

Key Points
N-Ras expression is essential for the proliferative advantage of acute myeloid leukemias with oncogenic NRAS/Nras mutations.
Mitogen-activated protein kinase kinase inhibition prolongs survival in Nras-mutant AML by reducing proliferation, but fails to undergo apoptosis.
Abstract
Oncogenic NRAS mutations are highly prevalent in acute myeloid leukemia (AML). Genetic analysis supports the hypothesis that NRAS mutations cooperate with antecedent molecular lesions in leukemogenesis, but have limited independent prognostic significance. Using short hairpin RNA–mediated knockdown in human cell lines and primary mouse leukemias, we show that AML cells with NRAS/Nras mutations are dependent on continued oncogene expression in vitro and in vivo. Using the Mx1-Cre transgene to inactivate a conditional mutant Nras allele, we analyzed hematopoiesis and hematopoietic stem and progenitor cells (HSPCs) under normal and stressed conditions and found that HSPCs lacking Nras expression are functionally equivalent to normal HSPCs in the adult mouse. Treating recipient mice transplanted with primary NrasG12D AMLs with 2 potent allosteric mitogen-activated protein kinase kinase (MEK) inhibitors (PD0325901 or trametinib/GlaxoSmithKline 1120212) significantly prolonged survival and reduced proliferation but did not induce apoptosis, promote differentiation, or drive clonal evolution. The phosphatidylinositol 3-kinase inhibitor GDC-0941 was ineffective as a single agent and did not augment the activity of PD0325901. All mice ultimately succumbed to progressive leukemia. Together, these data validate oncogenic N-Ras signaling as a therapeutic target in AML and support testing combination regimens that include MEK inhibitors.
Introduction
Although the genomic landscape of acute myeloid leukemia (AML) has been characterized extensively, translating our knowledge of common molecular lesions into more effective and less toxic therapies remains a fundamental challenge. The rationale for developing chemical inhibitors of oncogenic “driver” proteins relies on 2 broad principles: (1) the mutant protein should be required for cancer maintenance and (2) inhibiting its activity must have a beneficial therapeutic index and not result in unacceptable toxicity to normal cells. Recent work suggests that early lesions in a preleukemic stem cell harboring mutations in epigenetic modulators such as DNMT3A or TET2 may evade therapy and serve as a reservoir for disease relapse persisting even at leukemic remission.1-3 Additional molecular analysis supports the notion that late acquisition of signaling mutations in oncogenes such as NRAS or FLT3 often represent secondary events in AML pathogenesis,1,4-6 and the prognostic significance of NRAS mutations in particular is uncertain.7-10 Furthermore, the observation that NRAS and FLT3 mutations detected at diagnosis may disappear at relapse raises questions about the therapeutic benefits of targeting signaling aberrations in AML.4 Importantly, however, clinical trials of potent and selective Flt3 inhibitors such as quizartinib (also known as AC220) showed that monotherapy induced remission in some patients with refractory AML,11 suggesting that targeting oncogenic signaling mutations in AML may have therapeutic benefit. Interestingly, “gatekeeper” FLT3 mutations occurred after quizartinib treatment in relapsed patients.12 These compelling genetic data indicate strong selective pressure for primary AML cells to restore aberrant Flt3 signaling in vivo.
NRAS is one of the most common targets of oncogenic signaling mutations in blood cancers, including in 10% to 15% of AML.13,14 Ras proteins transduce signals from activated growth factor receptors and other extracellular stimuli to the nucleus, thereby regulating cellular behaviors that include proliferation, differentiation, and survival.15 Cancer-associated RAS mutations encode proteins that accumulate in the active, guanosine triphosphate–bound conformation because of defective intrinsic hydrolysis and resistance to guanosine triphosphatase–activating proteins.15 Directly targeting oncogenic Ras is an extremely challenging problem for rational drug design,16,17 and no clinically available mechanism-based therapies for the ∼30% of human cancers with oncogenic RAS mutations currently exist. Given this, there is intensive interest in developing anticancer drugs targeting components of the Ras-regulated phosphoinositide 3-kinase (PI3K)/Akt cascade and the mitogen-activated protein kinase (MAPK) pathway, which includes Raf, MAPK kinase (MEK), and extracellular signal-regulated kinase (ERK).16
Here we examine the functional consequence of NRAS/Nras knockdown in AML cells, characterize the effect of Nras inactivation on normal hematopoiesis, and investigate the preclinical efficacy of MEK and PI3K inhibitors in primary Nras-mutant AML. We show that NRAS expression is required for the proliferative advantage of human AML cell lines in vitro and for the maintenance of Nras-mutant AML in vivo. To investigate if targeting N-Ras might be deleterious for normal hematopoietic cells, we assessed hematopoietic stem and progenitor cell (HSPC) function in homozygous Nras knockout mice. Importantly, hematopoietic stem cells (HSCs) lacking Nras expression exhibited normal differentiation and proliferative potential, suggesting that targeting AML with NRAS mutations will have a beneficial therapeutic index. We next transplanted primary AML generated in NrasG12D “knock in” mice18 and performed preclinical trials of the MEK inhibitors PD0325901 (PD901),19 trametinib (also known as GlaxoSmithKline 1120212),20,21 or the PI3K inhibitor GDC-0941.22 Both MEK inhibitors significantly improved the survival of recipient mice by inhibiting AML proliferation, but did not induce apoptosis, promote differentiation, or drive clonal selection. PI3K inhibition was ineffective in this model, and combining PD901 and GDC-0941 was no better than MEK inhibition alone.
