2-GFP line with fish injected with the cmlc2:caAHR-2AtRFP, using immunostaining to examine GFP, RFP, and caAHR-induced expression of cyp1a. From these images, it is clear that cmlc2 expression, whether from a stable transgenic line or from transient DNA injection, is restricted to the heart.
FIG. 5.
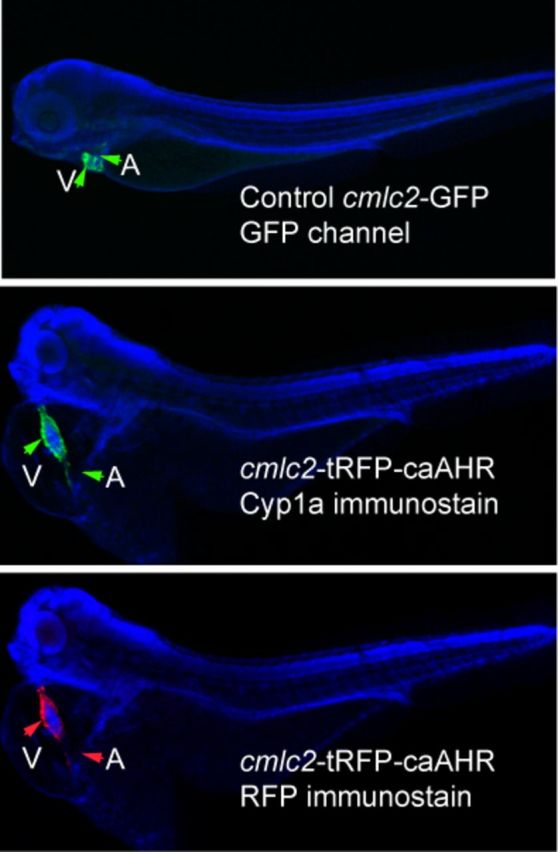
Tissue specificity of cmlc2 promoter. The figures show DAPI-mounted 72 hpf zebrafish. In all panels, the ventricle (V) and atrium (A) are indicated by arrows. The top panel shows a wild-type embryo expressing GFP from the cmlc2 promoter. The middle panel shows an embryo injected with the cmlc2:caAHR-2AtRFP DNA, stained with anti-cyp1a antibodies shown as green. The same fish is shown in the bottom panel with the tRFP indicated by red fluorescence. Close examination shows the signal restricted to the heart in both specimens.
AHR Activation in Cardiac Myocytes Disrupts Peripheral Blood Flow
We found that AHR activation within cardiomyocytes caused loss of blood flow similar to that produced by TCDD. In these experiments, blood flow was measured in posterior arteries of the trunk at 96 hpf, counting the number of red blood cells passing a marker within a 10-s interval (Carney et al., 2006; Prasch et al., 2003). Although TCDD produced almost entire circulation collapse, the caAHR expressed in cardiomyocytes was even more effective (Fig. 6). In fish treated with TCDD at 1 ng/ml, circulation completely halts very soon after the 96 hpf point. In this experiment, many had already reached a complete collapse, whereas others retained flow at a measurable level. This increased the variability in flow in the TCDD-exposed group. A higher dose of TCDD has been shown to completely halt circulation by 96 hpf (Belair et al., 2001; Henry et al., 1997); however, we chose the dose to be consistent with our extensive collection of embryo cardiotoxicity data at this level.
FIG. 6.

Cardiomyocyte expression of caAHR causes circulation failure. Immediately after fertilization, embryos were treated with DMSO, or TCDD in water, or they were injected with cmlc2:caAHR−dbd-2AtRFP or cmlc2:caAHR-2AtRFP DNA as described in the Materials and Methods. Blood flow was measured at 96 hpf as the number of red blood cells perfusing an intersegmental artery in the posterior trunk within 10 s. White bar, DMSO; black bar, TCDD; light gray bar, caAHR−dbd; and dark gray bar, caAHR. Lowercase letters indicate a significant difference between samples with other letters (P < 0.05), n = 5 for each treatment. Error bars represent standard error of the mean.
As expected, blood flow rates in vehicle-exposed control fish and those expressing the caAHR−dbd were similar. This indicates that the DNA binding is essential for the effects of the caAHR in the heart.
AHR Activation in Cardiac Myocytes Reproduces Multiple Non-Cardiac Endpoints of TCDD Exposure
Although the heart malformations and circulation collapse were consistent with our hypothesis that AHR affects the heart directly, we were surprised to find that the overall appearance of caAHR expressing fish matched that of TCDD-exposed embryos (Fig. 7). Heart-specific caAHR expression caused pericardial and yolk sac edema, loss of the swim bladder, failed CCV regression, and craniofacial malformation.
FIG. 7.
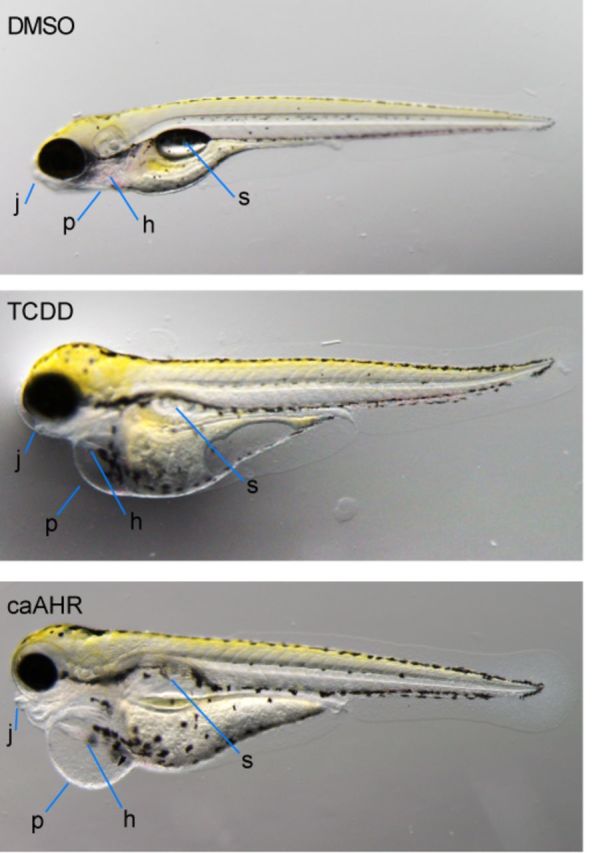
Cardiomyocyte expression of caAHR reproduces many of the effects of TCDD. Wild-type AB embryos were exposed to vehicle (A) or TCDD (B) immediately after fertilization. Tg(cmlc2:caAHR-2AtRFP) embryos (C) were collected from in-crossing founders. Representative individuals are shown, photographed at 96 hpf, n = 5 for each condition. Jaw, j; pericardium, p; heart, h; swim bladder, s.
