

Abstract
Endocrine disrupting chemicals (EDCs) pose a significant threat to human health, society, and the environment. Many EDCs elicit their toxic effects through nuclear hormone receptors, like the estrogen receptor α (ERα). In silico models can be used to prioritize chemicals for toxicological evaluation to reduce the amount of costly pharmacological testing and enable early alerts for newly designed compounds. However, many of the current computational models are overly dependent on the chemistry of known modulators and perform poorly for novel chemical scaffolds. Herein we describe the development of computational, three-dimensional multi-conformational pocket-field docking, and chemical-field docking models for the identification of novel EDCs that act via the ligand-binding domain of ERα. These models were highly accurate in the retrospective task of distinguishing known high-affinity ERα modulators from inactive or decoy molecules, with minimal training. To illustrate the utility of the models in prospective in silico compound screening, we screened a database of over 6000 environmental chemicals and evaluated the 24 top-ranked hits in an ERα transcriptional activation assay and a differential scanning fluorimetry-based ERα binding assay. Promisingly, six chemicals displayed ERα agonist activity (32nM–3.98μM) and two chemicals had moderately stabilizing effects on ERα. Two newly identified active compounds were chemically related β-adrenergic receptor (βAR) agonists, dobutamine, and ractopamine (a feed additive that promotes leanness in cattle and poultry), which are the first βAR agonists identified as activators of ERα-mediated gene transcription. This approach can be applied to other receptors implicated in endocrine disruption.
Keywords: estrogen receptor α, bisphenol A (BPA), endocrine disrupting chemicals, green chemistry, molecular docking, virtual ligand screening
Endocrine disrupting chemicals (EDCs) are synthetic or natural compounds that mimic or alter the synthesis, metabolism, mechanism of action or excretion of hormones in humans or other organisms. Exposure to EDCs, particularly during developmental stages, can lead to permanent health effects (Colborn et al., 1993), including reproductive deformities, cancer, and obesity in humans (Diamanti-Kandarakis et al., 2009). Additionally, EDCs can have detrimental effects on the endocrine systems of wildlife (Colborn et al., 1993). EDCs are found in numerous sources including pharmaceuticals, pesticides, and plasticizers, and act at a variety of proteins nuclear hormone receptors (NRs), such as the estrogen, androgen, and thyroid hormone receptors. Small molecule binding to the ligand-binding domains (LBD) of NRs promotes a sequence of molecular events ultimately resulting in the transcription of genes linked to specific hormone response elements (Heldring et al., 2007). The estrogen receptors (ER) α and β are among the most commonly activated receptors by EDCs, including the plasticizer bisphenol A (BPA) (Diamanti-Kandarakis et al., 2009).
EDCs pose the biggest threat once they are in widespread use; therefore, the early identification of EDC potential of newly synthesized compounds is crucial. In the past century over 80,000 chemicals were introduced into the human environment. Although programs like ToxCast (Dix et al., 2007) and Tox21 (Collins et al., 2008) are currently screening ∼2000 and 8000 chemicals, respectively, thorough toxicological evaluation of each new chemical is unfeasible. Thus new methods need to be developed, like the Tiered Protocol for Endocrine Disruption (TiPED) that proposes a five-tiered approach from computational to in vivo tests, to prioritize and evaluate chemicals (Schug et al., 2013).
Computational models are increasingly employed for chemical prioritization, such as in the initial screening tier of the TiPED protocol. Historically, quantitative structure-activity relationship (QSAR) models that correlate the physicochemical properties of chemicals with biological activity have been the predominant models for compound toxicity predictions (e.g., Shi et al., 2000). The major drawback of QSAR models is their limited ability to identify novel chemotypes because they are typically derived from small chemical databases (Agatonovic-Kustrin et al., 2011), or focused on specific chemotypes (Papa et al., 2010). Promisingly, a recent study by Zhang et al. (2013) retrospectively developed both QSAR and structure-based models for ERα and ERβ, and the VirtualToxLab has developed a workflow incorporating multi-dimensional QSAR and docking for 16 toxicity targets (Vedani et al., 2012). However, robust computational techniques, with chemical training-independent sets, are required to prospectively identify new EDCs.
Docking-based database screening can be employed as a chemically unbiased method for the identification of novel EDCs (Kufareva et al., 2012a; Park et al., 2010; Rueda et al., 2012). Screening against a pocket-based model is an exceptionally powerful method for chemical activity prediction, whose predictive power can be further increased by considering the conformational plasticity of the binding pockets via the ensemble docking approach (Rueda et al., 2009, 2012). Alternatively, compounds can be screened against the three-dimensional (3D) chemical fields of active ligands in their crystallographic conformations (Wolber and Langer, 2004). This can be implemented using Atomic Property Fields (APF), with seven grid potentials describing the preference for the chemical properties of ligand atoms at each point in space, averaged across multiple superimposed ligands (Totrov, 2008). This approach, though ligand-based, is less biased toward known chemistry than QSAR, because it captures the important interaction features of the active compounds in their bound conformation (but not their chemical structure) and accentuates the key features of the superimposed chemicals.
It is essential that benchmarking be employed to ensure high prediction accuracy of all compound activity models, by testing their ability to retrospectively discriminate known active compounds from inactive or decoy compounds. By using diverse benchmarking sets, this procedure not only estimates the predictive power of each model but also helps to determine the parameters and cutoffs for prospective compound activity predictions.
Herein, we describe the development, benchmarking, and application of chemical field-based and pocket-based models for the chemically unbiased computational identification of novel EDCs that act via the LBD of ERα. The models were used to prospectively screen the Tox21 database that interact with ERα, but these techniques could be easily applied to other toxicity targets. Pharmacological evaluation of the top scoring previously uncharacterized compounds confirmed the accuracy of the computational predictions and led to the identification of six chemicals with agonist activity against the human ERα and two compounds with moderately stabilizing effects against ERα.
MATERIALS AND METHODS
Ligand preparation, preparation of APF maps, virtual ligand screening (VLS), and analyses were carried out in ICM version 3.7-3 (Molsoft L.L.C., La Jolla, CA) (Abagyan and Totrov, 1994; Abagyan et al., 1994).
Collection and preparation of ERα LBD complex structures for docking and VLS
Co-crystal structures of the ERα were obtained from the Pocketome database (Kufareva et al., 2012b). The ERα pocketome entry contained 64 unique co-crystal structures, including 52 unique co-crystalized ligands, which were used as seed ligands for the development of the APF model. Complexes were prepared for docking and screening in ICM. During the preparation, crystallographic water molecules were deleted, missing or disordered side chains rebuilt and optimized, formal charges assigned to the ligand molecules at pH 7.4 using the pKa prediction model in ICM, hydrogen atoms added, rotatable polar hydrogen optimized, and optimal rotamers/tautomer forms of His, Pro, Asn, and Gln determined.