Methods
Mice, enumeration of hematopoietic populations, and competitive repopulation
All studies were approved by the Committees on Animal Research at the University of California, San Francisco, and the University of Michigan and conducted in accordance with the GlaxoSmithKline Policy on the Care, Welfare and Treatment of Laboratory Animals. Nrasfl/f, NrasG12D/+, and Mx1-Cre mice were maintained on a C57BL/Ka-CD45.2:Thy-1.1 background. Recipients in reconstitution assays were C57BL/Ka-CD45.1:Thy-1.2 mice, at least 8 weeks old at the time of irradiation. pIpC (Amersham) was administered at 0.5 µg/g body mass per day by intraperitoneal injection every other day, for a total of 6 doses. 5-bromo-2′-deoxyuridine (BrdU, Sigma) was administered as a single dose of 200 mg/kg body mass by intraperitoneal injection followed by 1 mg/mL BrdU in the drinking water. Bone marrow (BM) cells were prepared and CD150+CD48− Lin−Sca1+c-Kit+ (LSK) HSCs and CD150−CD48− LSK multipotent progenitors (MPPs) were analyzed as previously described.23 Common myeloid progenitors, granulocyte-macrophage progenitors, megakaryocyte-erythroid progenitors, and lineage cells were analyzed as previously described.18 Nonviable cells were excluded from analyses using the viability dye 4′,6-diamidino-2-phenylindole (1 μg/mL). BrdU incorporation in vivo was measured by flow cytometry using the APC BrdU Flow Kit (BD Biosciences). Long-term competitive repopulation assay was performed as previously described.23 To identify CD45.2+ cells, antibodies against CD45.2 (104-FITC, BioLegend) and CD45.1 (A20-APC780, BioLegend) were used.
Preclinical trials
MOL4070LTR mutagenesis in Lox-STOP-Lox-NrasG12D mice was performed on an F1 C57BL/6 × 129Sv/Jae strain background.18 Primary cryopreserved AML cells (5 × 106) were injected intravenously into sublethally irradiated (600 rad) 8- to 12-week-old recipients that were then treated by oral gavage with control 0.5% hydroxypropyl methylcellulose vehicle, PD901 (5 mg/kg per day)18,24 or GDC-0941 (125 mg/kg per day). For combination treatment, mice received PD901 (5 mg/kg per day) daily, and GDC-0941 (100 mg/kg per day) was cycled 4 days, on followed by 4 days off until death. We independently evaluated trametinib (0.5 mg/kg per day) in a subsequent trial. Recipients injected with 2 transplantable myeloproliferative neoplasms (MPNs) were monitored with weekly complete blood counts, enrolled when blood leukocyte counts reached 25 000 cells/µL, and euthanized when they appeared moribund or when weekly leukocyte counts exceeded 180 000 cells/µL. Transplant recipients of 3 aggressive AML lines were enrolled 4 days posttransplant and euthanized when they appeared moribund.
shRNA experiments
Cultured AML cell lines were obtained from ATCC (THP-1, HL-60), DSMZ (OCI-AML3, NB4, and MV4-11), and Dr Neil Shah (MOLM-14). Lines infected with the corresponding lentiviral short hairpin RNA (shRNA) construct at equivalent multiplicity of infection were assayed for percentage mCherry-positive cells by flow cytometry. Data are represented as a normalized percentage of shRNA-expressing cells over time divided by the maximum percentage of infected cells for each culture. Primary AML cells were harvested and plated in short-term culture, spin-infected for 2 hours, transplanted in bulk to recipient animals or sorted for mCherry-positive cells 2 days after infection, and transplanted into recipient animals. See supplemental Methods on the Blood Web site for additional information.
Results
NRAS/Nras expression is required for maintenance of AMLs harboring oncogenic mutations
We generated lentiviral shRNA constructs targeting NRAS and infected 6 human AML cell lines (Figure 1A), including 3 with oncogenic NRAS mutations (OCI-AML3, THP-1, and HL60). Two independent hairpins that markedly reduced N-Ras protein levels (Figure 1B) inhibited the growth of NRAS-mutant AML cells compared with a control shRNA targeting Renilla luciferase (Figure 1A). Importantly, these effects were genotype-specific because AML cell lines with mutations in FLT3 or KRAS were insensitive to NRAS knockdown (Figure 1A).
Figure 1.
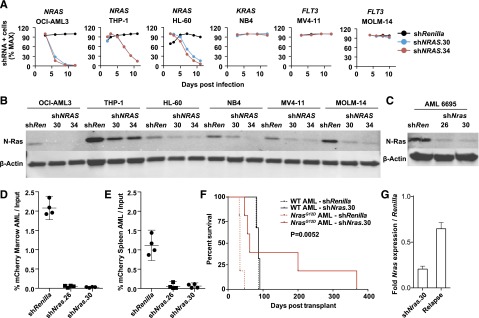
NRAS/Nras expression is required for maintenance of AML harboring oncogenic mutations. (A) Human AML cell lines with mutations in NRAS (OCI-AML3, HL60, THP-1), KRAS (NB4), or FLT3 (MV4-11 and MOLM-14) were infected with control shRenilla targeting Renilla luciferase and 2 independent NRAS shRNAs shown in panel B. Note depletion of NRAS-mutant AML cells expressing either NRAS hairpin. Max, maximum. (B) Western blot demonstrating N-Ras protein knockdown in each of the human AML cell lines using shNRAS.30, 34, and shRenilla control. (C) Western blot demonstrating N-Ras protein knockdown in primary murine NrasG12D AML #6695 expressing shNras.26, shNras.30, or the control shRenilla. We infected AML #6695 with shNras.26, shNras.30, or shRenilla at equivalent multiplicities of infection (input mCherry-positive cells 17% to 29% of total AML), bulk transplanted into recipient mice (n = 4 mice per shRNA), and measured the percentage of mCherry-positive blasts (D) in BM and spleen (E) 3 weeks later. Note the selective dropout of shNras-expressing cells with 2 independent shRNAs (P < .0001). (F) Survival of mice transplanted with NrasG12D AML #6695 (red lines, n = 10) or with a control AML generated in a WT mouse (black lines, n = 6). AMLs were infected with shNras.30 (solid lines) or control shRenilla (broken lines) and sorted to near purity before transplant. Nras knockdown prolongs survival in Nras-mutant AML (P = .0052). (G) Nras message knockdown in primary mouse AML cells before transplant (shNras.30) assessed by quantitative polymerase chain reaction. Early relapse (day 62) shNras.30 mCherry-positive leukemia (relapse) partially restored Nras expression relative to Renilla controls.