Although all of these could be secondary to reduced blood flow, it has been previously shown that TCDD has a direct effect on the key regulator of chondrogenesis in the jaw, sox9b (Xiong et al., 2008). Fish expressing the caAHR showed yolk sac and pericardial edema that was at least as severe as that observed in TCDD-exposed embryos; however, caAHR-induced reduction in jaw development and length was milder than that caused by TCDD.
caAHR Expression in the Heart Prevents Epicardium Formation
Although we focused on transient cmlc2:caAHR expression, we carried some injected fish to adulthood to screen for cmlc2:caAHR-2AtRFP founder fish. Our assumption that fish carrying germline cmlc2:caAHR-2AtRFP would not be able to produce a viable line proved to be correct: those offspring expressing the transgene died in early larval development. This prevented us from establishing a stable transgenic line. Nonetheless, we made use of these individual fish in crosses with transgenic pard3:EGFP, and flk1:EGFP reporter lines to follow development of the epicardium, bulbus arteriosus (BA), and cardiac valves.
TCDD has been shown to block epicardium formation in zebrafish embryos (Plavicki et al., 2013). We found that AHR activation within cardiomyocytes was sufficient to reproduce this effect with striking similarity. At 96 hpf, epicardial cells have normally formed over the ventricle surface, evident as flat, elongated GFP-positive cells (Fig. 8A). In this control image, the myocardium was made visible in red by ALCAM immunostaining, outlining each cardiomyocyte, whereas the surrounding epicardial cells are indicated by arrows. The BA is also GFP positive and has developed and expanded in the control embryo.
FIG. 8.

Effect of caAHR expression in cardiomyocytes on epicardium, BA, and valve development. (A–C) Transgenic caAHR founders (cmlc2:caAHR-2AtRFP) or wild-type controls (AB) were crossed with Tg(pard3:EGFP) reporter fish marking the epicardial cells. (A) Wild-type x Tg(pard3:EGFP) offspring treated with DMSO, (B) wild-type x Tg(pard3:EGFP) treated with TCDD as described in the Materials and Methods. Embryos were collected and stained with antibodies against ALCAM (red) and stained with DAPI (blue), and representative images are shown, n = 7; 120 hpf. (C) Offspring of cmlc2:caAHR-2AtRFP founder crossed with Tg(pard3:EGFP) reporter fish. caAHR expression in the myocardium is indicated by the red RFP signal; epicardial and BA cells are green. The filled arrows indicate the BA; white solid arrows indicate normal epicardial cells. (D–E) Wild-type (AB) or transgenic caAHR founders (cmlc2:caAHR-2AtRFP) were crossed with Tg(flk1:EGFP) reporter fish. (D) Wild-type x Tg(flk1:EGFP) offspring. (E) Offspring of cmlc2:caAHR-2AtRFP founder crossed with Tg(flk1:EGFP) reporter fish. The filled arrows indicate the BA; white solid arrows indicate normal valve cushions. n = 7; 96 hpf.
As previously reported, TCDD prevented the formation of the epicardium, and no GFP signal was observed on the myocardium surface (Fig. 8B). TCDD also prevented the development and expansion of the GFP-positive BA, consistent with previous reports for TCDD and the AHR agonist PCB-126 (Grimes et al., 2008; Mehta et al., 2008; Plavicki et al., 2013). The image in Figure 8C showing the offspring of one of the caAHR founder fish crossed with the pard3:EGFP reporter line is remarkably similar to that shown for TCDD. Note that in this case it was not necessary to counter stain the cardiomyocytes as the tRFP reporter was faithfully and specifically expressed in these cells and not in surrounding tissues. The tRFP signal was indistinguishable from the ALCAM stain. As these are not mosaic, in each of the offspring with any tRFP expression, the myocardium was uniformly labeled with the tRFP marker, and the hearts were deformed in a stereotypical fashion matching the effects of TCDD. This included a compacted ventricle, elongated atrium, an underdeveloped BA, failure of CCV regression (Bello et al., 2004), and no epicardial cells expressing the pard3:EGFP marker.
In normal valve development the endothelial cells marked with a flk1-GFP reporter in Figure 8D converge to form rings at the junctions between chambers, the most notable being the bulboventricular (BV) and the atrioventricular (AV) junctions. These rings of endothelial cells further develop into primitive valve cushions and then into valve leaflets. In the figure, GFP marks the valve cushions as they are just beginning to form valve leaflets.
As with TCDD-exposed embryos, the caAHR founder offspring expressing tRFP in the heart did not form valves (Fig. 8E). When crossed with the flk1:EGFP reporter fish the endocardial cells labeled by flk1:EGFP, while present, never formed the organized rings at the junctions, and failed to form normal valve cushions or leaflets at the BV or AV junctions. The presumptive sites of valve formation are indicated by the dashed line boxes.
Effects of Mosaic caAHR Expression in the Heart
Although the mosaic expression in transient experiments produces limitations, it also provides opportunities. As described, caAHR expression varied due to stochastic mosaic incorporation that in some cases made a boundary across the heart itself. The patterns of tRFP and Cyp1a expression always and consistently coincided, as indicated by the confocal images shown in Figure 9.
FIG. 9.

