

Background: Retroviruses package a genomic RNA dimer. In vitro studies suggested kissing loop and extended dimer (ED) RNA-RNA interactions.
Results: HIV-1 mutants with ED defects are replication-impaired, and revertant viruses with compensatory RNA mutations were selected.
Conclusion: We describe a correlation between in vitro ED defects and poor virus replication.
Significance: We present, to our knowledge, the first in vivo evidence for the importance of the ED.
Keywords: HIV-1 Protease, Molecular Evolution, Reverse Transcription, RNA Structure, Virology, HIV-1 RNA, HIV-1 RNA Packaging, RNA Dimerization, Virus Evolution
Abstract
The 5′ leader of the HIV-1 RNA genome encodes signals that control various steps in the replication cycle, including the dimerization initiation signal (DIS) that triggers RNA dimerization. The DIS folds a hairpin structure with a palindromic sequence in the loop that allows RNA dimerization via intermolecular kissing loop (KL) base pairing. The KL dimer can be stabilized by including the DIS stem nucleotides in the intermolecular base pairing, forming an extended dimer (ED). The role of the ED RNA dimer in HIV-1 replication has hardly been addressed because of technical challenges. We analyzed a set of leader mutants with a stabilized DIS hairpin for in vitro RNA dimerization and virus replication in T cells. In agreement with previous observations, DIS hairpin stability modulated KL and ED dimerization. An unexpected previous finding was that mutation of three nucleotides immediately upstream of the DIS hairpin significantly reduced in vitro ED formation. In this study, we tested such mutants in vivo for the importance of the ED in HIV-1 biology. Mutants with a stabilized DIS hairpin replicated less efficiently than WT HIV-1. This defect was most severe when the upstream sequence motif was altered. Virus evolution experiments with the defective mutants yielded fast replicating HIV-1 variants with second site mutations that (partially) restored the WT hairpin stability. Characterization of the mutant and revertant RNA molecules and the corresponding viruses confirmed the correlation between in vitro ED RNA dimer formation and efficient virus replication, thus indicating that the ED structure is important for HIV-1 replication.
Introduction
The untranslated 5′ leader region (Fig. 1A) is the most conserved part of the HIV-1 RNA genome when comparing the nucleotide sequence of different virus isolates. The leader can fold multiple stem-loop structures that regulate various steps of the viral replication cycle, including the poorly understood processes of RNA dimerization and RNA packaging in virion particles. The DIS3 hairpin folds a stable stem-loop structure that is sufficient to produce HIV-1 RNA dimers in vitro (1–5). It harbors a palindrome sequence in the loop (GCGCGC for HIV-1 subtype B), such that two DIS hairpins can engage in kissing loop (KL) base pairing interaction (Fig. 1, B and C), thus forming “loose” RNA dimers (6–8). Heat treatment or incubation with the viral nucleocapsid protein triggers opening of the hairpin stem to allow extended interstrand base pairing and formation of a more stable extended dimer (ED) (Fig. 1, B and C) (9–11). Such ED RNA forms have only been described in vitro, and their in vivo relevance remains unclear.
FIGURE 1.

The HIV-1 RNA genome and dimer formation. A, schematic of the HIV-1 RNA genome with enlarged leader domain (nt 1–368), which encodes multiple regulatory signals: trans-acting response (TAR) element, polyadenylation signal (poly(A)), primer activation signal (PAS), primer binding site (PBS), dimerization initiation site (DIS), splice donor (SD) site, packaging signal (Ψ), and AUG start codon (AUG) of the Gag open reading frame (Gag). B, HIV-1 RNA dimer formation. The leader adopts the LDI conformation that cannot dimerize but can be triggered to fold the branched multiple hairpin (BMH) conformation that exposes the DIS hairpin for dimerization. The KL dimer is formed and can be converted into the ED. C, nucleotide resolution of the KL and ED dimers. The two DIS binding partners are shown in different colors. The 6-nt palindromic DIS sequence is boxed in pink and black. The numbers indicate the nucleotide position in HIV-1 genomic RNA relative to the transcriptional start site.
We previously reported that structural rearrangements in the leader regulate RNA dimerization in vitro (12–14). In vitro RNA dimerization of HIV-1 transcripts was prevented by formation of the so-called long distance interaction (LDI) conformation in which the DIS sequences are engaged in an intramolecular, long distance base pairing interaction with the upstream poly(A) hairpin (Fig. 1). As a result, the DIS palindrome is not available for initiating RNA dimer formation. Alternative HIV-1 RNA folding schemes have recently been proposed (15–17), but most models converge on the concept of regulation of RNA dimerization through RNA conformational changes, with one of the conformations being dimerization-incompetent because of the absence of the DIS hairpin or shielding of the DIS palindrome. This inhibition of RNA dimerization could be relieved by heat treatment or incubation with the HIV-1 nucleocapsid chaperone protein (9, 18). Both treatments trigger refolding of the LDI conformation into the dimerization-competent BMH (branched multiple hairpin) structure, which exposes the DIS hairpin (Fig. 1B).
Infectious HIV-1 particles contain two genomic RNA molecules that are noncovalently linked near their 5′ end, possibly at more than one position (19). It is thought that the native dimer structure of the RNA genome is important for its efficient reverse transcription into DNA, a complex process that includes two strand transfer events (20). RNA genome dimers are also thought to facilitate recombination during reverse transcription, which contributes to the generation of HIV genetic diversity (21, 22). RNA recombination rates are indeed significantly reduced when the DIS palindrome sequence is altered (22–24).