Injecting neonatal Mx1-Cre; Lox-STOP-Lox-NrasG12D mice with the MOL4070LTR retrovirus generated leukemias with striking similarities to human AML types with somatic NRAS mutations, including M4/M5-like morphology and were associated with Evi1 dysregulation.18 Importantly, NrasG12D is a secondary mutation in this system, which models the common pathogenic sequence in human AML.25,26 To test whether an oncogenic Nras mutation that occurs as a cooperating event in AML pathogenesis is required for leukemia maintenance, we infected primary NrasG12D leukemia #6695 and a control AML induced by infecting wild-type (WT) mice with the MOL4070LTR retrovirus with lentiviral vectors expressing shRNA targeting either Nras (shNras.26 or shNras.30) or Renilla luciferase (shRenilla) linked to an mCherry reporter and transplanted these cells into secondary recipients. Both Nras shRNA constructs efficiently reduced N-Ras protein levels in AML #6695 (Figure 1C), and we observed a dramatic reduction in the percentage of cells expressing the mCherry reporter 3 weeks after transplant in the BM (Figure 1D; supplemental Figure 1A) and spleens (Figure 1E) of recipient mice relative to the shRenilla control. By contrast, control WT AML cells infected with either Nras shRNA or with shRenilla exhibited similar levels of engraftment (supplemental Figure 1B-C).
We next infected AML #6695 and the WT control AML with a lentiviral construct expressing shNras.30 or shRenilla, sorted these cells to near purity, and transplanted them into sublethally irradiated mice (n = 10 mice with AML #6695, n = 6 mice with control AML). Nras knockdown significantly delayed the onset of AML in recipients of Nras-mutant AML #6695 (P = .0052) but did not alter the survival of WT control AML recipients (Figure 1F). All of the secondary recipient mice transplanted with NrasG12D AML infected with shRenilla died within 47 days (n = 5). Of the recipients of NrasG12D AML cells expressing shNras.30 (n = 5), 2 mice (surviving 47 and 56 days) were outgrowths of mCherry-negative cells lacking shNras.30 expression. One mouse surviving 62 days retained mCherry expression, but restored Nras expression to 65% of control levels (Figure 1G). The 2 remaining shNras.30 transplant recipients survived 200 and 368 days posttransplant.
Effects of Nras inactivation on HSPC populations
Mice homozygous for a germ line Nras “knockout” allele are phenotypically normal,27 with only modest defects in CD8+ thymocytes.28 To directly investigate the consequences of Nras inactivation in HSPC, we used the Mx1-Cre transgene29 to inactivate Nras and analyzed hematopoiesis under normal and stressed conditions. Efficient Nras inactivation was confirmed by western blot (supplemental Figure 2A); we hereafter refer to these mice as NrasΔ/Δ. Blood leukocyte counts (supplemental Figure 2B) and spleen sizes were normal in NrasΔ/Δ mice. The frequencies and absolute numbers of phenotypic signaling lymphocyte activation molecule HSCs,23 MPPs, and LSK progenitor cells were similar in NrasΔ/Δ and control WT littermates (Figure 2A). NrasΔ/Δ mice also had normal numbers and frequencies of myeloid and erythroid progenitor cells (supplemental Figure 2C). In addition, Nras inactivation did not alter the numbers of differentiated leukocytes or the proportions of less and more mature erythroid and lymphoid cells within the marrow (supplemental Figure 2C-D). BrdU labeling revealed an equal percentage of HSCs in S-phase in NrasΔ/Δ and WT BM (Figure 2B), and BM and spleen cellularity was similar in both genotypes (Figure 2C). We conclude that NrasΔ/Δ mice exhibit normal steady-state hematopoiesis.
Figure 2.

Nras is dispensable for normal function of HSCs. (A) Total number of CD150+CD48−LSK HSCs, CD150−CD48−LSK MPPs, and LSK cells in BM and spleens (SP) of WT (Nras+/+) and Mx1-cre; Nrasfl/fl (NrasΔ/Δ) mice 2 weeks after pIpC treatment (n = 10). (B) BrdU incorporation in WT and NrasΔ/Δ HSCs after 24-hour BrdU incorporation (n = 6). SLAM, signaling lymphocyte activation molecule. (C) BM and spleen cellularity in WT and NrasΔ/Δ mice (n = 10). (D) 5 × 105 donor BM cells from Mx1-cre; Nrasfl/fl (NrasΔ/Δ) or littermate control mice at 2 weeks after pIpC treatment were transplanted into irradiated recipient mice with 5 × 105 recipient BM cells. Donor cell reconstitution in the myeloid (Gr-1+ or Mac-1+ cells), B- (B220+), and T- (CD3+) cell lineages was monitored for 4 to 20 weeks after transplantation (n = 5 recipients/genotype). Only the week 4 time point is significant for B- (P < .001) and T-cell (P = .024) repopulation. (E) Competitive repopulation of Mx1-cre; NrasG12D/fl; (NrasG12D/Δ), Mx1-cre; NrasG12D/+ (NrasG12D/+), or littermate control BM cells (n = 5 recipients/genotype). Two-tailed Student t tests were used to assess statistical significance and P < .001 between NrasG12D/+ or NrasG12D/Δ and control at 8, 12, 16, and 20 weeks with no significant difference between NrasG12D/+ and NrasG12D/Δ.
Competitive repopulation is a robust assay for comparing the relative fitness of immature hematopoietic cells under stress conditions. We mixed equal numbers of NrasΔ/Δ or control CD45.2+ BM cells with WT CD45.1+ competitors, transplanted them into lethally irradiated recipients, and monitored the percentage of donor-derived cells over time. Nras-deficient HSCs retained the capacity to repopulate the myeloid (Gr-1+, Mac-1+), B-cell (B220+), and T-cell (CD3+) compartments. Importantly, although there is a transient reduction of lymphoid reconstituting potential (B and T cells at 4 weeks, P < .05) in NrasΔ/Δ cells, there is no difference in short-term myeloid reconstitution or long-term multilineage reconstitution after adoptive transfer (Figure 2D). Together, these data indicate that HSCs lacking Nras expression are functionally equivalent to normal HSCs in the adult mouse.