Mosaic expression of caAHR in cardiomyocytes and heart malformation. Embryos were injected with cmlc2:caAHR-2AtRFP as described in the Materials and Methods and collected at 96 hpf for immunostaining with antibodies against tRFP (red) and Cyp1a (green) and stained with DAPI (blue). (A) Low magnification confocal images of four representative individuals merging all three colors. Lateral views; specimens are positioned such that the heart is at center with the eye at the left edge and the yolk sac is at right. The arrow points to the heart; the merge between RFP and Cyp1a shows as yellow. Higher magnification images of the hearts are shown below each individual. (B) RFP indicating mosaic regions of caAHR expression. (C) Cyp1a immunostaining for comparison. The atrium is indicated by A and the ventricle by V. The images represent examples from sets of n = 7 for mosaic and n = 7 for whole-heart expression.
Although expression varied in different individuals, we found that even partial coverage of the heart could have profound effects. The images in Figure 9 show caAHR expression patterns that vary from patches to large regions of the ventricle. It appears that even when AHR activation is restricted to a region of the myocardium, the entire heart can be affected, especially when the expression was found in the ventricle and at the AV junction.
Wild-Type AHR2 and Cardiotoxicity
Our caAHR required a chimeric fusion, adding the AHR carboxy-terminal activation domain from a specific mouse allele to the amino-terminal DNA binding and dimerization domains of zebrafish AHR2, and lacking the PAS B domain of both. As with similar constructs, we expected to retain the normal gene targets, but this is unproven. Even if it were, it would not rule out the possibility of some unknown gain of function.
Wild-type AHR in cell culture has long been known to produce considerable agonist-independent activity as measured with reporter plasmids (Garrison et al., 1996). This has been noted in previous work with both fish and mammalian AHR (Abnet et al., 1999; Andreasen et al., 2002a; McGuire et al., 2001; Tanguay et al., 1999, 2000). The reasons for this basal activity have never been proven, but it is known that the activity is DBD dependent, that it is ARNT dependent, and that it requires a reporter with normal AHRE sequences. Because high basal expression is associated with AHR overexpression from heterologous promoters, a popular hypothesis is that the higher than normal concentration of AHR in the cytoplasm simply allows escape of some of the AHR proteins from the chaperones and movement into the nucleus for interaction with ARNT.
This raised the possibility that cmlc2 expression of wild-type AHR2 might produce heart-specific, TCDD-independent activity in vivo. In this case, the activity would be due to a normal AHR2, rather than the altered caAHR. We tested this using the methods described above for transient overexpression of the caAHR with tRFP as a marker and a DBD mutation in AHR2 as a control.
We found that the wild-type AHR2 could also induce cardiotoxicity when overexpressed in cardiomyocytes (Fig. 10A). However, the incidence and severity of the cardiotoxicity was not as great as that seen with the caAHR (Fig. 10B). Approximately 35% of those tRFP-positive AHR2 overexpressing embryos showed the severe heart malformations produced by TCDD. As before, all of the embryos expressing the control AHR2−dbd construct were normal in appearance, showing that the observed effects required the AHR2 DNA binding activity.
FIG. 10.
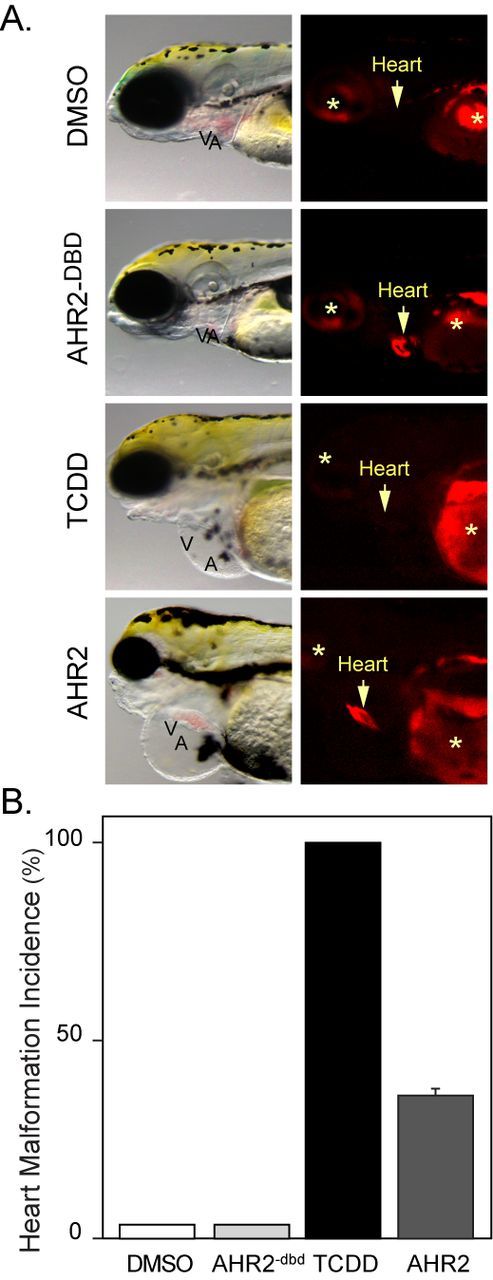
Cardiomyocyte overexpression of AHR2 and heart malformation. Embryos were exposed to vehicle or TCDD just after fertilization or injected with cmlc2:AHR2–2AtRFP or cmlc2:AHR2−dbd-2AtRFP expression constructs. (A) Brightfield and fluorescent images showing lateral views with anterior to the left. The arrow indicates position of the heart and asterisks indicate autofluorescence. (B) The incidence of heart malformation following each treatment: white bar, vehicle (n = 5); black bar, TCDD (n = 5); light gray bar, AHR−dbd (n = 18); and dark gray bar, AHR2 (n = 42). Error bar represents standard error of the mean.
We found that the expression of AHR2 was confined to the heart and was accompanied by cyp1a induction (Supplementary fig. 1). Expression of the control AHR2−dbd construct produced no cyp1a expression (not shown).