Studies on the conformation of genomic RNA inside virus particles are intrinsically difficult. Early electron microscopy studies indicated multiple RNA-RNA contacts along the dimeric genome, with the most stable interaction located near the 5′ ends (25). Stabilization of the RNA dimer was reported during maturation of newly assembled virion particles, but details on the molecular events remained obscure (26, 27). The HIV-1 DIS motif is considered critical for RNA dimerization (3, 5, 28, 29), but it may also regulate RNA packaging (30). Surprisingly, some studies suggested that the DIS palindrome is dispensable for RNA dimerization in vivo: its deletion or alteration to a smaller palindromic sequence does not affect RNA dimer stability inside virus particles, although RNA packaging was reduced (6). In addition, cell type-specific DIS defects were reported (31, 32). Additional RNA contact sites have been proposed for the upstream HIV-1 TAR hairpin and other leader motifs, including the SL3 region downstream (33–37). However, such studies require a careful analysis because leader mutations may induce RNA misfolding and conformational changes that indirectly trigger or impede RNA dimerization (38, 39).
Full-length HIV-1 RNA genomes are selected from an excess of cellular and subgenomic viral RNA during the virus assembly process and encapsidated by the Gag proteins. RNA dimerization and packaging are tightly linked processes: if proper RNA dimerization is compromised, the virus particles contain less genomic RNA and are less infectious (6, 40–42). The initial RNA-RNA dimer is formed in the virus-producing cell and is subsequently packaged. During virus particle maturation the stability of the HIV-1 RNA dimer increases, which depends on viral Protease activity and processing of the Gag-Pol polyprotein (27, 40, 43). In general, not much is known about the actual RNA-RNA contacts during virion assembly and subsequent maturation. Possibly, this dimer maturation process is analogous to the stabilizing RNA rearrangements that are observed in vitro when the KL dimer is converted into the ED. Unfortunately, classical RNA probing studies cannot discriminate between the KL and ED conformations because the same nucleotides are involved in these intra- and intermolecular base pairing schemes. Therefore, the molecular nature of the structural transition from fragile to stable RNA dimers inside virions remains elusive. Moore et al. (24) created DIS mutants that, at least in vitro, can form KL dimers, but not ED. Their experiments indicated that ED formation is not required for the initial RNA partner selection, subsequent RNA packaging, and the process of recombination. This does not exclude other roles for the ED in HIV-1 replication. The initial interaction between Gag and viral RNA during assembly and packaging has been studied in detail (44, 45).
To investigate the role of ED formation on virus replication, we analyzed a set of HIV-1 variants with mutations that affect the DIS hairpin (13). Virus replication was severely affected by alteration of a three-nucleotide motif (GGA triplet) immediately upstream of the DIS hairpin. Further analysis demonstrated that this mutation reduced in vitro ED formation and the amount of RNA dimers in virions. The crippled HIV-1 mutants were allowed to evolve by selection of faster replicating revertant viruses. Characterization of these variants confirmed the importance of the GGA triplet in both ED formation and viral replication.
EXPERIMENTAL PROCEDURES
DNA Constructs
The 5′ leader mutations (J8, J9, and J10) were introduced in the HIV-1 molecular clone pLAI (46) and the pLAI-R37 derivative (13). pLAI-R37 has a deletion in the R-U5 region of the 3′ LTR. The WT 3′ LTR is restored during the reverse transcription process in the first round of virus replication (47).
Virus Production
C33A human cervix carcinoma cells were cultured in DMEM (Invitrogen) supplemented with 10% FBS, nonessential amino acids (Invitrogen), 20 mm glucose, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C and 5% CO2. The cells were grown in 2 cm2 wells and transfected with 1 μg of WT or mutant pLAI-R37 DNA by the calcium phosphate method, as previously described (48). Two days post-transfection, the supernatant was harvested, and the cells were lysed in PBS containing 1% Empigen-BB. The intracellular and supernatant CA-p24 level was determined by ELISA (49).
Virus Replication and Evolution Studies
SupT1 T cells were cultured in RPMI 1640 medium supplemented with 10% (v/v) FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C and 5% CO2. Cells (10 × 106) were transfected with 250 ng or 1 μg of pLAI-R37 DNA by electroporation (250 V, 975 microfarad) using a Bio-Rad Gene Pulser II as previously described (48). Cells were split 1 to 10 twice a week. The CA-p24 level in the culture medium was determined by ELISA. For virus evolution, 15 × 106 cells were transfected with 40 μg of pLAI-R37 DNA by electroporation and immediately split into six cultures. The protocol for virus evolution by prolonged cell-free passage of virus onto fresh, uninfected SupT1 cells was described previously (50). Isolation of total cellular DNA was performed by Tween 20/proteinase K treatment (51). The LTR leader region was PCR-amplified with primers T7-1 and TA014 (13). The PCR products were sequenced directly, thus providing the population sequence of the viral quasispecies.
PCR products were also cloned in the pCRII-TOPO vector (Invitrogen). The HindIII-ClaI fragment of selected clones was used to replace the corresponding fragment in pBlue-5′ LTR (52). The plasmid contains the XbaI-ClaI fragment of the infectious pLAI clone, including the 5′ LTR promoter sequence, the full-length leader sequence, and part of the Gag open reading frame (−454/+376, relative to the transcriptional start site at +1). Subsequently, the progeny XbaI-ClaI fragments were cloned into pLAI-R37. The constructs were verified by BigDye terminator sequencing (Applied Biosystems, Foster City, CA).
Single Cycle Infection
TZM-bl cells (53, 54) were maintained in DMEM supplemented with 10% FBS, nonessential amino acids (Invitrogen), 20 mm glucose, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C and 5% CO2. TZM-bl cells were grown to 60% confluency in 2-cm2 wells and infected with WT or mutant C33A-produced virus (corresponding to 10 ng of CA-p24) in the presence of 80 μg/ml DEAE dextran. The cells were washed with PBS 2 days after infection and lysed in passive lysis buffer (Promega). The firefly luciferase was measured in cell lysates with the luciferase assay kit (Promega).