Collection and preparation of chemical datasets
For model benchmarking, compound structures (in SMILES format) and activity data against ERα were obtained from ChEMBL (Target ID: CHEMBL206) (Gaulton et al., 2012). Bioactivity data in the forms of IC50, ED50, EC50, and Ki were combined and used without differentiating between the type of activity. For benchmarking purposes, compounds were artificially divided into two classes, active (activity of < 1μM) and inactive (activity of > 10μM); the gray zone compounds with activity between 1μM and 10μM were discarded.
The National Center for Toxicological Research Estrogen Receptor (NCTRER) binding database (NCTRER_v4b_232_15Feb2008) (Blair et al., 2000; Branham et al., 2002; Fang et al., 2001) ligand dataset was combined with the ChEMBL dataset, as well as the ERα library from the Database of Useful Decoys (DUD) (Huang et al., 2006), to create a benchmarking library. For compounds with multiple data points, the data points were averaged if they were consistent or removed from consideration otherwise. Duplicate compounds were removed and the final benchmark dataset for ERα contained 1691 active and 4785 inactive/decoy compounds.
For prospective screening, the Tox21 database (Collins et al., 2008) was used. In its March 2012 release (Tox21_v2a_8193_22Mar2012), it contained over 8000 potentially hazardous chemicals. Duplicate compounds, chemical mixtures, polymers, inorganic, and organometallic compounds, as well as chemicals with less than seven atoms or molecular weight below 120 Da or above 650 Da were removed. In total 6885 chemical structures were retained for VLS.
Database molecules in the SMILES format converted to two-dimensional (2D) structures using ICM. They were then standardized by removing salts and explicit hydrogens, standardizing chemical group topology, enumerating stereoisomers of racemic compounds, and assigning formal charges at pH 7.4 using the pKa prediction model in ICM. For the ChEMBL, DUD, and NCTRER databases, an activity column was added, with active compounds set to 1 and decoys or inactive compounds set to 0. Compounds were converted from 2D to 3D using ICM.
Construction and optimization of pocket-based model for compound activity prediction
Automatic Ligand-guided Backbone Ensemble Receptor Optimization (Katritch et al., 2012; Rueda et al., 2012) docking protocol was used for ensemble docking to identify the optimal ERα crystal structure ensemble. The binding site was defined by residues within 4 Å of all co-crystallized ligands. The optimal recognition for the benchmark was achieved by an ensemble consisting of five ERα crystal structures with PDB IDs 1L2I, 2QE4, 2JFA, 3DT3, and 3OS9.
Construction and optimization of 3D chemical fields for compound activity prediction
Fifty-two unique co-crystallized ligands were extracted from the ERα Pocketome entry (the highest resolution crystal structure was retained for duplicated, co-crystallized ligands) and were used to build the APF maps as described in Abagyan et al. (2012) and Totrov (2008). Benchmark compounds were docked into the APF grid potential allowing torsional variable and flexible ring sampling, with a thoroughness setting of 4 and a steric factor was calculated (which penalizes atoms docked outside the APF cloud). For the best compound pose, the APF score was calculated as a sum of the scores in the seven APF potential grid maps. A steric factor with a coefficient of 2, which represents the envelope fit, was added to the sum of the seven APF scores, yielding the final APF score.
Data analysis of VLS results
VLS performance, for both pocket- and chemical field-based methods, was evaluated using receiver operating characteristic (ROC) curves, where the rate of true positives (TPs) is plotted versus the false positive (FP) rate for all compounds in the ranked list (Kirchmair et al., 2008). The area under the ROC curve (AUC) is a metric that can be used to evaluate VLS performance. For an ideal discrimination, where all the TPs rank higher than the FPs, the AUC is 100, whereas for random discrimination the AUC is 50. The TP rate can also be plotted versus the square root of the FP rate (x = √FP) for all compounds in the ranked list, to give the square root ROC curve. Similar to AUC, the normalized square-root AUC (NSQ_AUC) metric can be used to evaluate VLS performance, where AUC* is obtained from a plot of the TP versus the square root of the false positive rate (x = √FP). An NSQ_AUC is 100 for ideal discrimination and 0 for random discrimination. The NSQ_AUC metric emphasizes early VLS enrichment, and as a result, was used to assess VLS performance (Katritch et al., 2010).
Normalization of the docking scores
In all models, the predicted scores (docking or APF scores) were converted to a universal scale, which approximates the probability of compound being inactive (Supplementary fig. 1). The score transformation was calculated from the distribution of the scores of inactive and decoy compounds (from the benchmarking dataset). Assuming that a lower value for the score corresponds to a higher likelihood of compound activity, the cumulative distribution was built for the lowest 25%-ile of inactive compound scores. This results in a cumulative distribution function, F:R→[0,1], where F(S) represents the fraction of inactive compounds with binding scores not exceeding S. Preliminary studies show that this function is usually closely approximated by an exponential function, and therefore that F′(S) = log10F(S) is approximately linear. Linear regression is used to approximate F′(S) as A×S+B. Finally, the derived values of A and B can be used to approximate, for every new compound score, the probability that this score was produced by an inactive compound, i.e., the p-value for the hypothesis about compound activity (p-value = 10−F’(S)). For the pocket-based method A = 0.12 and B = 2.4 and for the chemical field-based method A = 0.053 and B = 5.2. These values were used to normalize the docking and APF scores for the Tox21 database, respectively.
Cutoff criteria for prospective VLS
Chemicals in pocket-based VLS with calculated p-values of less than 0.1 (207 chemicals) and chemicals in chemical field-based VLS with calculated p-values of less than 0.1 (188 chemicals) were considered for pharmacological evaluation.
Chemicals
Bisphenol A and 4,4'-thiodiphenol were purchased from ACROS Organics. C.I. basic violet 14, fendiline hydrochloride and p-xylenol blue were purchased from Alfa Aesar. Seratrodast was purchased from Enzo Life Sciences. Bromadiolone, chlorophacinone, lasalocid sodium (in acetonitrile), ractopamine, sulfan blue, and 2',4',5',7'-tetrabromofluorescein were purchased from Fluka. Amiodarone, C.I. basic red and dobutamine were purchased from MP Biomedicals. Ezetimibe was purchased from Selleck Chemicals. Dimethyl sulfoxide (DMSO), 17β-estradiol, lercanidipine, melengestrol acetate, and 2,2',2'',2'''-[1,2-ethanediylidenetetrakis(4,1-phenyleneoxymethylene)]tetrakis-oxirane were purchased from Sigma-Aldrich. Hydralazine HCl was purchased from Spectrum Chemical Manufacturing Corp. Brilliant green, 4,4'-butylidenebis(6-tert-butyl-m-cresol), diphenolic acid, 4,4'-(4-methylpentane-2,2-diyl)diphenol, and pamoic acid were purchased from TCI America. CAS numbers are provided in Supplementary table 1.