Murine NrasG12D AMLs frequently exhibit loss of the WT Nras allele because of somatic uniparental disomy that also results in duplication of oncogenic NrasG12D.30 We performed competitive repopulation experiments to assess the relative fitness of Mx1-Cre, NrasG12D/Δ, and Mx1-Cre; NrasG12D/+ BM cells in vivo. Inactivating WT Nras did not enhance the in vivo competitive advantage previously reported for heterozygous NrasG12D-mutant BM cells (Figure 2E).31 These data are consistent with a recent study that failed to uncover tumor suppressor activity of WT Nras, but showed that increasing Nras oncogene dosage promotes cell growth and modulates Ras effector activation.30
MEK inhibition reduces the growth of NrasG12D AML lines in vivo and prolongs survival
Given the lack of mechanistic-based therapies for cancers with NRAS mutations, we asked if NrasG12D murine AMLs are dependent on canonical Ras effector pathways in vivo. We transplanted 5 independent myeloid malignancies into cohorts of sublethally irradiated recipients and assigned these mice to treatment with control vehicle, a potent allosteric MEK inhibitor (PD901), a pan-PI3K inhibitor (GDC-0941), or a combination of both compounds. PD901 and GDC-0941 were administered at the maximum tolerated doses previously established in this strain background (F1 C57BL/6 × 129Sv/Jae).24,32-34 Leukemias #6730 and #8064 are classified as transplantable MPNs35 because recipient mice develop progressive leukocytosis, retain residual erythropoiesis, and have a median survival of 70 days (supplemental Figure 3A-B). These MPNs represent a more aggressive disease than the indolent MPNs developed in Nras-mutant mice without retroviral insertions that are not transplantable to sublethally irradiated recipients.18 By contrast, AML lines #6695, #6606, and #6768 induce aggressive AML characterized by severe anemia and a median survival of 20 days after transplantation (supplemental Figure 3A-B). In addition to expressing NrasG12D from the endogenous locus, each of these leukemias contains different clonal retroviral integrations (supplemental Figure 3D).18 As described in our “Methods” section, the different biological behaviors of NrasG12D-mutant MPNs and AML lines (supplemental Figure 3B) necessitated using different enrollment criteria for preclinical trials.
Treatment with PD901 extended the survival of recipients transplanted with NrasG12D-transplantable MPNs (P = .0009), but all of the mice ultimately developed progressive disease (Figure 3A). The PI3K inhibitor was ineffective as a single agent (P = .55). Administering both PD901 and GDC-0941 modestly extended survival compared with PD901 alone, but this difference did not reach statistical significance (P = .143, Figure 3A). Treatment with PD901 also significantly extended the survival of recipients transplanted with aggressive primary NrasG12D AML lines (Figure 3B). Again, GDC-0941 was ineffective (P = .82), whereas recipient mice that received both MEK and PI3K inhibitors showed reduced survival compared with mice given the control vehicle, which was likely because of severe anemia resulting from suppression of residual normal hematopoiesis (Figure 3B; supplemental Figure 3C).
Figure 3.
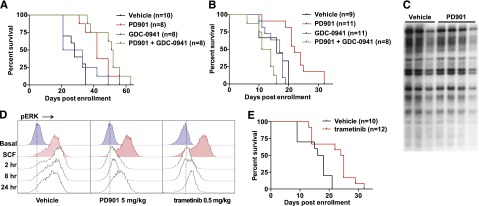
MEK inhibition prolongs survival in mice transplanted with NrasG12D AML. (A) Survival of secondary recipient mice engrafted with 2 transplantable MPNs that were treated with PD901 (n = 8), GDC-0941 (n = 8), a combination of PD901 and GDC-0941 (n = 8), or control vehicle (n = 10). PD901 significantly prolonged the survival of transplant recipients (P = .0009). (B) Survival of recipient mice transplanted with 3 independent aggressive NrasG12D AML lines treated with PD901 (n = 11), GDC-0941 (n = 11), a combination of PD901 and GDC-0941 (n = 8), or the control vehicle (n = 9). PD901 significantly prolonged the survival of transplant recipients (P = .0003). (C) Southern blot analysis of AML #6768 reveals an identical pattern of retroviral integrations in recipients treated with PD901 compared with controls. (D) WT mice treated with vehicle, PD901 (5 mg/kg), or trametinib (0.5 mg/kg) were euthanized at time 0 and 2, 8, and 24 hours, BM was collected, and c-Kit+ cells were assayed for ERK phosphorylation (pERK) under basal conditions and after stimulation with stem cell factor (SCF). (E) Survival of recipient mice transplanted with the 3 aggressive NrasG12D AMLs shown in panel C treated with 0.5 mg/kg per day trametinib (n = 12) or the control vehicle (n = 10) demonstrating a significant improvement in overall survival (P = .0063).
To ask if these AML lines develop phenotypic resistance to MEK inhibition, we isolated BM at euthanasia, transferred it into secondary recipients, and re-treated a cohort of mice. In the case of AML #6606, the PD901-treated, relapsed AML (#6606-R) showed a statistically significant and similar median improvement in overall survival upon retreatment with PD901 compared with the parental leukemia (10.5 days and 6 days, respectively; supplemental Figure 4A). Interestingly, the relapsed #6606-R clone demonstrated a longer time to death in untreated animals compared with #6606, which could indicate a reduction in the leukemia initiating cell fraction resulting from prior PD901 treatment. MPN #6730 also responded similarly to PD901 after secondary transplantation and retreatment (supplemental Figure 4B). Despite the survival improvement in all myeloid neoplasms receiving PD901 (median 3 to 12.5 days; supplemental Table 1), we did not detect clonal evolution at the time of death as assessed by Southern blotting with a probe that detects MOL4070 integrations (Figure 3C; supplemental Figure 4C). Furthermore, exposing AML cells harvested from transplant recipients of AML lines #6606 or #6606-R to PD901 revealed equivalent inhibition of cytokine-induced ERK phosphorylation (supplemental Figure 4D). Together, these data indicate that MEK inhibition reduces NrasG12D AML growth in vivo without inducing phenotypic drug resistance or eradicating the dominant clone.