In Vitro Transcription and RNA Dimerization
The pLAI-based plasmids were used as template in a PCR with primers T7–2 and R:A368-A347 (complementary to nt +368 to +347 of the HIV-1 LAI genome with the transcription start site at +1). The PCR products were ethanol-precipitated and in vitro transcribed with the megashortscript T7 transcription kit (Ambion) in the presence of 1 μl of [α-32P]UTP (0.33 MBq/μl; PerkinElmer Life Sciences). Transcription reaction mixtures were incubated for 3 h at 37 °C, and the reactions were stopped by DNase treatment. The RNA was ethanol-precipitated, dissolved in water, and quantified by scintillation counting. To test the quality of the RNA, 20 ng of 32P-labeled RNA was incubated for 5 min at 85 °C in formamide-containing loading buffer (Ambion) and analyzed on a denaturing 6% polyacrylamide gel. Dimerization was performed with 20 ng of 32P-labeled RNA in 24 μl of dimerization buffer (83 mm Tris-HCl, pH 7.5, 125 mm KCl, and 5 mm MgCl2). The mixture was heated for 2 min at 85 °C, incubated for 10 min at 65 °C, and slowly cooled to room temperature for renaturing and dimerization. Samples were mixed with 12 μl of nondenaturing sample buffer (30% glycerol with bromphenol blue) and analyzed on nondenaturing 4% polyacrylamide gels in 0.25× TBE (22.5 mm Tris, 22.5 mm boric acid, 0.5 mm EDTA, pH 8.0) or 0.25× TBM (22.5 mm Tris, 22.5 mm boric acid, 0.1 mm MgCl2). Electrophoresis was performed for 3 h at 150 V at room temperature. Gels were dried and subjected to autoradiography, and the signals were quantified using a PhosphorImager (Molecular Dynamics).
RNA Secondary Structure Prediction
RNA secondary structure prediction was performed with the RNAstructure algorithm, version 5.3, offered by the Mathews Lab (55). The HIV-1 leader RNA (nt 1–368) and the DIS hairpin (nt 237–281) were folded with standard settings (37 °C). The most stable structure was selected. The structure was listed as LDI when the poly(A) and DIS hairpins were masked or as BMH when both hairpins were present.
Reverse Transcription Analysis in Infected SupT1 T Cells
293T cells were seeded in a 6-well format. The following day, at 80% confluency, the cells were transfected with 2.5 μg of LAI plasmid of WT, mutant, and revertant viruses using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. After 6 h, the cells were washed, and 48 h after transfection the virus-containing supernatant was harvested. Cells and cell debris were removed by centrifugation at 211 × g for 5 min and by passing the supernatant through a 0.2-μm pore size filter. Titers of the virus stocks were determined using CA-p24 ELISA. Similar amounts of virus (equivalent to 100 ng of CA-p24) were treated with 100 units of DNaseI (Roche) for 1 h at 37 °C in the presence of 10 mm MgCl2. The virus was immediately used to infect 2.5 × 106 SupT1 T cells. To selectively isolate the proviral HIV-1 DNA integrated in the host chromosome and to remove DNA present in other cellular fractions, a user-developed protocol of Qiagen was used. All buffers are derived from the Qiagen Genomic DNA or QIAfilter Plasmid Midi kit. 7 h after infection, the cells were washed three times to remove unbound virus. The cells were harvested 24 h after infection and washed three times with PBS. The cell pellet was dissolved in PBS to 107 cells/ml, and the cell membrane was lysed by addition of one volume ice-cold C1 buffer (1.28 m sucrose, 40 mm Tris-HCl, pH 7.5, 20 mm MgCl2, 4% Triton X-100) and three volumes of ice-cold distilled water. The cells were incubated for 10 min on ice and centrifuged for 15 min at 21,120 × g at 4 °C. The pellet was resuspended in one volume of C1 buffer and three volumes of distilled water. The nuclei were again pelleted, and the resulting nuclear pellet was resuspended in 300 μl of buffer P1 (50 mm Tris-HCl, pH 8.0, 10 mm EDTA, 100 μg/ml RNase A). The nuclei were lysed by addition of 300 μl of buffer P2 (200 mm NaOH, 1% SDS (w/v)) and incubated for 5 min at room temperature. After addition of 300 μl of buffer P3 (3 m KAc, pH 5.5), the sample was centrifuged at 21,120 × g for 10 min. The chromosomal DNA-containing pellet was dissolved in 1 ml of buffer G2 (800 mm guanidine-HCl, 30 mm Tris-HCl, pH 8.0, 30 mm EDTA, 5% Tween 20, 0.5% Triton X-100) in the presence of 500 μg/ml protease K and incubated at 50 °C for 1 h. DNA was precipitated in the presence of 0.7 volume isopropanol and centrifuged for 30 min, at 21,120 × g and 4 °C. The pellet was resuspended in 100 μl of TLE (10 mm Tris-HCl, pH 8.0, 0.1 mm EDTA). 1 ml of buffer QBT (750 mm NaCl, 50 mm MOPS, pH 7.0, 15% isopropanol, 0.15% Triton X-100) was added to equilibrate the sample for loading on a Genomic tip 20/G (Qiagen). The purification was performed according to the manufacturer's protocol. Genomic DNA was dissolved in PCR grade water.
The ability of HIV-1 variants to perform reverse transcription was analyzed by measuring the level of integrated HIV-1 DNA using real time PCR. DNA of 400,000 cells was used as input in a preamplification PCR of 10 cycles that specifically amplifies a short 5′ Gag region. This product was used as template in a TaqMan PCR (AbiPrism 7000; Applied Biosciences). Both PCRs were performed as described before (56). Gag levels were normalized to the β-actin gene as determined by TaqMan PCR (Invitrogen).