For the transactivation assays, test compounds were prepared at 1000-fold the final well concentrations, across seven concentrations using 1:10 serial dilutions from the highest concentration (highest stock concentration was usually 10mM). For the differential scanning fluorimetry (DSF) experiment, 10mM stock solutions of test compounds were used. All chemicals were diluted in DMSO except for hydralazine HCl and lasalocid sodium, which were diluted in water. All stock solutions were stored at −20°C.
Plasmids
The pCXN2-hERα vector encoding full-length human ERα (Hitoshi et al., 1991) and an MTV-ERE-Luc vector encoding firefly luciferase under an MTV promoter and an estrogen response element (Umesono and Evans, 1989) were kindly provided by Prof. B. Blumberg (University of California, Irvine), and the pET15b-His6-ERα(302-552) plasmid (Eiler et al., 2001) was kindly provided by Prof. D. Moras (University of Strasbourg). These vectors were propagated in XL10 Gold competent cells and purified with NucleoBond Xtra Midi kit (Clontech).
ER transcriptional activation assays
COS-7 cells were maintained in phenol-red free Dulbecco's Modified Eagle Medium (Invitrogen), supplemented with 10% charcoal/dextran stripped fetal bovine serum (Gemini Bio-products) in humidified conditions with 5% CO2 at 37°C. Cells were seeded at a concentration of 2 × 106 cells/10 cm dish and 24 h later transiently co-transfected with pCXN2-hERα (12 μg) and MTV-ERE-Luc (12 μg) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Five hours following transfection, cells were re-plated in 90 μl/well of the culture media at a density of 4 × 104 cells/well into 96-well black-walled microtiter plates (BD Falcon). 1000× DMSO stocks of chemicals were diluted 100-fold in culture media and 10 μl of the resulting dilution was added to 90 μl of cell suspension in triplicate, resulting in 1× desired final concentration of the chemicals and 0.1% of DMSO (vol/vol) per well. Vehicle (0.1% DMSO (vol/vol)) and 17β-estradiol (seven concentrations ranging from 1μM to 1pM) were included on each plate for quality control. Following stimulation, the cells were cultured for another 16–24 h and then visually inspected for evidence of cytotoxic or cytostatic effects of the test compound. Luciferase activity was determined by adding 100 μl of Steady-Glo reagent (Promega) per well, according to manufacturer's instructions, and measuring luminescence using a Victor X Light Luminescence Plate Reader (Perkin–Elmer). Each compound was tested in triplicate at seven concentrations + DMSO vehicle control, and each experiment was repeated at least twice on different days.
Data analysis of transcriptional activation experiments
Transactivation experiments were analyzed using a non-linear regression (Prism 6, GraphPad Software, La Jolla, CA). Data were normalized to the maximal response observed for 17β-estradiol. A sigmoidal-dose response curve (variable slope) was used as a model for data analysis and EC50 value calculation.
ER production and purification
BL21(DE3)pLysS competent cells were transformed with a plasmid encoding His6-ERα(302-552) (Eiler et al., 2001). Luria Broth medium (2.5% w/v) with carbenicillin (100 μg/ml) was inoculated with the transformed cells and grown at 37°C to an OD600nm of 0.6-0.8. The culture was induced with 0.5mM isopropyl β-D-1-thiogalactopyranoside (Mediatech) for 4 h at 37°C in the presence of 10μM 17β-estradiol. Cells were harvested by centrifugation at 4500 × g at 4°C for 20 min stored at −20°C.
Cells were resuspended in ice-cold buffer (500 μl, pH 7.5; 50mM Tris-HCl, 50mM NaCl, 20mM β-mercaptoethanol) and lysed using FastBreak Cell Lysis Reagent (Promega) in the presence of protease inhibitor cocktail (Sigma-Aldrich; P8849) and DNase I (Roche; 04536282001). The lysate was clarified by centrifugation at 14,000 × g for 40 min at 4°C. The cleared cell lysate was purified using a Micro Bio-Spin (Bio-Rad) chromatography column containing Profinity immobilized metal affinity chromatography nickel-charged resin (Bio-Rad), using the following buffer (pH 7.5; 50mM sodium phosphate, 500mM NaCl), with varying concentrations of imidazole; binding (10mM), wash (20mM), and elution (250mM) and analyzed with sodium dodecyl sulfate polyacrylamide gel electrophoresis. The protein was dialyzed overnight into the running buffer (pH 7.5; 50mM HEPES, 500mM NaCl, 10mM dithiothreitol (DTT), 5% glycerol) (DeSantis et al., 2012).
DSF
DSF experiments were undertaken using a Rotor-Gene Q 6-plex (Qiagen) to evaluate stability of purified His6-ERα LBD in the presence or absence of test compounds. Samples were prepared in a buffer of HEPES (50mM), NaCl (500mM), DTT (10mM), and glycerol (5%) at pH 7.5 (DeSantis et al., 2012). Experiments were performed in triplicate with a final volume of 50 μl and contained chemicals at a final concentration of 10μM (with a maximum DMSO concentration of 0.1%), protein at a final concentration of 1μM, and SYPRO Orange dye (Sigma-Aldrich) at a final concentration of 2×. The increase in fluorescence intensity of the latter was used to monitor the thermal denaturation of the protein, using the yellow channel (excitation 530 nm and emission 557 nm; gain 10). For the DSF assay, the test samples were heated gradually from 28°C to 95°C at a rate of 1°C/step, recording fluorescence at every 1°C increase, waiting 5 s between each step. The melting temperature (Tm) was determined from the first derivative plot of the denaturation curve, as calculated using the Rotor-Gene Q – Pure Detection software (version 2.0.3).