DISCUSSION
caAHR and AHR2
The finding that transcription factors contained activation domains that could be to some extent considered modular was a major event (Eilers et al., 1989; Picard et al., 1988). This allowed manipulation of transcription in many ways, including the creation of dominant negative or constitutively active proteins by the addition of repressor or transactivation domains onto the parent DBD. A constitutively active transcription factor such as caAHR could be made by as simple a manner as adding a highly active viral TAD from VP16, or as subtle as point mutations disabling a normal repressive activity. The desired effect, and a limitation to use, is that one seeks a gain of function that does not normally exist. Although useful as a biological probe, by definition the protein has abnormal characteristics. Therefore, interpreting the results requires caution.
In our case, we made the most conservative change that we could find, using an AHR TAD that had previously been shown to produce a caAHR. In previous work, the mouse Ahb1 TAD had been used before to create a potent caAHR (McGuire et al., 2001), and this proved effective with our chimeric zebrafish caAHR as well.
We have several reasons for concluding that our caAHR resembled a TCDD-activated zebrafish AHR2. First, in vitro reporter activity required ARNT1. Second, in vivo caAHR expression produced induction of cyp1a, a normal AHR2 target gene. Third, mutation of the DBD on the caAHR blocked all effects, indicating that the protein was working through DNA binding. Finally, we found that overexpression of the wild-type AHR2 reproduced the effects of the caAHR. The effects of the caAHR were stronger than those of the normal AHR, in vivo and in vitro; however, the wild-type AHR2 in this comparison is without TCDD. This shows that the caAHR needs no function that AHR2 lacks in producing the responses we observed. Furthermore, we saw no effects that are not produced by TCDD. Any off target effects produced by the caAHR were restricted to mimicking the TCDD response, not something one would expect from a non-specific response.
Normally the complete recapitulation of a biological response is a very compelling piece of evidence. Unfortunately, many different mutations and environmental insults produce pericardial edema in the embryo. Clearly, zebrafish and other fish are prone to pericardial edema in the embryo/early larval stages. We suspect that different stressors can activate a self-destructive feed forward loop, analogous to the reflex mechanisms triggered during human congestive heart failure. In both instances, distinct causes produce what is only superficially the same disease.
Looking more closely, the cardiotoxicity of TCDD has many distinct signs not replicated by other stressors. TCDD produces heart failure and edema, and also a block of epicardium formation, a failure of the endothelial cells to form rings and cushions at the presumptive valve sites, regurgitation of blood at the valves and halt in blood flow, a halt in BA expansion, and failure of CCV regression. During our long study of TCDD-induced cardiotoxicity, we have not found any non-AHR agonist that replicates TCDD toxicity. As examples, sox9b MOs produce heart failure and edema that by casual examination matches that produced by TCDD. However, the shape of the heart and the degree of circulation collapse are never the same as those produced by TCDD (Hofsteen et al., 2013). The loss of contractility produced by the silent heart sih mutant causes a superficially similar heart failure, but does not prevent epicardial progenitor formation as TCDD does (Plavicki et al., 2014).
AHR2 is present and activated in the hearts of zebrafish embryos exposed to TCDD. We conclude from our results that this activation of AHR within the heart itself is sufficient to explain the cardiovascular collapse that ensues.
AHR Activation in the Heart Produces Cardiotoxicity
Although the importance of TCDD cardiotoxicity has long been recognized, the cellular target has remained elusive because AHR is widely expressed, and TCDD is distributed throughout the body (Dong et al., 2004; Henry et al., 1997; Incardona et al., 2004). Collectively, our experiments demonstrate that AHR activation in cardiomyocytes is sufficient to cause most, if not all, of the developmental cardiotoxicity in TCDD-exposed zebrafish. Expression of the caAHR in cardiomyocytes replicated the cardiotoxicity of TCDD exposure: elongated heart, failure to form an epicardium, undeveloped BA, lack of CCV regression, extensive pericardial edema, and circulatory collapse. Expression of the caAHR throughout the myocardium was sufficient to disrupt valve formation, as observed with TCDD exposure (Mehta et al., 2008). This strongly supports the simplest model of TCDD-induced cardiotoxicity: direct activation of AHR within the heart leads to heart failure.
It is known that an important effect of TCDD on the developing zebrafish heart is blockade of epicardium formation (Plavicki et al., 2013). Hearts expressing caAHR throughout the myocardium had no epicardium. It is unclear at this time if AHR activation in the myocardium is disrupting the formation of the proepicardial organ, or whether it is acting at a later stage to prevent attachment and migration of epicardial cells across the myocardial surface.
In some instances, we show that the caAHR could produce edema and circulation collapse that were more severe than what we observed in TCDD-exposed fish. This is not surprising given that the mouse caAHR has very high activity in cell-based assays (McGuire et al., 2001).
However, this is not evidence of an unusual response. TCDD produces the same response as seen with the caAHR at 5 ng/ml, only fivefold higher than our standard 1 ng/ml dose (Belair et al., 2001; Henry et al., 1997). Continuous exposure to PCB-126 also produces an AHR2-dependent phenotype in zebrafish that occurs earlier and is more severe than what we observe with this TCDD exposure protocol (Grimes et al., 2008). Therefore, the response we observed is indistinguishable from the AHR2-dependent cardiotoxicity produced by TCDD.
Transgenic Versus Transient caAHR Expression Using cmlc2a
We used both transient and transgenic expression experiments to express caAHR in cardiomyocytes. One reason for this is that much of the transient work was completed before we were able to isolate transgenic founders. Because those offspring derived from caAHR founders that actually carried the transgene all died with cardiovascular failure we never had access to a transgenic line that was uniform and stable. Rather we had a few individual founders of limited fecundity.
Although not ideal, the situation also produced some advantages. We found that both transgenic and transient expression of caAHR in the cardiomyocytes caused heart failure and edema: the two approaches reinforced the conclusion. In addition, transient expression allowed observation of the effects of mosaic expression. Although we focused our attention on embryos that expressed the transgene throughout most or all of the heart, we were nevertheless struck by the appearance of some embryos that expressed the transgene in only parts of the heart.