Viral RNA Isolation
Human embryonic kidney 293T cells were cultured at conditions similar to that of C33A cells. One day prior to transfection, 293T cells were seeded and cultured in 15 ml of complete medium without antibiotics in T75 flasks. The following day, at a confluency of 80%, the cells were transfected with 20 μg of WT or mutant pLAI plasmid using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Culture supernatant was harvested after 48 h, centrifuged at 211 × g for 10 min to remove cells, and passed through a 0.2-μm filter to remove cell debris. Virion particles were purified over a 20% sucrose cushion in PBS by centrifugation at 32,000 rpm (175,000 × g) for 2 h at 4 °C in a Beckman SW32 Ti rotor. The virion pellet was resuspended in 400 μl of lysis buffer (50 mm Tris-HCl, pH 7.4, 10 mm EDTA, 100 mm NaCl, 1% SDS (w/v), 100 μg/ml proteinase K, and 100 μg/ml yeast tRNA (Ambion)) and incubated at 37 °C for 30 min. Viral RNA was purified by two subsequent phenol-chloroform-isoamylalcohol (25:24:1 v/v; Invitrogen) extractions at 4 °C. The RNA was precipitated at −20 °C with 1/10 volume 3 m NaAc, pH 5.2, and 2.5 volume 100% ethanol and pelleted by centrifugation at 21,120 × g for 30 min at 4 °C. The pellet was washed once with 70% ethanol, resuspended in 50 μl of TENS buffer (10 mm Tris-HCl, pH 7.5, 1 mm EDTA, 100 mm NaCl, 1% SDS), aliquotted, and stored at −80 °C.
Primer Extension Assay
Viral RNA was quantified by primer extension as previously described (57). RNA isolated from an equal amount of virion particles (corresponding to 50 ng of CA-p24) was precipitated with 0.3 m NaAc, pH 5.2, 80% ethanol and subsequently dissolved in 12 μl of 83 mm Tris-HCl, pH 7.5, and 125 mm KCl with 20 ng of primer CN1 (GGTCTGAGGGATCTCTAGTTACCAGAGTC, nt 123–151 of LAI). The samples were heated at 85 °C for 2 min, at 65 °C for 10 min, followed by slow cooling to room temperature in 1 h to anneal the primer. After addition of 6 μl of 3× RT buffer (9 mm MgCl2, 30 mm DTT, 150 μg/ml actinomycin D, 30 μm dCTP, 30 μm dGTP, 30 μm dTTP, 1.5 μm dATP), 0.3 μl of [α-32P]dATP (0.33 MBq/μl; PerkinElmer Life Sciences) and 2.5 units of HIV-1 RT (22 nm; p51/p66 heterodimer; kindly provided by D. Stammers, Glaxo Wellcome Research Laboratories, Medical Research Council), the samples were incubated at 37 °C for 30 min to extend the annealed primer. The cDNA was precipitated in 25 mm EDTA, 0.3 m NaAc, pH 5.2, and 70% ethanol at −20 °C. cDNA pellets were washed once with 70% EtOH and dissolved in gel loading buffer II (Ambion). The cDNA product was heated at 85 °C for 3 min and analyzed on a denaturing 6% polyacylamide-urea sequencing gel. The signals were quantified using a PhosphorImager (Amersham Biosciences).
Northern Blot Analysis
For nondenaturing Northern blot analysis, viral RNA isolated from equal amounts of virions (equivalent to 100 ng of CA-p24) was diluted in 10 μl of TENS buffer, mixed with 5 μl of sample buffer (30% glycerol with 0.25% bromphenol blue dye), and analyzed by electrophoresis on a 0.9% agarose gel in 1× TBE buffer at 72 V, 4 °C for 6 h. The gel was soaked in 10% formaldehyde at 65 °C for 30 min to denature the RNA. The RNA was transferred overnight by capillary force onto a positively charged nylon membrane (Roche Applied Science) using 20× SSC (3.0 m NaCl, 0.3 m sodium citrate, pH 7.0). A UV cross-linker (Stratagene) was used to cross-link the RNA to the membrane. A 1014-bp DNA fragment covering the Nef, U3, and R regions of pLAI (positions 8770–9784) was produced by PCR with primers TTA1 (ACAGCCATAGCAGTAGCTGAG) and CN1 and pLAI as template. The fragment was 32P-labeled by random-primed labeling (High Prime DNA labeling kit; Roche Diagnostics) through incorporation of [α32P]CTP (0.33 MBq/μl; PerkinElmer Life Sciences). The membrane was prehybridized in ULTRAhyb buffer (Ambion) for 1 h at 55 °C and subsequently hybridized with the labeled probe for 16 h at 55 °C. The membrane was washed four times for 5 min in low stringency buffer (2× SSC, 0.2% SDS) at room temperature and twice for 15 min in high stringency buffer (0.1× SSC, 0.2% SDS) at 50 °C. The blot was analyzed using a PhosphorImager (Amersham Biosciences) and the ImageQuant software package.
For denaturing Northern blot analysis, the viral RNA was mixed with denaturing loading dye (final concentrations, 40 mm MOPS, pH 7.0, 10 mm sodium acetate, 5% formaldehyde, 0.05 mg/ml ethidium bromide, 0.5 mg/ml orange G, 7 g/ml sucrose) and electrophoresed on a 0.9% agarose gel in MOPS buffer (40 mm MOPS, 10 mm sodium acetate, pH 7.0) with 7% formaldehyde at 100 V for 4 h. The RNA was blotted and cross-linked to the membrane, washed, and analyzed as described above for the nondenaturing blot.