RESULTS
Two Types of in Silico 3D Models for Prediction of Compound Estrogenic Activity: The Pocket-Based Model and the Chemical Field-Based Model
In this work, we constructed two classes of 3D models that can accurately predict the potential estrogenic activity of a new chemical. An optimal ensemble of receptor crystal structures of ERα, each converted into a set of potential grid maps, forms the pocket-based model. Prediction of the estrogenic activity for a new chemical is performed by flexible docking of that chemical to the pocket maps, with subsequent full-atom re-scoring of the energetically favorable poses to determine their steric and electrostatic complementarity to the pocket. Ensembles of receptor conformations are used because they have been shown to improve the VLS recognition of active chemicals (Park et al., 2010; Rueda et al., 2009). The second method is an aggregated potential field representing the preferred chemical features of high-affinity ERα ligands at each 3D point inside the binding pocket. For compound activity prediction, each compound is also conformationally sampled in this field and each pose is scored in terms of its similarity to the field features. In both types of models, the statistical significance of the obtained scores is calculated, thus approximating the likelihood of the corresponding compound being inactive.
Model Benchmarking and Parameterization
Prior to undertaking large-scale VLS of chemical libraries with unknown activity at ERα, the performance of the models was evaluated using a chemical library of known active and inactive/decoy compounds. The library was screened against both models and the preferential ranking of active ligands over inactive/decoy compounds was used as a measure of the model performance.
The optimal pocket-based model consisting of five crystal structures (PDB IDs: 1L2I, 2QE4, 2JFA, 3DT3, and 3OS9) was able to efficiently separate known active compounds from inactive/decoy molecules with an AUC of 83.9 and an NSQ_AUC of 69.5 (Supplementary fig. 2B). These crystal structures were co-crystallized with agonists, antagonists, and partial agonists, and should, therefore, be able to capture a range of ligands in the pocket-based model. To further avoid the conformational bias of the receptor, chemical field-based models were also developed. The recognition performance of the cumulative 3D chemical field model was even higher than the pocket-based model, with an AUC of 94.5 and the NSQ_AUC of 88.1 (Supplementary fig. 5B). Based on the benchmarking performance, we believe that this chemical field-based model could be used for predictive purposes to identify compounds that interact with the LBD of ERα.
The docking or APF scores for all benchmark decoy and inactive compounds were used to derive parameters for conversion of the scores into a normalized universal scale approximating the probability of a compound with a given score being inactive (see Materials and Methods).
Application of the Models to Prospective Identification of Novel EDCs
Following the development of pocket-based and chemical field-based models that both displayed high recognition for known active chemicals in the benchmarking dataset, we began prospective screening for novel EDCs that interact with the LBD of ERα. The Tox21 database contains over 8000 structurally diverse, potentially hazardous chemicals (Collins et al., 2008), including industrial chemicals, pesticides, food-additives, and pharmaceuticals. As such, Tox21 was an ideal dataset for prospective screening to evaluate the predictive ability of the pocket- and chemical field-based models. The results predicted active compounds from the pocket-based and chemical field-based models were combined to select compounds for pharmacological evaluation. Some overlap was observed between the two types of models, however, not all active compounds were identified by both models making these techniques complementary. Promisingly, 34% of the compounds identified were known to be active at ERα, with a further 11% comprising vitamins and endogenous compounds, and only 8% of the chemicals identified were known to be inactive (Fig. 3). Of the 47% of chemicals that had not previously been evaluated at ERα, a diverse range of chemicals (7%) were selected for pharmacological evaluation, with the purpose of confirming the in silico predictions and prioritizing chemicals for further toxicological studies.
FIG. 3.

The proportion of the predicted active compounds (from both the pocket-based and chemical field-based models) in the Tox21 database that were known to be active, inactive, or had been untested at ERα. 7% of the compounds that were predicted to be active, yet were untested, were selected for pharmacological evaluation.
The majority of the chemicals in the Tox21 database scored poorly in the pocket-based VLS, indicating a lack of activity against ERα (Fig. 1A). Promisingly, approximately half of the top-ranked chemicals identified by the pocket-based model were already known to interact with ERα (91/207 chemicals) or were vitamins that had not been evaluated pharmacologically at ERα (4/207 chemicals). These chemicals included agonists such as diethylstilbestrol and equilin, antagonists including fulvestrant and selective estrogen receptor modulators (SERMs), such as raloxifene and bazedoxifene (Supplementary table 2). This method also identified known EDCs that interact with ERα, including the naturally occurring polyphenol resveratrol, and the mycotoxins zearalanol and zearalenone (Supplementary fig. 4). Additionally, this method also identified potential BPA replacements, such as bisphenol S, as potentially active compounds. Only 14 out of the 207 compounds identified in the pocket-based VLS had previously been demonstrated to be inactive at ERα. Out of the 207 predicted EDCs identified in the pocket-based VLS, 98 chemicals were potentially novel because their estrogenic activity had not been previously evaluated pharmacologically (Supplementary fig. 6).
FIG. 1.
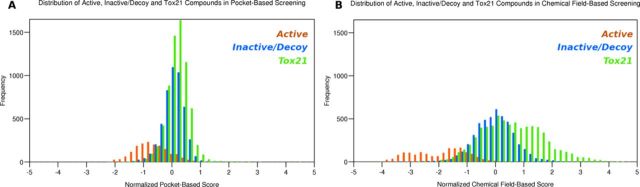
Distribution of the pocket-based VLS (A) and chemical field-based VLS (B) for the Tox21 database (green), compared with the benchmarking library (actives—orange; inactives—blue).
Similar to the pocket-based model, the majority of the Tox21 chemicals were predicted to be inactive molecules at ERα in the chemical field-based model (Fig. 1B). It was also promising to note that many of the top-ranked chemicals identified by the chemical field-based model were already known to interact with ERα (60/188 chemicals) or were steroid-based compounds (39/188 chemicals). Specifically, active chemicals included known agonists such as estriol, known antagonists such as fulvestrant, and known SERMs such as 4-hydroxytamoxifen and raloxifene (Supplementary table 3). Furthermore, the method also identified known EDCs such as BPA, an EDC, and phenol red, which is a pH indicator and a weak ERα agonist (Fig. 2A). Only 14 of the compounds identified in the chemical field-based VLS had previously been identified as inactive at ERα. Out of the 188 chemicals identified in the chemical field-based VLS, 75 compounds had not been previously evaluated pharmacologically at ERα (Supplementary fig. 6).
FIG. 2.