To extend these observations to a structurally distinct and Food and Drug Administration–approved MEK inhibitor, we established that administering a single 0.5 mg/kg dose of trametinib20,21 by oral gavage led to measurable serum concentrations for 24 hours in mice (supplemental Figure 5). Pharmacodynamic studies of mice treated with PD901 (5 mg/kg per day) or with trametinib (0.5 mg/kg per day) revealed sustained MAPK pathway inhibition in the BM of WT mice that was similar for both compounds (Figure 3D). Treating recipient mice transplanted with the aggressive primary NrasG12D AML with trametinib (0.5 mg/kg per day) significantly prolonged survival compared with vehicle control (P = .0057, Figure 3E). Statistical analyses of individual myeloid neoplasms treated with different inhibitors or combinations are presented in supplemental Tables 1 and 2.
MEK inhibition reduces proliferation in NrasG12D AML without inducing leukemic differentiation or apoptosis
We investigated the mechanism by which in vivo MEK inhibition prolonged the survival of recipients transplanted with myeloid malignancies driven by oncogenic N-RasG12D expression. PD901 treatment blunted the rise in leukocyte counts observed in recipient mice injected with the transplantable MPNs for approximately 3 weeks (Figure 4A). Mice engrafted with aggressive AML #6695 showed significant improvement in peripheral leukocyte counts (Figure 4B) and spleen size (Figure 4C) after only 4 days of treatment with PD901, and MEK inhibition dramatically reduced the proliferation of NrasG12D AMLs in short-term liquid cultures (Figure 4D). Biochemical analysis showed that PD901 potently inhibited cytokine-induced ERK phosphorylation in AML cells isolated from recipient mice, but had no effect on Akt phosphorylation (Figure 4E). Although the levels of PI3K/Akt pathway activation varied between individual leukemias, we did not detect a compensatory increase in phosphorylated Akt levels upon MAPK pathway inhibition.
Figure 4.
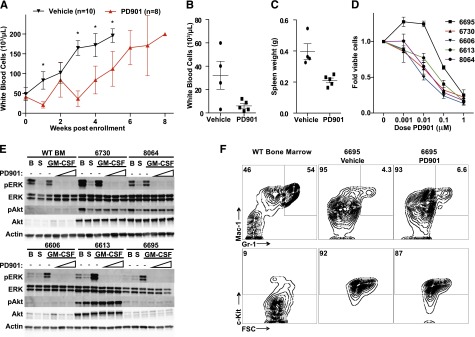
MEK inhibition reduces proliferation in Nras-mutant AML without inducing differentiation. (A) Serial white blood cell counts in secondary transplant recipients of myeloid neoplasms #6730 and #8064 treated with PD901 (red line, n = 8) or a control vehicle (black line, n = 10), with error bars representing the 95% confidence interval (*statistical significance by unpaired t test). Next, mice transplanted with AML #6695 were treated with 4 daily doses of vehicle (n = 4) or PD901 (n = 5) starting 10 days posttransplant. (B-C) PD901 treatment effectively reduced white blood cell counts (P = .045) and spleen size (P = .0057). (D) Normalized viable cell count after 4 days of growth in liquid culture with increasing doses of PD901 in 5 independent N-RasG12D myeloid neoplasms (n ≥ 3 mice per leukemia). (E) Western blot analysis of granulocyte macrophage colony-stimulating factor (GM-CSF) stimulated pERK and phosphorylated Akt (pAkt) in BM cells isolated from transplant recipients of NrasG12D myeloid neoplasms and WT BM control. BM from moribund mice was assayed for level of pERK, total ERK, pAkt, total Akt, and β-actin at B (basal conditions), following S (starvation), and subsequent GM-CSF stimulation in the absence or presence of increasing concentrations of PD901 (0.01, 0.1, and 1 μM). (F) Mice engrafted with AML #6695 were treated with PD901 (n = 5) or vehicle (n = 4) for 4 days (as in panels B and C) and total BM was stained for c-Kit, Mac-1 (CD11b), and Gr-1 surface expression. WT untreated BM is shown as a control. Representative plots are shown, and numbers represent the mean percentage for each gate across replicates.
Next we asked whether treatment with PD901 promoted the differentiation of AML blasts in vivo. Blinded pathological review of peripheral smears and BM cytospins from the preclinical trials conducted with PD901 (from Figure 3A) suggested that there was an increase in differentiated cells in the PD901-treated animals compared with vehicle controls. However, this observation could be due to a reduced disease burden at the time of death and a larger contribution of residual normal hematopoiesis in sublethally irradiated recipient animals. To directly assess differentiation in vivo, we transplanted mice with AML #6695, treated them with PD901 for 4 days, isolated BM, and assessed myeloid lineage marker expression (Figure 4F). The immunophenotype of the AML #6695 blast population is c-Kit+/Mac-1int/Gr-1lo, and this did not change after treatment with PD901. We observed a small increase in the mature granulocyte double-positive Mac-1hi/Gr-1hi population (from 4.3% to 6.6%) and slight reduction in the c-Kit+ fraction (from 92% to 87%) with PD901 treatment (Figure 4F), despite a dramatic reduction in disease burden (Figure 4B-C). The staining for individual transplant recipients is shown in supplemental Figure 6. Although we cannot exclude subtle effects on differentiation, we conclude that MEK inhibition does not promote bulk differentiation of the leukemia blasts into mature granulocytes.