When the caAHR was expressed uniformly across the heart, we always observed severe heart malformation. Occasionally, mosaicism resulted in only partial expression of the caAHR across the heart. In some cases, especially when the caAHR was expressed in the ventricle or at the AV junction, even a mosaic only partially covering the heart produced the complete response. It is not clear why this would be so, but we note that the boundaries between heart chambers, near the presumptive sites of valve formation, are important as organizer sites for cell migration, electrical stimulation, mechanical coupling of the chambers, valve formation, and haemodynamic forces needed for blood propulsion. In all cases, expression of Cyp1a coincided perfectly with tRFP expression.
The most well-known heart-specific promoter in zebrafish drives cmlc2and the promoter fragment we used has been used in many studies, producing the pattern shown in Figure 5. The inclusion of the 2AtRFP reporter should have identified any extra-cardiac sites of expression, yet none were observed.
Nonetheless, it is very difficult to demonstrate absolute tissue specificity, and it can be argued that the effects of our constructs were due to expression in some hypothetical tissue outside the heart. However, the mosaic expression produced in the transient studies bolsters the idea that the toxic effects were due to heart-specific expression. Had the effects been due to leaky expression in some other tissue, there should have been mosaic fish carrying the transgene in these hypothetical tissues but not in the heart. In other words, we should have observed toxicity in fish not expressing the transgene in the heart. The transgene DNA was dispersed into mosaic tissues, yet toxicity was only observed in fish expressing tRFP in cardiomyocytes. The fact that we only observed toxicity when we also saw caAHR expressed in the heart argues strongly against the possibility that low-level expression in another tissue drives toxicity.
Beyond the Cardiovascular System
Our initial assumptions were that AHR activation in cardiomyocytes would explain the heart failure caused by TCDD, whereas other aspects of TCDD-induced toxicity would be caused by AHR activation in other tissues. We were therefore surprised by the large number of TCDD-like effects produced by the caAHR and the wild-type AHR expressed from cmlc2. These included craniofacial malformations, massive edema, and failure to develop the swim bladder.
Reduction in blood flow has been suggested as a cause of jaw malformation and has been noted in sih embryos lacking heart function (Incardona et al., 2004). Given the demands of developing tissues, this is not surprising. However, there is also abundant evidence for a direct effect of TCDD on cartilage development as well (Abbott et al., 1989; Dong et al., 2012; Planchart and Mattingly, 2010; Xiong et al., 2008). Although this was not our focus, our observations suggested that in contrast to edema and heart malformation, the jaw defects caused by the caAHR in the heart were not as severe as those caused by TCDD. This is notable given that blood flow is reduced more rapidly and severely in embryos expressing myocardial caAHR than in TCDD-exposed embryos. We speculate that TCDD affects craniofacial development through both direct and indirect mechanisms.
Previous work has established a requirement for blood flow in swim bladder development. Using sih embryos, Winata et al. showed that blood flow is required for normal growth and inflation of the swim bladder (Winata et al., 2010). A reduction in blood flow was also postulated as a potential mechanism for the loss of swim bladder inflation in embryos exposed to the AHR agonist PCB-126 (Jonsson et al., 2012). Swim bladder inflation occurs between 96 and 120 hpf in zebrafish, a time by which both TCDD and cmlc2:caAHR essentially halt blood flow. The simplest explanation is that AHR activity in the cardiomyocytes causes swim bladder failure secondary to circulation loss.
There is evidence to support the hypothesis that the endothelium is involved in DLC-induced toxicity: brain haemorrhage is observed in adult mice exposed to acute doses of the AHR agonist 3-MC within two days of exposure (Chang et al., 2012), similar to TCDD-exposed embryonic zebrafish (Dong et al., 2004). The haemorrhage produced by TCDD appears to precede circulation loss. Although TCDD has been reported to increase vascular permeability and cause haemorrhage (Dong et al., 2004; Guiney et al., 2000), caAHR expression in the heart did not cause haemorrhage, suggesting that effects on the blood vessels are more direct.
The elongated hearts associated with either TCDD exposure or caAHR expression appear so small that one suspects cell loss and possible cell lysis leading to the release of cytokines or other intracellular mediators of the responses we observed. Although we do not rule this out, we have studied TCDD-exposed embryonic hearts extensively and have never observed signs of necrosis, and apoptosis assays are consistently negative in these heart cells.
TCDD exposure affects proper development of the cardiovascular system in fish, birds, and mammals (Antkiewicz et al., 2005; Canga et al., 1988, 1993; Ivnitski et al., 2001). Our results support a model whereby TCDD acts directly on cardiomyocytes to disrupt normal heart function and development. The resulting reduction in cardiac output then acts systemically to cause edema and disrupt blood flow-dependent developmental processes. This work highlights the importance of the vertebrate heart as a target organ of DLCs and identifies a single cellular site of action for TCDD that can account for multiple endpoints of toxicity, including early life stage mortality.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
The National Institute of Environmental Health Sciences (NIEHS) (R01 ES012716 to W.H. and R.E.P.); the University of Wisconsin Sea Grant Institute, National Sea Grant College Program, National Oceanic and Atmospheric Administration, U.S. Department of Commerce (NA 16RG2257), Sea Grant Project R/BT-25 (to W.H. and R.E.P.); and NIEHS support from T32 ES007015 (K.A.L.).
Supplementary Material
Acknowledgments
We would like to thank Dorothy Nesbit, Maggie Shuda, B. Gibbons, D. Hill, and F. Beard for outstanding technical assistance.