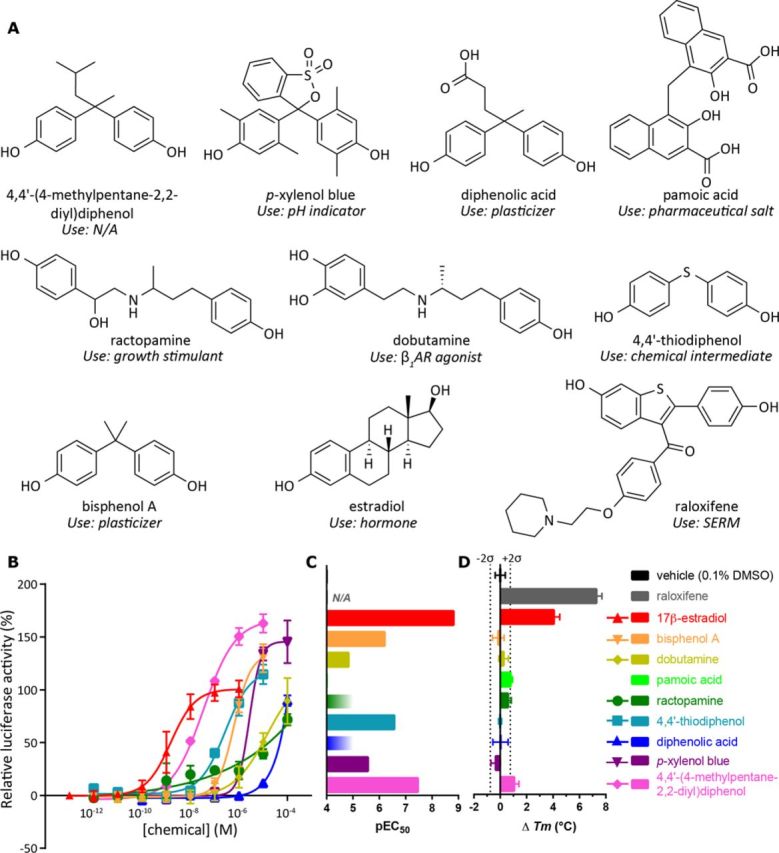
Active chemicals identified in the transactivation and/or thermal shift assays, as well as the compounds used as controls (A), the dose-response curves from the transactivation assays (B), the EC50 and Tm values for active compounds (C), and the change in Tm with respect to the vehicle (D).
Six Compounds Displayed Agonist Activity in an ERα Transcriptional Activation Assay
From the sets of chemicals identified in the pocket-based and the chemical field-based VLS, 24 chemicals (Supplementary table 1) were selected for preliminary pharmacological evaluation at ERα based on their potential for widespread use and commercial availability. Their ability to cause ERα-mediated activation of transcription of a reporter gene (firefly luciferase) was studied in a cell-based assay.
Of the 24 compounds, six chemicals displayed agonist activity (Fig. 2, Supplementary table 1). The chemicals that had ERα agonist activity included three compounds that closely resemble BPA, specifically 4,4′-(4-methylpentane-2,2-diyl)diphenol, 4,4'-thiodiphenol, and diphenolic acid. p-Xylenol blue showed weak ERα agonist activity and is structurally related to phenol red, which is also known to be a weak ERα agonist. Finally, two chemically similar β-adrenergic receptor (βAR) agonists, a β1AR agonist dobutamine, and a growth stimulant ractopamine, were also weakly active at ERα. Full dose-response curves could not be obtained for all chemicals due to low compound solubility at high chemical concentrations, thus their dose-response curves were incomplete (Fig. 2B). Judging by the shape of the dose-response curves for ractopamine and p-xylenol blue, we cannot exclude the possibility that the interaction stoichiometry between ERα-LBD and these compounds is different from 1:1. It is worth noting that each of ractopamine, dobutamine, and p-xylenol blue was tested in at least two independent transactivation assays, each performed in triplicates, and in all cases we observed robust increase in reporter expression in response to increasing concentrations of the chemicals. Such increase was not observed in the absence of receptor (supplementary fig. 9A) and thus was ERα-mediated. No compound aggregation was observed in any of the assays. Cell viability was not affected by even the highest concentration of the tested chemicals. We are therefore convinced that these chemicals do act as ERα agonists.
DSF evaluation of the predicted EDCs
The 24 chemicals identified as potential EDCs by the in silico screening were also assessed for direct binding to purified ERα using DSF (Supplementary table 1). The ERα melting temperature (Tm) in the presence of 0.1% DMSO vehicle was 61.2 ± 0.4°C. 4,4'-(4-methylpentane-2,2-diyl)diphenol, which was identified as the most active compound in the functional assay, had the most stabilizing effect on ERα (Tm = 62.2 ± 0.4°C). Pamoic acid (Tm = 61.9 ± 0.1°C) also had a moderate stabilizing effect upon ERα. The potent estrogenic hormone, 17β-estradiol, had a significantly stabilizing effect upon ERα (Tm = 65.2 ± 0.4°C), as did the SERM raloxifene (Tm = 68.4 ± 0.4°C).
Several compounds that were active in the transcriptional activation assay showed no significant temperature shift in this binding assay. This was the case, for example, for the known endocrine disruptor BPA (Tm = 61.0 ± 0.4°C). This exemplifies the known observation that although a significant positive melting temperature shift indicates a binder, the opposite is not necessarily true, i.e., not all binders cause significant Tm shifts. Conversely, some compounds lacking transcriptional activation activity showed noticeable increase in the Tm, indicating their ERα LBD binding potential. This may be due to their antagonistic (rather than agonistic) action at ERα. Additionally, the compounds were tested in the binding assay at a single concentration of 10μM which, for weak ERα activators, may be too close to the functional EC50 to observe a significant shift in the Tm.
Eosin Y (2',4',5',7'-tetrabromofluorescein) could not be evaluated in the DSF assay because it is a fluorescent red dye that absorbs and emits light at the same wavelengths as the SYPRO Orange dye, which was used to monitor the protein denaturation. Likewise, lasalocid was not evaluated because it was dissolved in acetonitrile, which is known to cause protein denaturation. No other non-specific interactions between the dye and test compounds were observed.
DISCUSSION
For the design of safer chemicals, it is crucial that any potential EDCs, regardless of their potency, are identified early in the chemical development and prioritized for further toxicological evaluation. This study describes the development and evaluation of in silico models for the identification of novel EDCs that act through the LDB of ERα, an NR that is commonly targeted by EDCs, followed by a preliminary pharmacological evaluation of the predicted EDCs in ERα transcriptional activation and DSF assays; stages 1 and 2 of the TiPED workflow (Schug et al., 2013), respectively.
The prioritization of chemicals using in silico techniques, exemplified herein using the ERα, aims to reduce the amount of toxicological evaluations required and could be applied to the prediction of endocrine disruption at numerous other targets involved in hormone synthesis, action, or metabolism, for which endocrine disruption is known to be a result of a specific target-chemical interaction, and there are target crystal structures available. These techniques will enable the evaluation of chemicals even at the design stage to assist in the prevention of potential EDCs early and actively by directing a chemist into safer chemical space.