RESULTS
Characteristics and Virus Replication of DIS Mutants
We previously tested RNA transcripts of three HIV-1 leader mutants (J8, J9, and J10; Fig. 2A) for their impact on in vitro RNA dimerization (13) and now investigated in vivo ED RNA dimerization and virus replication. Short purine-rich segments were mutated that flank the DIS stem on the 5′ side (240GGA242 triplet mutated to CCC) and 3′ side (278GGG280 to UCC) in J8 and J9 to extend the DIS stem with three additional base pairs (Fig. 2A). The impact on the predicted thermodynamic stability of the DIS hairpin, the LDI and BMH conformations, and the LDI-BMH equilibrium are plotted in Table 1. As expected, DIS stabilization shifts the equilibrium toward the BMH folding (ΔΔGLDI-BMH in kcal/mol: −5.6 (WT), 6.8 (J8), and 2.5 (J9)). The J10 double mutant combines the J8 and J9 changes, resulting in a normal DIS hairpin with altered sequences in both flanking triplets. The overall RNA folding and the LDI-BMH equilibrium were only marginally affected (Table 1).
FIGURE 2.

Characterization of the HIV-1 DIS mutants J8, J9, and J10. A, predicted secondary structure of the WT and mutant DIS hairpins. The structures of WT and the mutant J8 were confirmed by SHAPE RNA probing (results not shown). The DIS palindrome is boxed in pink, and the mutated nucleotides are boxed in black. B, replication capacity of the WT and mutant viruses. SupT1 T cells were transfected with 1 μg of WT or mutant DNA construct. Virus spread was monitored by measurement of the supernatant CA-p24 level. A representative experiment is shown (n = 2). C, virus production by DNA-transfected C33A cells. The CA-p24 levels in cell extracts (closed bars) and the culture supernatant (open bars) are shown as the means with the S.D. (WT arbitrarily set at 1, n = 3). D, TZM-bl reporter cells were infected with C33A-produced virus (equivalent to 10 ng of CA-p24). Luciferase levels in cell lysates were measured (means with S.D., n = 2). E, cDNA produced by primer extension on viral RNA isolated from equal amounts of virions (determined by CA-p24). Shown is the mean with the S.D. (WT set at 1, n = 2).
TABLE 1.
Evolution of HIV-1 DIS mutants
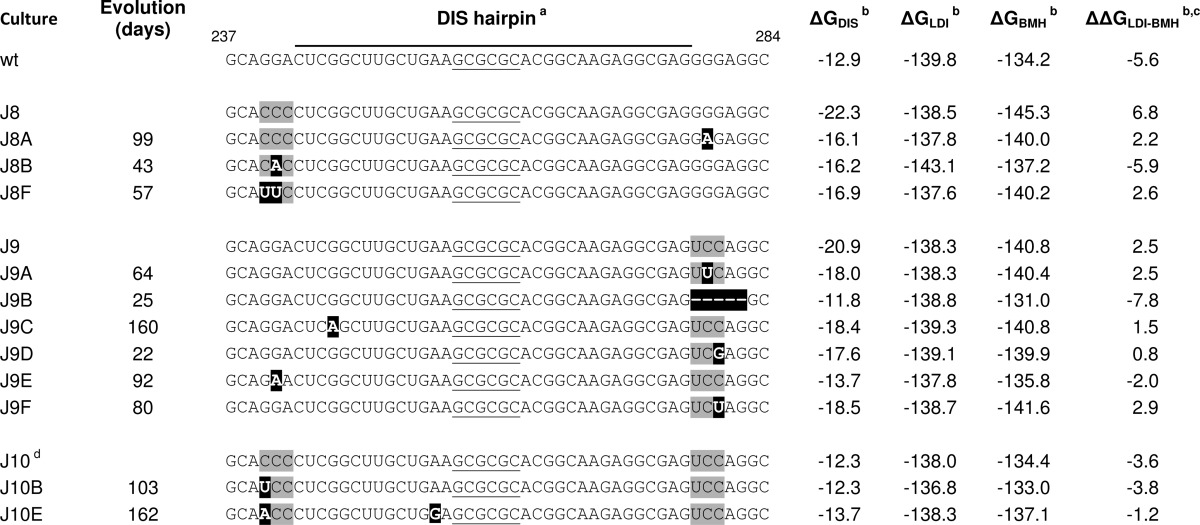
a DIS palindrome is underlined, mutated nucleotides are shaded gray, and reversions are in black boxes, − indicates deletion.
b RNAstructure predictions, ΔG and ΔΔG in kcal/mol.
c ΔΔG values < 0 represent LDI, and ΔΔG values > 0 represent BMH folding.
d Shows a rearranged DIS base pairing scheme in RNAstructure, repaired by forcing positions 257–262 single-stranded and positions 105–115 base paired to 334–344.
The WT RNA folds predominantly the LDI conformation in vitro, thus not exposing the DIS hairpin and preventing KL dimerization (13). However, J8 and J9 fold the BMH conformation because of the stabilized DIS hairpin and undergo spontaneous KL RNA dimerization (13). Evidence that RNA structure changes and not sequence changes dictate this phenotype comes from inspection of J10 that dimerizes in a restricted manner as WT. In contrast to WT and J9, J8 and J10 formed negligible amounts of ED (13). For J8, this may be due to DIS stem stabilization. J10 has a WT-like DIS stem, and nevertheless ED formation is reduced. In both J8 and J10, the GGA triplet was mutated, indicating that this triplet is important for ED formation.