REFERENCES
- Abbott B. D., Diliberto J. J., Birnbaum L. S. 2,3,7,8-tetrachlorodibenzo-p-dioxin alters embryonic palatal medial epithelial cell differentiation in vitro. Toxicol. Appl. Pharmacol. 1989;100:119–131. doi: 10.1016/0041-008x(89)90096-3. [DOI] [PubMed] [Google Scholar]
- Abnet C. C., Tanguay R. L., Heideman W., Peterson R. E. Transactivation activity of human, zebrafish, and rainbow trout aryl hydrocarbon receptors expressed in COS-7 cells: Greater insight into species differences in toxic potency of polychlorinated dibenzo-p-dioxin, dibenzofuran, and biphenyl congeners. Toxicol. Appl. Pharmacol. 1999;159:41–51. doi: 10.1006/taap.1999.8719. [DOI] [PubMed] [Google Scholar]
- Akimenko M. A., Johnson S. L., Westerfield M., Ekker M. Differential induction of four msx homeobox genes during fin development and regeneration in zebrafish. Development. 1995;121:347–357. doi: 10.1242/dev.121.2.347. [DOI] [PubMed] [Google Scholar]
- Andersson P., McGuire J., Rubio C., Gradin K., Whitelaw M. L., Pettersson S., Hanberg A., Poellinger L. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc. Natl Acad. Sci. U.S.A. 2002;99:9990–9995. doi: 10.1073/pnas.152706299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen E. A., Hahn M. E., Heideman W., Peterson R. E., Tanguay R. L. The zebrafish (Danio rerio) aryl hydrocarbon receptor type 1 is a novel vertebrate receptor. Mol. Pharmacol. 2002a;62:234–249. doi: 10.1124/mol.62.2.234. [DOI] [PubMed] [Google Scholar]
- Andreasen E. A., Spitsbergen J. M., Tanguay R. L., Stegeman J. J., Heideman W., Peterson R. E. Tissue-specific expression of AHR2, ARNT2, and CYP1A in zebrafish embryos and larvae: Effects of developmental stage and 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure. Toxicol. Sci. 2002b;68:403–419. doi: 10.1093/toxsci/68.2.403. [DOI] [PubMed] [Google Scholar]
- Andreasen E. A., Tanguay R. L., Peterson R. E., Heideman W. Identification of a critical amino acid in the aryl hydrocarbon receptor. J. Biol. Chem. 2002c;277:13210–13218. doi: 10.1074/jbc.M200073200. [DOI] [PubMed] [Google Scholar]
- Antkiewicz D. S., Burns C. G., Carney S. A., Peterson R. E., Heideman W. Heart malformation is an early response to TCDD in embryonic zebrafish. Toxicol. Sci. 2005;84:368–377. doi: 10.1093/toxsci/kfi073. [DOI] [PubMed] [Google Scholar]
- Antkiewicz D. S., Peterson R. E., Heideman W. Blocking expression of AHR2 and ARNT1 in zebrafish larvae protects against cardiac toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 2006;94:175–182. doi: 10.1093/toxsci/kfl093. [DOI] [PubMed] [Google Scholar]
- Baba T., Mimura J., Nakamura N., Harada N., Yamamoto M., Morohashi K., Fujii-Kuriyama Y. Intrinsic function of the aryl hydrocarbon (dioxin) receptor as a key factor in female reproduction. Mol. Cell. Biol. 2005;25:10040–10051. doi: 10.1128/MCB.25.22.10040-10051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beischlag T. V., Luis Morales J., Hollingshead B. D., Perdew G. H. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot. Gene Expr. 2008;18:207–250. doi: 10.1615/critreveukargeneexpr.v18.i3.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belair C. D., Peterson R. E., Heideman W. Disruption of erythropoiesis by dioxin in the zebrafish. Dev. Dyn. 2001;222:581–594. doi: 10.1002/dvdy.1213. [DOI] [PubMed] [Google Scholar]
- Bello S. M., Heideman W., Peterson R. E. 2,3,7,8-tetrachlorodibenzo-p-dioxin inhibits regression of the common cardinal vein in developing zebrafish. Toxicol. Sci. 2004;78:258–266. doi: 10.1093/toxsci/kfh065. [DOI] [PubMed] [Google Scholar]
- Brunnberg S., Andersson P., Poellinger L., Hanberg A. The constitutively active Ah receptor (CA-AhR) mouse as a model for dioxin exposure - effects in reproductive organs. Chemosphere. 2011;85:1701–1706. doi: 10.1016/j.chemosphere.2011.09.015. [DOI] [PubMed] [Google Scholar]
- Bunger M. K., Glover E., Moran S. M., Walisser J. A., Lahvis G. P., Hsu E. L., Bradfield C. A. Abnormal liver development and resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in mice carrying a mutation in the DNA-binding domain of the aryl hydrocarbon receptor. Toxicol. Sci. 2008;106:83–92. doi: 10.1093/toxsci/kfn149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canga L., Levi R., Rifkind A. B. Heart as a target organ in 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity: Decreased beta-adrenergic responsiveness and evidence of increased intracellular calcium. Proc. Natl Acad. Sci. U.S.A. 1988;85:905–909. doi: 10.1073/pnas.85.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canga L., Paroli L., Blanck T. J., Silver R. B., Rifkind A. B. 2,3,7,8-tetrachlorodibenzo-p-dioxin increases cardiac myocyte intracellular calcium and progressively impairs ventricular contractile responses to isoproterenol and to calcium in chick embryo hearts. Mol. Pharmacol. 1993;44:1142–1151. [PubMed] [Google Scholar]
- Carney S. A., Chen J., Burns C. G., Xiong K. M., Peterson R. E., Heideman W. Aryl hydrocarbon receptor activation produces heart-specific transcriptional and toxic responses in developing zebrafish. Mol. Pharmacol. 2006;70:549–561. doi: 10.1124/mol.106.025304. [DOI] [PubMed] [Google Scholar]
- Chang C. C., Lee P. S., Chou Y., Hwang L. L., Juan S. H. Mediating effects of aryl-hydrocarbon receptor and RhoA in altering brain vascular integrity: The therapeutic potential of statins. Am. J. Pathol. 2012;181:211–221. doi: 10.1016/j.ajpath.2012.03.032. [DOI] [PubMed] [Google Scholar]
- Cook P. M., Robbins J. A., Endicott D. D., Lodge K. B., Guiney P. D., Walker M. K., Zabel E. W., Peterson R. E. Effects of aryl hydrocarbon receptor-mediated early life stage toxicity on lake trout populations in Lake Ontario during the 20th century. Environ. Sci. Technol. 2003;37:3864–3877. doi: 10.1021/es034045m. [DOI] [PubMed] [Google Scholar]
- Dong W., Hinton D. E., Kullman S. W. TCDD disrupts hypural skeletogenesis during medaka embryonic development. Toxicol. Sci. 2012;125:91–104. doi: 10.1093/toxsci/kfr284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong W., Teraoka H., Tsujimoto Y., Stegeman J. J., Hiraga T. Role of aryl hydrocarbon receptor in mesencephalic circulation failure and apoptosis in zebrafish embryos exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 2004;77:109–116. doi: 10.1093/toxsci/kfh023. [DOI] [PubMed] [Google Scholar]