A major concern about previously developed computational models, specifically QSAR models, is that they are often biased toward known chemistry and are unable to identify novel chemotypes, which is the key to recognizing new EDCs (Schug et al., 2013). Here, we propose two types of in silico 3D models that are significantly less chemically biased than traditional QSAR models. Specifically, the pocket-based models have no memory of the co-crystallized chemicals except for (potentially) the induced fit; to combat this potential chemical bias, an ensemble of five receptor conformations was used in model generation. For the chemical field-based model, the fields were aggregated across all of the seed ligands to eliminate potential bias toward individual chemotypes. As a result, we believe that the developed in silico 3D models are less reliant upon known chemistry, which should enable the identification of novel chemotypes of EDCs. Additionally, benchmarking the models using a challenging dataset in which many of the active and inactive/decoy chemicals were structurally related demonstrated the independence of the model predictions on known chemotypes.
The main caveat for the presented models is that they are only predictive of ligand binding to the LBD of ERα. Specifically, these models are currently unable to predict chemicals that bind to allosteric sites or the mode of action of the test compounds that bind to the LBD (i.e., agonist, antagonist, SERM). Additionally, the prediction of chemical metabolism, which may result in the increased activity of the compounds at the receptor of interest, and assessment of the downstream effects of the compounds are beyond the scope of these models.
Benchmarking of the presented models against diverse sets of active, inactive, and decoy compounds demonstrated their high recognition potential: from the top 5% of the ranked chemicals in the pocket-based VLS benchmarking, 93% of the compounds were known actives and in the chemical field-based-VLS benchmarking, 96% of the compounds had known activity at ERα. In a similar study, Zhang et al. developed QSAR and structure-based models for ERα and ERβ yet it lacked a prospective screening element (Zhang et al., 2013). Although the results cannot be directly compared with the study by Zhang and co-workers, due to the different ligand and protein datasets employed, we were pleased to note that we also obtained good enrichment in our benchmarking studies. Similarly, when screening the Tox21 database in silico, many of the top-scoring hits were known ERα modulators. For example, although screening the Tox21 database against the pocket-based model, both zearalenone and α-zearalanol (Supplementary fig. 4) were identified as active compounds. Zearalenone is a non-steroidal estrogenic mycotoxin produced by fungi that contaminates cereal crops and zearalenone and its metabolites, such as zearalanol, bind to ERα. Upon the consumption of contaminated cereals, zearalenone has been demonstrated to cause reproductive abnormalities and decrease fertility in farm animals, yet α-zearalanol is commonly used as a growth promoter (Le Guevel and Pakdel, 2001). Currently, there are no crystal structures of zearalenone or its active metabolites in complex with ERα. This illustrates that the in silico models are able to identify chemicals with different chemotypes compared with their source co-crystallized ligands (Fig. 4).
FIG. 4.

The growth promoter α-zearalanol, docked into the crystal structure of ERα (PDB ID: 2JFA), displaying polar residues in the LDB as sticks (A). α-Zearalanol displayed hydrogen bonds to Glu353, Arg394, and Gly521 (B).
Screening of the Tox21 database against the in silico models in this study also identified chemicals that were previously unknown to bind to ERα. Out of the 24 predicted active compounds, six demonstrated agonist activity in the ERα transactivation assay (Fig. 2, Supplementary table 1); 4,4'-(4-methylpentane-2,2-diyl)diphenol, 4,4'-thiodiphenol, diphenolic acid, p-xylenol blue, dobutamine, and ractopamine. Furthermore in the DSF assay, two compounds (4,4'-(4-methylpentane-2,2-diyl)diphenol and pamoic acid) had moderately stabilizing effects on ERα (ΔTm +1.05°C and +0.78°C, respectively, Figs. 2C and 2D).
Interestingly, diphenolic acid has been proposed as a suitable replacement for BPA (Guo et al., 2008), yet there have been limited toxicological evaluations (Blair et al., 2000). Here we demonstrate that, similar to BPA, diphenolic acid is also a weak ERα agonist, which suggests that further toxicological evaluations of diphenolic acid are required before it can be considered as a BPA replacement. A laboratory pH indicator, p-xylenol blue that was inconclusive when tested at ERα according to the PubChem BioAssay data (Wang et al., 2012), displayed weak ERα agonist activity and is structurally related to phenol red, another pH indicator that is also a weak ERα agonist. There are no crystal structures of phenol red in complex with ERα, which again illustrates the ability of the models to discover novel chemotypes. In addition to being the most active compound in the transactivation assay, 4,4'-(4-methylpentane-2,2-diyl)diphenol also had the greatest stabilizing effect on ERα in the thermal stabilization assay, increasing the melting temperature of ERα by 1.05°C.
The most non-trivial ERα agonists identified by our in silico screening (specifically, screening against the chemical field model) were the two chemically related compounds belonging to the class of βAR agonists: dobutamine (3.98μM) and ractopamine (micromolar activity). Ractopamine, which also had a moderate stabilizing effect upon ERα (+0.5°C) in the DSF assay, is a growth promoter that is often used to increase muscle and decrease fat in farm animals (Etherton, 2009) and dobutamine is used in the treatment of heart failure. Both dobutamine and ractopamine are secondary amines and carry a positive charge at physiological pH. As shown by the co-crystal structure of β1AR with dobutamine (PDB IDs: 2Y00 and 2Y01) (Warne et al., 2011), they bind to the orthosteric site of β1AR, which is solvent exposed and relatively polar (12/20 polar residues). In contrast, the binding site of the ERα LBD is relatively hydrophobic (13/18 non-polar residues) similar to its endogenous agonist, with the three key polar residues (Glu353, Arg394, and His524) clustered at opposite ends of the pocket (Figs. 5A and 5D). Despite the different properties of the β1AR and ERα pockets, dobutamine and ractopamine docked favorably into the chemical field models for ERα (Figs. 5B and 5E). When superimposed with a crystal structure of ERα (PDB ID: 2JFA), the catechol of dobutamine is positioned to form hydrogen bonds to Glu353 and Arg394 (Figs. 5B and 5E), which is consistent with the binding mode of 17β-estradiol in complex with ERα (Figs. 5A and 5D), but lacks the hydrogen bonding interaction to His524 (Figs. 5C and 5F). This phenomenon is apparently related to the flexible nature of the dobutamine molecule that reveals an extended and a folded conformation when bound to β1AR and ERα, respectively (Supplementary fig. 7). These predictions were confirmed in the binding and transactivation assays, with both dobutamine and ractopamine interacting weakly with ERα in the transactivation assay and ractopamine stabilizing ERα in the DSF assay (Supplementary table 1).