Because of their intriguing ED phenotype, we assessed the effect of the J8, J9, and J10 mutations on virus replication. The DIS mutations were introduced into the infectious HIV-1 molecular clone of the primary LAI isolate. We transfected this set of pLAI plasmids into the SupT1 T cell line and followed viral spread over time by monitoring CA-p24 accumulation in the culture supernatant. All mutants (J8–J10) exhibited dramatically reduced replication kinetics when compared with WT (Fig. 2B). The efficiency of virus replication was consistently ranked as follows: WT > J9 ≫ J10 ≈ J8. From this, we conclude that mutation of the 5′-flanking GGA triplet affects HIV-1 replication more severely than mutation of the 3′-flanking GGG triplet. Interestingly, the J8 and J10 mutants that display reduced ED formation in vitro show the most severe replication defect, providing a handle to study the importance of ED formation in HIV-1 biology.
Mapping the Replication Defect of KL-ED Mutants
We next set out to identify the nature of the replication defect of these DIS mutants. We first assessed whether the mutations affect the late phase of virus production. C33A cells were transfected with the WT and mutant DNA constructs. Virus particles produced by the transfected cells will not initiate a new round of infection because the receptors for virus entry are absent from these cervical carcinoma cells. We analyzed the production of the viral capsid protein CA-p24 in the cells and culture supernatant (Fig. 2C). None of the DIS mutant viruses were significantly affected in cell-associated and supernatant CA-p24 production, indicating that similar levels of virus particles were produced. This also indicates that the mutations do not exert an effect at the level of viral gene expression (e.g. transcription, splicing, mRNA nuclear export, translation, and mRNA stability). We next probed the early phase by infecting the TZM-bl reporter cell line, where Tat protein production upon successful infection (including cell entry, reverse transcription, integration, and subsequent viral protein production) will activate the LTR-luciferase reporter. Thus, any of the expected problems during HIV-1 RNA dimerization and/or RNA packaging will show up in this assay. With the same input (based on CA-p24) of WT and mutant viruses produced in C33A cells, we measured a severe reduction in infectivity for the J8, J9, and J10 mutant viruses (Fig. 2D). The ranking order of particle infectivity closely resembles that observed for the replication kinetics: WT > J9 > J10 ≈ J8.
We isolated virus particles from the supernatant of transfected C33A cells by ultracentrifugation and determined the level of viral RNA in virions using a primer extension assay. The WT and DIS mutant particles contain similar levels of viral RNA, suggesting that the DIS mutations do not affect HIV-1 RNA packaging (Fig. 2E). As will be described below, Northern blot analysis of virion-extracted RNA confirmed that the WT and mutant RNA genomes are packaged with similar efficiency.
Selection of Revertant Viruses
To gain further insight into how the DIS mutations affect virus replication, we performed forced HIV-1 evolution experiments. This approach was previously optimized by us to dissect the role of important regulatory RNA motifs in the HIV-1 genome (58). In short, we transfected SupT1 T cells with high amounts of the mutant DNA constructs and passaged the cells until the first signs of virus replication emerged (syncytia). From then on, the cell-free culture supernatant was serially passaged onto fresh SupT1 cultures, starting with large inocula and gradually going down in volume. For every mutant we performed six independent evolution cultures. The J9 mutant eventually replicated efficiently in all cultures, but the J8 and J10 mutants in only three and two cultures, respectively (Table 1). This evolution potential correlates with the intrinsic replication capacity of the original mutants.
Chromosal DNA from the infected cells was collected at each passage to analyze the sequence of the integrated HIV-1 proviruses. Specifically, the complete 5′ leader of the viruses was PCR-amplified and sequenced to identify the mutations that improved virus replication. This population-based sequence will only reveal the dominant changes that were selected during virus evolution. We frequently scored first site reversions inside the mutated stretch of nucleotides, thus inside the upstream triplet (GGA to CCC) for mutant J8 and the downstream triplet (GGG to UCC) in J9 (Table 1). Second site mutations on the opposing side of the DIS hairpin were also observed for J8 and J9. All these J8 and J9 reversions have in common that the thermodynamic stability of the stabilized DIS hairpin is reduced (see the ΔGDIS column in Table 1), which thus seems the driving force for virus evolution. The J8 hairpin with a ΔGDIS of −22.3 kcal/mol was destabilized to a ΔGDIS value varying from −16.1 to −16.9 kcal/mol. The J9 hairpin with a ΔGDIS of −20.9 kcal/mol was destabilized to ΔGDIS values between −11.8 and −18.5 kcal/mol. On the other hand, the J10 double mutant with a WT DIS hairpin stability did acquire reversions in the upstream triplet, but these mutations did not significantly affect the hairpin stability (range of ΔGDIS between −12.3 and −13.7 kcal/mol). Possibly, these mutations restored an important sequence motif, although the mutations did not recreate the WT sequence.
Overall, important sequence information seems to be present in both triplet motifs, with changes occurring most frequently in the first two positions of the GGA triplet and the last two positions of the GGG motif (Table 1). In some cultures, the viruses acquired a mutation in the DIS hairpin itself that did (J9C) or did not (J10E) affect the predicted stability of the stem. The mutations also affected the LDI-BMH equilibrium. The J8, J9, and J10 reversions partially restored the LDI-BMH equilibrium when analyzed in silico (see ΔΔGLDI-BMH column in Table 1). The WT and J10 sequences favor the LDI conformation (negative ΔΔGLDI-BMH). For the J8 and J9 mutants, the BMH conformation is favored (positive ΔΔGLDI-BMH), whereas most mutations selected during virus evolution move the equilibrium toward LDI folding (the WT pattern).
For each mutant, we selected two revertants for further testing, and molecular clones were constructed with these precise mutations to filter out possible effects of acquired mutations elsewhere in the HIV-1 genome (Fig. 3). We first assessed the replication potential in SupT1 cells (Fig. 4). All revertants except J9C showed a significantly improved replication capacity when compared with the original mutants. The replication capacity was ranked as follows: WT > J8F ≈ J9E > J8A ≈ J10E > J9 > J10B ≫J8 ≈ J10 ≈ J9C (summarized in Table 2). It is possible that J9C requires an additional mutation outside the leader region that was recloned. As will be described below, J9C does improve the in vitro RNA dimerization properties of the J9 mutant but apparently is not sufficient for recovery of virus replication.