Consistent with an antiproliferative effect on leukemia blasts, in vivo cell-cycle analysis showed that 24 hours of PD901 treatment increased the percentage of cells in the G0/G1 fraction (P = .0071) with a corresponding reduction in the G2/M fraction (P = .0012)(Figure 5A). There was no difference in the sub-G0 fraction, which suggested that PD901 treatment does not induce apoptosis (Figure 5A). Consistent with this hypothesis, similar percentages of AML cells isolated from vehicle and PD901-treated recipients stained positive for activated Caspase-3 or Annexin V/7-AAD (Figure 5B-C). Exposing NrasG12D AML cells to 1 μM of PD901 in short-term liquid culture dramatically reduced proliferation and ERK phosphorylation, but did not reduce viability or increase the percentage of apoptotic cells (Figures 4D-E and 5D). As expected, basal levels of apoptosis were higher in primary cells grown in liquid culture (Figure 5D) than after transplantation (Figure 5B-C). However, exposure to PD901 did not induce a significant increase in apoptosis ex vivo or in vivo. Together, these studies support the hypothesis that the beneficial therapeutic responses of primary NrasG12D AML to PD901 are due to reduced proliferation.
Figure 5.
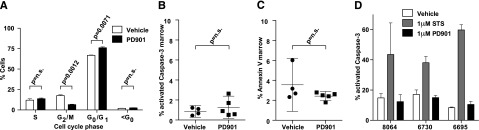
MEK inhibition does not induce apoptosis in Nras-mutant AML. (A) Cell-cycle analysis using in vivo BrdU incorporation comparing vehicle- (n = 2) and PD901- (n = 3) treated NrasG12D AML #6730 after 24 hours. (B-C) The percentage of BM cells collected from recipients of AML #6695 after PD901 (n = 5) or control vehicle (n = 4) treatment expressing cleaved Caspase-3 (P = .43) and Annexin V/7-AAD (P = .18). (D) Induction of apoptosis in short-term ex vivo culture measured by percentage activated Caspase-3 in 3 independent NrasG12D AML lines (n = 2 per AML) treated with control vehicle, staurosporine (STS), or PD901.
Discussion
Recent deep genomic analyses of diagnostic AML specimens assessing mutant allele frequencies and interrogating serial samples obtained during remission and at subsequent relapse are consistent with earlier work suggesting that FLT3, NRAS, and CKIT mutations cooperate with antecedent transcription factor fusions and mutations in epigenetic modifiers to promote leukemogenesis.1-4,6 Indeed, HSPC from AML patients may retain DNMT3A or TET2 mutations during remission, yet exhibit relatively normal proliferation and differentiation in vivo. These data support the hypothesis that preleukemic HSPC that evade therapy may serve as a reservoir for subsequent disease relapse.1-3 Although these studies might be interpreted as indicating that targeting aberrant signaling in AML will ultimately prove ineffective, the absence of FLT3 and NRAS mutations during remission suggest that aberrant signal transduction is intrinsic to and essential for leukemic growth. This idea is consistent with the dramatic clinical responses of some FLT3-mutant AML lines to potent tyrosine kinase inhibitors followed by outgrowth of drug-resistant clones with “gatekeeper” FLT3 mutations.12 Our in vitro and in vivo studies of human AML cell lines and primary murine leukemias with NRAS/Nras mutations provide additional evidence that gain-of-function signaling mutations are required for AML maintenance and are thus attractive therapeutic targets. Moreover, in contrast to HSCs lacking Flt3,36 Nras-mutant HSCs are functionally normal, which suggests that blocking oncogenic N-Ras signaling will not adversely impact normal hematopoiesis.
Given our current inability to directly inhibit oncogenic N-Ras in vivo, we investigated MEK and PI3K as potential therapeutic targets. Previous data reported from analysis of the Cancer Cell Line Encyclopedia identified activating mutations in NRAS as a top predictor of sensitivity to MEK inhibition,37 and others have shown AML cell lines are particularly sensitive to trametinib in vitro.38 Consistent with observations in human cancer cell lines, we show that treating primary Nras-mutant AML cells with 2 structurally distinct MEK inhibitors demonstrated efficacy by reducing leukocytosis, improving splenomegaly, and prolonging survival across multiple independent and genetically heterogeneous leukemias. However, MEK inhibition did not trigger apoptosis, induce differentiation of AML blasts, or select for the outgrowth of drug-resistant subclones. We postulate that MEK inhibition may serve as an ideal “backbone” on which combination targeted therapy should be built but will be insufficient on its own to eradicate NRAS-mutant disease. Although the clinical development of PD901 has stalled because of toxicity concerns,39 there is emerging evidence that other MEK and MAPK pathway inhibitors may be clinically useful in Ras-driven AML.40-42 Our data support evaluating MEK inhibitors in combination with other conventional and targeted agents in preclinical models and identifying genes and pathways that act synergistically with MEK inhibition to kill NRAS-mutant AML cells.
The failure of MEK inhibition to induce apoptosis of leukemic blasts in our study is consistent with findings from other models. In KRAS-mutant lung cancer cell lines, treatment with MEK and PI3K inhibitors cause a uniform induction of growth arrest but variable apptotic response depending on the balance of specific BCL-2 family members.43 Intriguingly, a pooled shRNA–drug screen strategy identified that combined inhibition of MEK and the anti-apoptotic BH3 family member BCL-XL with navitoclax (ABT-263) induced marked regression of KRAS-mutant cancer cell line xenografts.44 These data raise the possibility of using MEK and BCL-2 family inhibitors in combination in NRAS-mutant AML to promote cell death.