FIG. 5.

APF fields, used in chemical field-based methods, superimposed with 17β-estradiol co-crystalized with ERα (PDB ID: 1GWR) (A), dobutamine interacting with ERα as identified with chemical field-based methods (B), and 17β-estradiol and dobutamine superimposed in the binding site of ERα (C and F). 17β-estradiol displayed hydrogen bonds to polar residues in the LDB of ERα; specifically Glu353, Arg394, and His524 (D), whereas dobutamine only formed hydrogen bonds to Glu353 and Arg394 (E).
Aside from the adrenergic activity of dobutamine and ractopamine, the estrogenic agonist activity of these compounds may be implicated in the development of endocrine-related disorders such as obesity and cancer (Diamanti-Kandarakis et al., 2009). Of additional concern, the extensive use of βAR antagonists for the treatment of hypertension has lead to β-blockers being detected in the aquatic environment (Massarsky et al., 2011). Although the endocrine disruption potential of βAR antagonists is most frequently related to its interactions with various adrenergic receptors, one study has also identified βAR antagonist binding to ERα (Manthey et al., 2010). However, to our knowledge, dobutamine and ractopamine are the first βAR agonists that have been identified as potential EDCs. This is surprising considering their structural similarity to βAR antagonists and their widespread use as growth promoters in livestock (i.e., ractopamine (Etherton, 2009)).
In conclusion, we have developed in silico 3D models that are able to identify chemicals that interact with the LBD of ERα. Significantly, these models are less biased toward known chemistry, compared with previously developed models. Using the models, we discovered estrogenic activity of six chemicals in the Tox21 database that had previously not been pharmacologically evaluated at ERα. We confirmed the estrogenic activity of these chemicals in transactivation assays, with the EC50s ranging from 32nM to 3.98μM, that is, 25% of the tested “unknown” chemicals showed some activity at ERα. These newly identified ERα agonists include two chemically related βAR agonists, dobutamine and ractopamine, which to our knowledge are the first βAR agonists with estrogenic activity. These encouraging results show the value and applicability of the models for identification of EDC with known and novel chemotypes. These in silico models could therefore be used in prioritization of chemical databases prior to in vitro and in vivo toxicological evaluations.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institutes of Health [grant numbers R01 GM071872, U01 GM094612, U54 GM094618].
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Prof. Bruce Blumberg for sharing his expertise regarding the ERα functional assays and for generously providing the pCXN2-hERα and MTV-ERE-Luc plasmids. The authors are also thankful to Prof. Dino Moras for kindly providing the His6-ERα(302-552) plasmid. Thanks are also due to Prof. Kendall Nettles for helpful discussions. The authors are grateful to Prof. Tracy Handel for providing access to a luminometer and Dr. Lauren Holden for assistance with DSF experiments. The authors thank Stephen Khuu, DooHyang Kwon, Jacqueline Le and Sean Hui for help with binding and functional assays. The authors are also thankful to Prof. Terry Collins, Prof. Cheryl Watson, Dr. Pete Myers, and Dr. Karen O'Brien for sharing their insights and knowledge on different aspects of endocrine disruption and the application of the TiPED workflow to ERα.
Footnotes
Present address: Schrödinger, Inc., 120 West 45th Street, New York, NY, 10036
REFERENCES
- Abagyan R., Chen W., Kufareva I. Chapter 5 docking, screening and selectivity prediction for small-molecule nuclear receptor modulators. In: Cozzini P., Kellogg G. E., editors. Computational Approaches to Nuclear Receptors. Cambridge, UK: The Royal Society of Chemistry; 2012. pp. 84–109. [Google Scholar]
- Abagyan R., Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- Abagyan R., Totrov M., Kuznetsov D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994;15:488–506. [Google Scholar]
- Agatonovic-Kustrin S., Alexander M., Morton D. W., Turner J. V. Pesticides as estrogen disruptors: QSAR for selective ER. Comb. Chem. High Throughput Screen. 2011;14:85–92. doi: 10.2174/138620711794474097. [DOI] [PubMed] [Google Scholar]
- Blair R. M., Fang H., Branham W. S., Hass B. S., Dial S. L., Moland C. L., Tong W., Shi L., Perkins R., Sheehan D. M. The estrogen receptor relative binding affinities of 188 natural and xenochemicals: Structural diversity of ligands. Toxicol. Sci. 2000;54:138–153. doi: 10.1093/toxsci/54.1.138. [DOI] [PubMed] [Google Scholar]
- Branham W. S., Dial S. L., Moland C. L., Hass B. S., Blair R. M., Fang H., Shi L., Tong W., Perkins R. G., Sheehan D. M. Phytoestrogens and mycoestrogens bind to the rat uterine estrogen receptor. J. Nutr. 2002;132:658–664. doi: 10.1093/jn/132.4.658. [DOI] [PubMed] [Google Scholar]
- Colborn T., vom Saal F. S., Soto A. M. Developmental effects of endocrine-disrupting chemicals in wildlife and humans. Environ. Health Persp. 1993;101:378–384. doi: 10.1289/ehp.93101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins F. S., Gray G. M., Bucher J. R. Transforming environmental health protection. Science. 2008;319:906–907. doi: 10.1126/science.1154619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis K., Reed A., Rahhal R., Reinking J. Use of differential scanning fluorimetry as a high-throughput assay to identify nuclear receptor ligands. Nucl. Recept. Signal. 2012;10:e002. doi: 10.1621/nrs.10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E., Bourguignon J.-P., Giudice L. C., Hauser R., Prins G. S., Soto A. M., Zoeller R. T., Gore A. C. Endocrine-disrupting chemicals: An endocrine society scientific statement. Endocr. Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix D. J., Houck K. A., Martin M. T., Richard A. M., Setzer R. W., Kavlock R. J. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci. 2007;95:5–12. doi: 10.1093/toxsci/kfl103. [DOI] [PubMed] [Google Scholar]
- Eiler S., Gangloff M., Duclaud S., Moras D., Ruff M. Overexpression, purification, and crystal structure of native ERα LBD. Protein Expr. Purif. 2001;22:165–173. doi: 10.1006/prep.2001.1409. [DOI] [PubMed] [Google Scholar]
- Etherton T. D. ASAS centennial paper: Animal growth and development research: Historical perspectives. J. Anim. Sci. 2009;87:3060–3064. doi: 10.2527/jas.2009-1805. [DOI] [PubMed] [Google Scholar]