FIGURE 3.
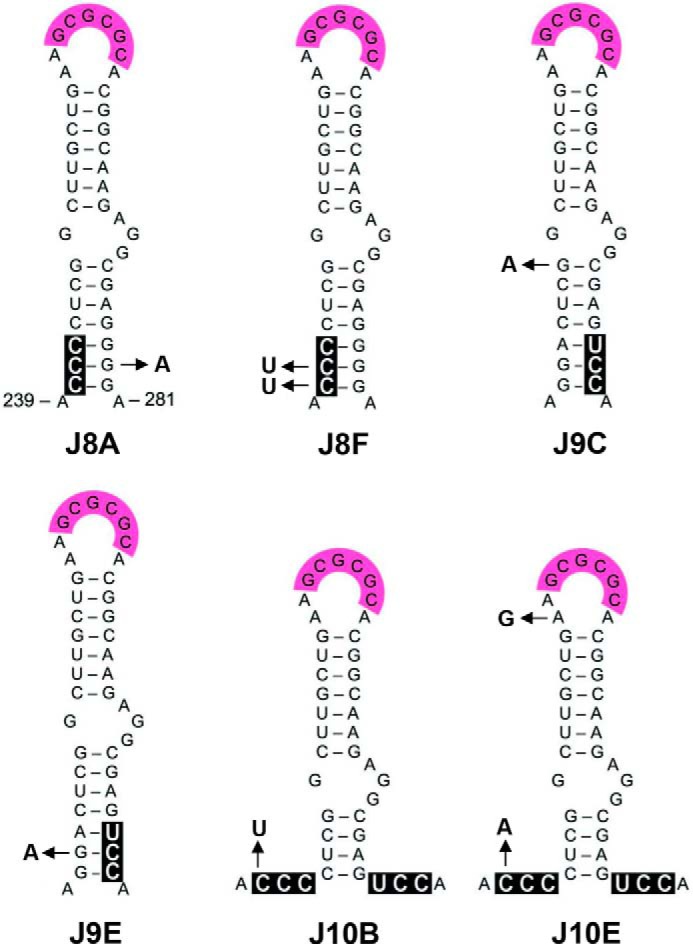
RNA structural changes upon evolution of mutant viruses. Virus variants were selected upon long term culturing of the J8, J9, and J10 mutated viruses. The mutations are boxed in black. The J8, J9, and J10 mutations acquired upon evolution in the DIS hairpin region are indicated by arrows.
FIGURE 4.
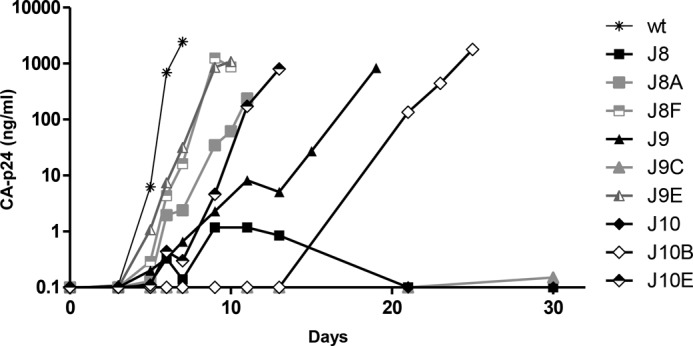
Replication capacity of WT, mutant, and revertant viruses. SupT1 T cells were transfected with 250 ng of WT, mutant, or revertant DNA constructs. Virus spread was assessed by monitoring the CA-p24 level in the culture supernatant. Shown is a representative experiment (n = 2).
TABLE 2.
Properties of DIS mutants and revertants
| HIV-1 variants | In silico |
In vitro |
In vivo |
||||
|---|---|---|---|---|---|---|---|
| RNA conformationa | RNA conformationb | KL + ED formationc | ED formationd | RNA packaginge | RNA monomerf | Replication capacityg | |
| % | |||||||
| WT | LDI | LDI | + | + | + | 14 | ++++ |
| J8 | BMH | BMH | +++ | − | + | 42 | − |
| J8A | BMH | LDI | ++ | + | + | 34 | ++ |
| J8F | BMH | BMH/LDI | +++ | + | + | 18 | +++ |
| J9 | BMH | BMH | +++ | + | + | 34 | + |
| J9C | BMH | BMH/LDI | +++ | + | − | 33 | − |
| J9E | LDI | LDI | ++ | + | + | 23 | +++ |
| J10 | LDI | LDI | ++ | − | + | 36 | − |
| J10B | LDI | LDI | + | − | + | 35 | + |
| J10E | LDI | LDI | ++ | + | + | 31 | ++ |
a ΔΔG values < 0 represent LDI, and ΔΔG values > 0 represent BMH folding (Table 1).
b As determined by TBE gel electrophoresis (Fig. 5).
c +, WT signal; ++, <2.5× WT signal; +++, >2.5× WT signal (TBM gel; Fig. 5), normalized for total RNA signal.
d +, measured signal ≥ WT signal; −, measured signal < WT signal −3% (TBE gel; Fig. 5), normalized for total RNA signal.
e +, mean similar to WT (Fig. 6B); −, significantly reduced compared to WT.
f As determined by native northern electrophoresis (Fig. 6D).
g ++++, WT replication; +++, <5 days delayed compared to WT; ++, <8 days delayed compared to WT; +, ≥8 days delayed; −, no detectable CA-p24 level within 30 days of culture.