Given our prior observation that PI3K/Akt signaling is activated in many NrasG12D AML lines,18 the lack of efficacy of the pan-PI3K inhibitor in all myeloid neoplasms tested was somewhat unexpected with the caveat that GDC-0941 does not durably inhibit the pathway at the maximum tolerated dose.34 The toxicity observed with combined MAPK and PI3K/Akt inhibition suggests that investigating isoform-specific PI3K inhibitors may be of value. Despite this, combined GDC-0941/PD901 was highly effective in MOL4070LTR-induced Kras-mutant T-cell acute lymphoblastic leukemia, inducing prolonged survival, clonal evolution, and phenotypic drug resistance.34 Furthermore, the single-agent efficacy of PD901 in NrasG12D AML was less pronounced than in Nf1-mutant AML,24 adding further evidence that targeting the aberrant signaling output of hyperactive Ras is highly isoform and context dependent.
Directly targeting mutant Ras oncoproteins remains a fundamental challenge in cancer therapy. Novel chemical approaches suggest mutation-specific inhibitors, as in the case of targeting KRASG12C, might be feasible.45 The disappointing clinical efficacy of farnesyl transferase inhibitors greatly reduced interest in therapeutically targeting posttranslational processing and subcellular trafficking of oncogenic Ras proteins in cancer.16 However, recent studies suggest that this approach might be successful. Small molecule inhibitors of the interaction between Ras and the prenyl-binding protein PDEδ disrupt signaling by mislocalizing farnesylated Ras to endomembranes.46 Inhibiting the palmitoylation/depalmitoylation cycle is an alternative approach for selectively targeting cancers with oncogenic NRAS mutations.47,48 Our data in normal hematopoietic and NRAS/Nras-mutant leukemia cells provide a strong rationale for pursuing these and other therapeutic strategies.
Acknowledgments
The authors thank Jessica Gannon, Tona Gilmer, and GlaxoSmithKline for providing trametinib and pharmacokinetic data.
This work was supported by the Rally Foundation for Childhood Cancer Research and The Truth 365; the Leukemia and Lymphoma Society of America Specialized Center of Research (LLS 7019-04); the National Institutes of Health, National Cancer Institute grants R37 CA72614 (K.S.), and T32 CA108462 (M.R.B.), grant K01 CA118425 (K.M.H.), and grant K08 CA134649 (Q.L.); an American Cancer Society–Hillcrest Committee Postdoctoral Fellowship (PF-14-146-01-LIB) (M.R.B.); and a Damon Runyon Cancer Research Foundation Fellowship (DRG-2149-13) (A.J.F.) S.L. is an Investigator of the Howard Hughes Medical Institute, and K.S. is an American Cancer Society Research Professor.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.R.B. designed experiments, performed research studies, analyzed data, and wrote the manuscript; E.H., A.J.F., T.H., and J.X. designed experiments, performed research studies, and analyzed data; J.Z., D.S., and S.L. provided reagents and technical advice; N.B. and T.W. designed experiments and performed research studies; S.C.K. analyzed pathology and provided technical advice; K.M.H. developed NrasG12D knock-in and conditional knockout mice and edited the manuscript; K.S. designed experiments, reviewed the data, and wrote the manuscript; and Q.L. generated and characterized Mx1-Cre; NrasG12D mice, generated the Nras-mutant AMLs used in these studies, designed experiments, reviewed the data, and wrote the manuscript.
Conflict-of-interest disclosure: D.S. is a full-time employee of Genentech, a member of the Roche group, manufacturer of GDC-0941. The remaining authors declare no competing financial interests.
Correspondence: Kevin Shannon, Helen Diller Family Cancer Research Building, University of California, San Francisco, 1450 3rd St, Room 240, San Francisco, CA 94158-9001; e-mail: shannonk@peds.ucsf.edu; and Qing Li, Department of Medicine, Division of Hematology/Oncology, University of Michigan, 109 Zina Pitcher Pl, BSRB 1520, Ann Arbor, MI 48109; e-mail: lqing@med.umich.edu.
References
- 1.Shlush LI, Zandi S, Mitchell A, et al. HALT Pan-Leukemia Gene Panel Consortium. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia [published correction appears in Nature. 2014;508(7496):420]. Nature. 2014;506(7488):328–333. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA. 2014;111(7):2548–2553. doi: 10.1073/pnas.1324297111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jan M, Snyder TM, Corces-Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4(149):149ra118. doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bachas C, Schuurhuis GJ, Hollink IH, et al. High-frequency type I/II mutational shifts between diagnosis and relapse are associated with outcome in pediatric AML: implications for personalized medicine. Blood. 2010;116(15):2752–2758. doi: 10.1182/blood-2010-03-276519. [DOI] [PubMed] [Google Scholar]
- 5.Farr CJ, Saiki RK, Erlich HA, McCormick F, Marshall CJ. Analysis of RAS gene mutations in acute myeloid leukemia by polymerase chain reaction and oligonucleotide probes. Proc Natl Acad Sci USA. 1988;85(5):1629–1633. doi: 10.1073/pnas.85.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowen DT, Frew ME, Hills R, et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood. 2005;106(6):2113–2119. doi: 10.1182/blood-2005-03-0867. [DOI] [PubMed] [Google Scholar]
- 8.Damm F, Heuser M, Morgan M, et al. Integrative prognostic risk score in acute myeloid leukemia with normal karyotype. Blood. 2011;117(17):4561–4568. doi: 10.1182/blood-2010-08-303479. [DOI] [PubMed] [Google Scholar]
- 9.Sano H, Shimada A, Taki T, et al. RAS mutations are frequent in FAB type M4 and M5 of acute myeloid leukemia, and related to late relapse: a study of the Japanese Childhood AML Cooperative Study Group. Int J Hematol. 2012;95(5):509–515. doi: 10.1007/s12185-012-1033-x. [DOI] [PubMed] [Google Scholar]
- 10.Schlenk RF, Döhner K, Krauter J, et al. German-Austrian Acute Myeloid Leukemia Study Group. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358(18):1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 11.Cortes JE, Kantarjian H, Foran JM, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31(29):3681–3687. doi: 10.1200/JCO.2013.48.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith CC, Wang Q, Chin CS, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260–263. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bacher U, Haferlach T, Schoch C, Kern W, Schnittger S. Implications of NRAS mutations in AML: a study of 2502 patients. Blood. 2006;107(10):3847–3853. doi: 10.1182/blood-2005-08-3522. [DOI] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7(4):295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 16.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 17.Braun BS, Shannon K. Targeting Ras in myeloid leukemias. Clin Cancer Res. 2008;14(8):2249–2252. doi: 10.1158/1078-0432.CCR-07-1005. [DOI] [PubMed] [Google Scholar]