- Fang H., Tong W., Shi L. M., Blair R., Perkins R., Branham W., Hass B. S., Xie Q., Dial S. L., Moland C. L., et al. Structure-activity relationships for a large diverse set of natural, synthetic, and environmental estrogens. Chem. Res. Toxicol. 2001;14:280–294. doi: 10.1021/tx000208y. [DOI] [PubMed] [Google Scholar]
- Gaulton A., Bellis L. J., Bento A. P., Chambers J., Davies M., Hersey A., Light Y., McGlinchey S., Michalovich D., Al-Lazikani B., Overington J. P. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., Li K., Yu X., Clark J. H. Mesoporous H3PW12O40-silica composite: Efficient and reusable solid acid catalyst for the synthesis of diphenolic acid from levulinic acid. Appl. Catal. B. 2008;81:182–191. [Google Scholar]
- Heldring N., Pike A., Andersson S., Matthews J., Cheng G., Hartman J., Tujague M., Ström A., Treuter E., Warner M., et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- Hitoshi N., Ken-ichi Y., Jun-ichi M. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Huang N., Shoichet B. K., Irwin J. J. Benchmarking sets for molecular docking. J. Med. Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V., Rueda M., Abagyan R. Ligand-guided receptor optimization. In: Orry A. J. W., Abagyan R., editors. Homology Modeling. Vol. 857. New York, USA: Humana Press; 2012. pp. 189–205. [DOI] [PubMed] [Google Scholar]
- Katritch V., Rueda M., Lam P. C.-H., Yeager M., Abagyan R. GPCR 3D homology models for ligand screening: Lessons learned from blind predictions of adenosine A2a receptor complex. Proteins. 2010;78:197–211. doi: 10.1002/prot.22507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchmair J., Markt P., Distinto S., Wolber G., Langer T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection - What can we learn from earlier mistakes? J. Comput. Aided. Mol. Des. 2008;22:213–228. doi: 10.1007/s10822-007-9163-6. [DOI] [PubMed] [Google Scholar]
- Kufareva I., Chen Y.-C., Ilatovskiy A. V., Abagyan R. Compound activity prediction using models of binding pockets or ligand properties in 3D. Curr. Top. Med. Chem. 2012a;12:1869–1882. doi: 10.2174/156802612804547335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I., Ilatovskiy A. V., Abagyan R. Pocketome: An encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 2012b;40:D535–D540. doi: 10.1093/nar/gkr825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guevel R., Pakdel F. Assessment of oestrogenic potency of chemicals used as growth promoter by in-vitro methods. Hum. Reprod. 2001;16:1030–1036. doi: 10.1093/humrep/16.5.1030. [DOI] [PubMed] [Google Scholar]
- Manthey D., Gamerdinger M., Behl C. The selective β1-adrenoceptor antagonist nebivolol is a potential oestrogen receptor agonist with neuroprotective abilities. Br. J. Pharmacol. 2010;159:1264–1273. doi: 10.1111/j.1476-5381.2009.00610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massarsky A., Trudeau V. L., Moon T. W. β-blockers as endocrine disruptors: The potential effects of human β-blockers on aquatic organisms. J. Exp. Zool. A. 2011;315A:251–265. doi: 10.1002/jez.672. [DOI] [PubMed] [Google Scholar]
- Papa E., Kovarich S., Gramatica P. QSAR modeling and prediction of the endocrine-disrupting potencies of brominated flame retardants. Chem. Res. Toxicol. 2010;23:946–954. doi: 10.1021/tx1000392. [DOI] [PubMed] [Google Scholar]
- Park S.-J., Kufareva I., Abagyan R. Improved docking, screening and selectivity prediction for small molecule nuclear receptor modulators using conformational ensembles. J. Comput. Aided. Mol. Des. 2010;24:459–471. doi: 10.1007/s10822-010-9362-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda M., Bottegoni G., Abagyan R. Recipes for the selection of experimental protein conformations for virtual screening. J. Chem. Inf. Model. 2009;50:186–193. doi: 10.1021/ci9003943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda M., Totrov M., Abagyan R. ALiBERO: Evolving a team of complementary pocket conformations rather than a single leader. J. Chem. Inf. Model. 2012;52:2705–2714. doi: 10.1021/ci3001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug T. T., Abagyan R., Blumberg B., Collins T. J., Crews D., DeFur P. L., Dickerson S. M., Edwards T. M., Gore A. C., Guillette L. J., et al. Designing endocrine disruption out of the next generation of chemicals. Green Chem. 2013;15:181–198. doi: 10.1039/C2GC35055F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L. M., Fang H., Tong W., Wu J., Perkins R., Blair R. M., Branham W. S., Dial S. L., Moland C. L., Sheehan D. M. QSAR models using a large diverse set of estrogens. J. Chem. Inf. Comp. Sci. 2000;41:186–195. doi: 10.1021/ci000066d. [DOI] [PubMed] [Google Scholar]
- Totrov M. Atomic property fields: Generalized 3D pharmacophoric potential for automated ligand superposition, pharmacophore elucidation and 3D QSAR. Chem. Biol. Drug Des. 2008;71:15–27. doi: 10.1111/j.1747-0285.2007.00605.x. [DOI] [PubMed] [Google Scholar]
- Umesono K., Evans R. M. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell. 1989;57:1139–1146. doi: 10.1016/0092-8674(89)90051-2. [DOI] [PubMed] [Google Scholar]
- Vedani A., Dobler M., Smieško M. VirtualToxLab—A platform for estimating the toxic potential of drugs, chemicals and natural products. Toxicol. Appl. Pharm. 2012;261:142–153. doi: 10.1016/j.taap.2012.03.018. [DOI] [PubMed] [Google Scholar]
- Wang Y., Xiao J., Suzek T. O., Zhang J., Wang J., Zhou Z., Han L., Karapetyan K., Dracheva S., Shoemaker B. A., et al. PubChem's BioAssay database. Nucleic Acids Res. 2012;40:D400–D412. doi: 10.1093/nar/gkr1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T., Moukhametzianov R., Baker J. G., Nehme R., Edwards P. C., Leslie A. G. W., Schertler G. F. X., Tate C. G. The structural basis for agonist and partial agonist action on a β1-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolber G., Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2004;45:160–169. doi: 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- Zhang L., Sedykh A., Tripathi A., Zhu H., Afantitis A., Mouchlis V. D., Melagraki G., Rusyn I., Tropsha A. Identification of putative estrogen receptor-mediated endocrine disrupting chemicals using QSAR- and structure-based virtual screening approaches. Toxicol. Appl. Pharm. 2013;272:67–76. doi: 10.1016/j.taap.2013.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.