- Dong P. D. S., Munson C. A., Norton W., Crosnier C., Pan X., Gong Z., Neumann C. J., Stainier D. Y. R. Fgf10 regulates hepatopancreatic ductal system patterning and differentiation. Nature genetics. 2007;39:397–402. doi: 10.1038/ng1961. [DOI] [PubMed] [Google Scholar]
- Eilers M., Picard D., Yamamoto K. R., Bishop J. M. Chimaeras of myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature. 1989;340:66–68. doi: 10.1038/340066a0. [DOI] [PubMed] [Google Scholar]
- Firestone D. Etiology of chick edema disease. Environ. Health Perspect. 1973;5:59–66. doi: 10.1289/ehp.730559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison P. M., Tullis K., Aarts J. M., Brouwer A., Giesy J. P., Denison M. S. Species-specific recombinant cell lines as bioassay systems for the detection of 2,3,7,8-tetrachlorodibenzo-p-dioxin-like chemicals. Fundam. Appl. Toxicol. 1996;30:194–203. doi: 10.1006/faat.1996.0056. [DOI] [PubMed] [Google Scholar]
- Gasiewicz T. A., Singh K. P., Casado F. L. The aryl hydrocarbon receptor has an important role in the regulation of hematopoiesis: Implications for benzene-induced hematopoietic toxicity. Chem. Biol. Interact. 2010;184:246–251. doi: 10.1016/j.cbi.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodale B. C., La Du J. K., Bisson W. H., Janszen D. B., Waters K. M., Tanguay R. L. AHR2 mutant reveals functional diversity of aryl hydrocarbon receptors in zebrafish. PLoS One. 2012;7:e29346. doi: 10.1371/journal.pone.0029346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes A. C., Erwin K. N., Stadt H. A., Hunter G. L., Gefroh H. A., Tsai H. J., Kirby M. L. PCB126 exposure disrupts zebrafish ventricular and branchial but not early neural crest development. Toxicol. Sci. 2008;106:193–205. doi: 10.1093/toxsci/kfn154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiney P. D., Smolowitz R. M., Peterson R. E., Stegeman J. J. Correlation of 2,3,7,8-tetrachlorodibenzo-p-dioxin induction of cytochrome P4501A in vascular endothelium with toxicity in early life stages of lake trout. Toxicol. Appl. Pharmacol. 1997;143:256–273. doi: 10.1006/taap.1996.8051. [DOI] [PubMed] [Google Scholar]
- Guiney P. D., Walker M. K., Spitsbergen J. M., Peterson R. E. Hemodynamic dysfunction and cytochrome P4501A mRNA expression induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin during embryonic stages of lake trout development. Toxicol. Appl. Pharmacol. 2000;168:1–14. doi: 10.1006/taap.2000.8999. [DOI] [PubMed] [Google Scholar]
- Henry T. R., Spitsbergen J. M., Hornung M. W., Abnet C. C., Peterson R. E. Early life stage toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in zebrafish (Danio rerio) Toxicol. Appl. Pharmacol. 1997;142:56–68. doi: 10.1006/taap.1996.8024. [DOI] [PubMed] [Google Scholar]
- Hofsteen P., Plavicki J., Johnson S. D., Peterson R. E., Heideman W. Sox9b is required for epicardium formation and plays a role in TCDD-induced heart malformation in zebrafish. Mol. Pharmacol. 2013;84:353–360. doi: 10.1124/mol.113.086413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incardona J. P., Collier T. K., Scholz N. L. Defects in cardiac function precede morphological abnormalities in fish embryos exposed to polycyclic aromatic hydrocarbons. Toxicol. Appl. Pharmacol. 2004;196:191–205. doi: 10.1016/j.taap.2003.11.026. [DOI] [PubMed] [Google Scholar]
- Ivnitski I., Elmaoued R., Walker M. K. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) inhibition of coronary development is preceded by a decrease in myocyte proliferation and an increase in cardiac apoptosis. Teratology. 2001;64:201–212. doi: 10.1002/tera.1065. [DOI] [PubMed] [Google Scholar]
- Jonsson M. E., Kubota A., Timme-Laragy A. R., Woodin B., Stegeman J. J. Ahr2-dependence of PCB126 effects on the swim bladder in relation to expression of CYP1 and cox-2 genes in developing zebrafish. Toxicol. Appl. Pharmacol. 2012;265:166–174. doi: 10.1016/j.taap.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan K. M., Fujimoto E., Grabher C., Mangum B. D., Hardy M. E., Campbell D. S., Parant J. M., Yost H. J., Kanki J. P., Chien C. B. The Tol2kit: A multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- Lahvis G., Lindell S., Thomas R., McCuskey R., Murphy C., Glover E., Bentz M., Southard J., Bradfield C. Portosystemic shunting and persistent fetal vascular structures in aryl hydrocarbon receptor-deficient mice. Proc. Natl Acad. Sci. U.S.A. 2000;97:10442–10447. doi: 10.1073/pnas.190256997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanham K. A., Prasch A. L., Weina K. M., Peterson R. E., Heideman W. A dominant negative zebrafish Ahr2 partially protects developing zebrafish from dioxin toxicity. PLoS One. 2011;6:e28020. doi: 10.1371/journal.pone.0028020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire J., Okamoto K., Whitelaw M. L., Tanaka H., Poellinger L. Definition of a dioxin receptor mutant that is a constitutive activator of transcription: Delineation of overlapping repression and ligand binding functions within the PAS domain. J. Biol. Chem. 2001;276:41841–41849. doi: 10.1074/jbc.M105607200. [DOI] [PubMed] [Google Scholar]
- Mehta V., Peterson R. E., Heideman W. 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure prevents cardiac valve formation in developing zebrafish. Toxicol. Sci. 2008;104:303–311. doi: 10.1093/toxsci/kfn095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe L. D. Proposed source of chick edema factor. J. Assoc. Off. Anal. Chem. 1972;55:542–546. [PubMed] [Google Scholar]
- Moennikes O., Loeppen S., Buchmann A., Andersson P., Ittrich C., Poellinger L., Schwarz M. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res. 2004;64:4707–4710. doi: 10.1158/0008-5472.CAN-03-0875. [DOI] [PubMed] [Google Scholar]
- Picard D., Salser S. J., Yamamoto K. R. A movable and regulable inactivation function within the steroid binding domain of the glucocorticoid receptor. Cell. 1988;54:1073–1080. doi: 10.1016/0092-8674(88)90122-5. [DOI] [PubMed] [Google Scholar]
- Planchart A., Mattingly C. J. 2,3,7,8-Tetrachlorodibenzo-p-dioxin upregulates FoxQ1b in zebrafish jaw primordium. Chem. Res. Toxicol. 2010;23:480–487. doi: 10.1021/tx9003165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plavicki J., Hofsteen P., Peterson R. E., Heideman W. Dioxin inhibits zebrafish epicardium and proepicardium development. Toxicol. Sci. 2013;131:558–567. doi: 10.1093/toxsci/kfs301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plavicki J., Lanham K. A., Hofsteen P., Peterson R. E., Heideman W. The beat goes on: Epicardium development in zebrafish. BMC Dev. Biol. 2014. in press.