- 18.Li Q, Haigis KM, McDaniel A, et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood. 2011;117(6):2022–2032. doi: 10.1182/blood-2010-04-280750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown AP, Carlson TC, Loi CM, Graziano MJ. Pharmacodynamic and toxicokinetic evaluation of the novel MEK inhibitor, PD0325901, in the rat following oral and intravenous administration. Cancer Chemother Pharmacol. 2007;59(5):671–679. doi: 10.1007/s00280-006-0323-5. [DOI] [PubMed] [Google Scholar]
- 20.Flaherty KT, Robert C, Hersey P, et al. METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 21.Gilmartin AG, Bleam MR, Groy A, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17(5):989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 22.Folkes AJ, Ahmadi K, Alderton WK, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51(18):5522–5532. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 23.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 24.Lauchle JO, Kim D, Le DT, et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature. 2009;461(7262):411–414. doi: 10.1038/nature08279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horiike S, Misawa S, Nakai H, et al. N-ras mutation and karyotypic evolution are closely associated with leukemic transformation in myelodysplastic syndrome. Leukemia. 1994;8(8):1331–1336. [PubMed] [Google Scholar]
- 26.Shih LY, Huang CF, Wang PN, et al. Acquisition of FLT3 or N-ras mutations is frequently associated with progression of myelodysplastic syndrome to acute myeloid leukemia. Leukemia. 2004;18(3):466–475. doi: 10.1038/sj.leu.2403274. [DOI] [PubMed] [Google Scholar]
- 27.Umanoff H, Edelmann W, Pellicer A, Kucherlapati R. The murine N-ras gene is not essential for growth and development. Proc Natl Acad Sci USA. 1995;92(5):1709–1713. doi: 10.1073/pnas.92.5.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pérez de Castro I, Diaz R, Malumbres M, et al. Mice deficient for N-ras: impaired antiviral immune response and T-cell function. Cancer Res. 2003;63(7):1615–1622. [PubMed] [Google Scholar]
- 29.Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 30.Xu J, Haigis KM, Firestone AJ, et al. Dominant role of oncogene dosage and absence of tumor suppressor activity in Nras-driven hematopoietic transformation. Cancer Discov. 2013;3(9):993–1001. doi: 10.1158/2159-8290.CD-13-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Kong G, Liu Y, et al. Nras(G12D/+) promotes leukemogenesis by aberrantly regulating hematopoietic stem cell functions. Blood. 2013;121(26):5203–5207. doi: 10.1182/blood-2012-12-475863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang T, Krisman K, Theobald EH, et al. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J Clin Invest. 2013;123(1):335–339. doi: 10.1172/JCI63193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dail M, Li Q, McDaniel A, et al. Mutant Ikzf1, KrasG12D, and Notch1 cooperate in T lineage leukemogenesis and modulate responses to targeted agents. Proc Natl Acad Sci USA. 2010;107(11):5106–5111. doi: 10.1073/pnas.1001064107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dail M, Wong J, Lawrence J, et al. Loss of oncogenic Notch1 with resistance to a PI3K inhibitor in T-cell leukaemia. Nature. 2014;513(7519):512–516. doi: 10.1038/nature13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kogan SC, Ward JM, Anver MR, et al. Hematopathology subcommittee of the Mouse Models of Human Cancers Consortium. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood. 2002;100(1):238–245. doi: 10.1182/blood.v100.1.238. [DOI] [PubMed] [Google Scholar]
- 36.Mackarehtschian K, Hardin JD, Moore KA, Boast S, Goff SP, Lemischka IR. Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity. 1995;3(1):147–161. doi: 10.1016/1074-7613(95)90167-1. [DOI] [PubMed] [Google Scholar]
- 37.Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jing J, Greshock J, Holbrook JD, et al. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol Cancer Ther. 2012;11(3):720–729. doi: 10.1158/1535-7163.MCT-11-0505. [DOI] [PubMed] [Google Scholar]
- 39.LoRusso PM, Krishnamurthi SS, Rinehart JJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16(6):1924–1937. doi: 10.1158/1078-0432.CCR-09-1883. [DOI] [PubMed] [Google Scholar]
- 40.Jain N, Curran E, Iyengar NM, et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: a University of Chicago phase II consortium trial. Clin Cancer Res. 2014;20(2):490–498. doi: 10.1158/1078-0432.CCR-13-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 2014;11(7):385–400. doi: 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
- 42.Abdel-Wahab O, Klimek VM, Gaskell AA, et al. Efficacy of intermittent combined RAF and MEK inhibition in a patient with concurrent BRAF- and NRAS-mutant malignancies. Cancer Discov. 2014;4(5):538–545. doi: 10.1158/2159-8290.CD-13-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hata AN, Yeo A, Faber AC, et al. Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS-mutant lung cancers. Cancer Res. 2014;74(11):3146–3156. doi: 10.1158/0008-5472.CAN-13-3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corcoran RB, Cheng KA, Hata AN, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23(1):121–128. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zimmermann G, Papke B, Ismail S, et al. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature. 2013;497(7451):638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 47.Dekker FJ, Rocks O, Vartak N, et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat Chem Biol. 2010;6(6):449–456. doi: 10.1038/nchembio.362. [DOI] [PubMed] [Google Scholar]
- 48.Xu J, Hedberg C, Dekker FJ, et al. Inhibiting the palmitoylation/depalmitoylation cycle selectively reduces the growth of hematopoietic cells expressing oncogenic Nras. Blood. 2012;119(4):1032–1035. doi: 10.1182/blood-2011-06-358960. [DOI] [PMC free article] [PubMed] [Google Scholar]