- Poon K. L., Liebling M., Kondrychyn I., Garcia-Lecea M., Korzh V. Zebrafish cardiac enhancer trap lines: New tools for in vivo studies of cardiovascular development and disease. Dev. Dyn. 2010;239:914–926. doi: 10.1002/dvdy.22203. [DOI] [PubMed] [Google Scholar]
- Prasch A. L., Tanguay R. L., Mehta V., Heideman W., Peterson R. E. Identification of zebrafish ARNT1 homologs: 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in the developing zebrafish requires ARNT1. Mol. Pharmacol. 2006;69:776–787. doi: 10.1124/mol.105.016873. [DOI] [PubMed] [Google Scholar]
- Prasch A. L., Teraoka H., Carney S. A., Dong W., Hiraga T., Stegeman J. J., Heideman W., Peterson R. E. Aryl hydrocarbon receptor 2 mediates 2,3,7,8-tetrachlorodibenzo-p-dioxin developmental toxicity in zebrafish. Toxicol. Sci. 2003;76:138–150. doi: 10.1093/toxsci/kfg202. [DOI] [PubMed] [Google Scholar]
- Provost E., Rhee J., Leach S. D. Viral 2A peptides allow expression of multiple proteins from a single ORF in transgenic zebrafish embryos. Genesis. 2007;45:625–629. doi: 10.1002/dvg.20338. [DOI] [PubMed] [Google Scholar]
- Safe S. Polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs), dibenzofurans (PCDFs), and related compounds: Environmental and mechanistic considerations which support the development of toxic equivalency factors (TEFs) Crit. Rev. Toxicol. 1990;21:51–88. doi: 10.3109/10408449009089873. [DOI] [PubMed] [Google Scholar]
- Spitsbergen J. M., Walker M. K., Olson J. R., Peterson R. E. Pathologic alterations in early life stages of lake trout, Salvelinus namaycush, exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin as fertilized eggs. Aquat. Toxicol. 1991;19:41–72. [Google Scholar]
- Tanguay R. L., Abnet C. C., Heideman W., Peterson R. E. Cloning and characterization of the zebrafish (Danio rerio) aryl hydrocarbon receptor. Biochim. Biophys. Acta. 1999;1444:35–48. doi: 10.1016/s0167-4781(98)00252-8. [DOI] [PubMed] [Google Scholar]
- Tanguay R. L., Andreasen E., Heideman W., Peterson R. E. Identification and expression of alternatively spliced aryl hydrocarbon nuclear translocator 2 (ARNT2) cDNAs from zebrafish with distinct functions. Biochim. Biophys. Acta. 2000;1494:117–128. doi: 10.1016/s0167-4781(00)00225-6. [DOI] [PubMed] [Google Scholar]
- Thackaberry E. A., Nunez B. A., Ivnitski-Steele I. D., Friggins M., Walker M. K. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on murine heart development: Alteration in fetal and postnatal cardiac growth, and postnatal cardiac chronotropy. Toxicol. Sci. 2005;88:242–249. doi: 10.1093/toxsci/kfi302. [DOI] [PubMed] [Google Scholar]
- Tillitt D. E., Cook P. M., Giesy J. P., Heideman W., Peterson R. E. Reproductive impairment of Great Lakes lake trout by dioxin-like chemicals. In: Di Giulio R. T., Hinton D., editors. The Toxicology of Fishes, Unit IV, Case Studies. Boca Raton, FL: CRC Press, Taylor and Francis; 2008. pp. 819–876. [Google Scholar]
- Walker M. K., Spitsbergen J. M., Olson J. R., Peterson R. E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) toxicity during early life stage development of lake trout (Salvelinus namaycush) Can. J. Fish Aquat. Sci. 1991;48:875–883. [Google Scholar]
- Westerfield M. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio) 4th ed. Eugene, OR: University of Oregon Press; 2000. [Google Scholar]
- Winata C. L., Korzh S., Kondrychyn I., Korzh V., Gong Z. The role of vasculature and blood circulation in zebrafish swimbladder development. BMC Dev. Biol. 2010;10:3. doi: 10.1186/1471-213X-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K. Blue-sac disease of fish. 1969. No. 15, 4.
- Xiong K. M., Peterson R. E., Heideman W. AHR-mediated downregulation of Sox9b causes jaw malformation in zebrafish embryos. Mol. Pharmacol. 2008;74:1544–1553. doi: 10.1124/mol.108.050435. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.