In Vitro RNA Dimerization Properties
Because J8 and J10 transcripts exhibited the ED defect in vitro (13), we assessed the dimerization properties of the revertants. We analyzed in vitro transcribed RNAs corresponding to the complete leader (nt 1–368) by polyacrylamide gel electrophoresis in the presence or absence of Mg2+ (TBM and TBE gel, respectively; Fig. 5). As previously described, both KL and ED dimers are detected with Mg2+, whereas EDs are exclusively detected without Mg2+ (14, 35). The TBM gel confirmed that J8 and J9 produce much more total RNA dimers (KL + ED) than WT and J10 (Fig. 5, middle panel). This pattern of excessive RNA dimerization was (partially) reversed for some of the tested revertants (J8A, J9C, and J9E; summarized in Table 2).
FIGURE 5.

In vitro dimerization of WT, mutant, and revertant RNA transcripts. The leader RNA transcripts (nt 1–368) were heat-denatured and analyzed on a denaturing urea-containing gel to check for integrity (upper panel). Alternatively, the RNAs were allowed to fold and analyzed on nondenaturing TBM gel (middle panel) or TBE gel (lower panel) to separate the different RNA conformations, which are indicated along the gels. M, monomer.
The TBE gel confirmed that WT and J9 do form EDs, whereas J8 and J10 do not form detectable ED levels (Fig. 5, bottom panel). This ED defect was repaired for the revertant transcripts J8A with a single substitution in the GGG triplet and J8F with a double substitution in the GGA triplet, correlating with increased virus replication capacity (Fig. 4 and Table 2). In fact, the U241 nucleotide selected in J8F was also frequently observed in viral SELEX experiments (59). We also tested two J10 revertants that substitute the first position of the GGA triplet, either to a pyrimidine (C240U in J10B) or purine (C240A in J10E). We observed improved ED formation only for J10E (Fig. 5). This ED forming capacity correlates with the replication results in Fig. 4: J10B hardly replicates, but J10E shows greatly improved viral spread compared with mutant J10.
The LDI and BMH conformations of the HIV-1 RNA monomer can be distinguished on a TBE gel by differential migration (35). Whereas the WT and J10 transcripts were predominantly present in the LDI conformation, J8 and J9 adopt the BMH conformation (Fig. 5, bottom panel). The J8 and J9 revertants partially or completely restore the WT phenotype of LDI formation, and these results are in general agreement with the in silico RNA structure predictions (Table 2). The WT, J9E, J10, J10B, and J10E RNAs with a negative ΔΔGLDI-BMH adopt the LDI conformation (Fig. 5). The J8 mutant with high positive ΔΔGLDI-BMH folds the BMH conformation, and the other mutants and revertants with a ΔΔGLDI-BMH between 1.5 and 2.6 adopt either LDI or BMH or a mixed phenotype. Revertants that acquired two mutations generally repaired ED formation more efficiently than transcripts with a single acquired mutation (J8F > J8A and J10E > J10B), confirming the correlation between ED forming capacity and HIV-1 replication efficiency.
The Dimeric State of RNA Genomes Packaged in Virion Particles
We investigated the dimer status of the mutant and revertant RNA genomes inside virion particles. A correlation between the RNA dimerization in virions and the viral replication capacity has been described for HIV-1 and HIV-2 (6, 60–63). To obtain a high virus yield, we transfected 293T cells with the relevant plasmids and pelleted the produced virions by ultracentrifugation. Analysis of the virion RNA, isolated from an equal amount of particles (based on CA-p24), on a denaturing Northern blot indicated similar levels of packaged HIV-1 RNA for WT, all mutants and revertants except for J9C and J9E (Fig. 6, A and B, and Table 2). These results confirm that the DIS-flanking mutations do not affect the RNA packaging process.
FIGURE 6.

Virion RNA analysis of WT, mutant, and revertant HIV-1 variants. A and C, viral RNA was extracted from virions and analyzed on a denaturing gel (A) or nondenaturing gel (C), followed by Northern blotting. The position of the full-length, 9-kb HIV-1 transcript and monomer (M) and dimer (D) forms are indicated. In the 56 °C lane, the WT RNA sample was heat-denatured prior to gel loading to identify the RNA monomer position. B, quantitation of the 9-kb genomic RNA signals from A. Shown are the means and S.D. (n = 3). Virion RNA content did not vary significantly from the WT level (p > 0.05, Student's t test), except for J9C and J9E. D, quantitation of the RNA dimer and monomer signals from C with the mean and S.D. (n = 3). All RNA signals were quantitated by ImageQuant analysis. Statistical analysis demonstrated that all levels, except for J8F, varied significantly (p < 0.05) from the WT level. J8A and J8F levels varied significantly from the J8 level. The J9E level varied significantly from the J9 level. E, histogram profiles of the lanes in C, monomer (M) and dimer (D) forms are indicated.
We next analyzed the virion-derived RNA genome on a nondenaturing Northern blot to analyze the dimeric state (Fig. 6, C–E, and Table 2). The viral RNA from WT virions was predominantly present as a slow migrating dimer, and ∼14% of the RNA was present as fast migrating monomer. The mutants J8, J9, and J10 showed a severe reduction in RNA dimer yield and an increase in the RNA monomer level up to ∼40%. J8 revertants J8A and J8F both largely restore RNA dimer formation (J8F > J8A), which mimics the replication kinetics (J8F > J8A) and the observed restoration of the in vitro dimer-monomer pattern. J9C did not restore the WT pattern, likely linked to its inability to replicate. J9E displays a monomer-dimer phenotype almost similar to WT, which can be correlated to nearly complete restoration of in vitro RNA dimerization and virus replication. J10B does not restore the RNA dimer pattern, and this matches with the inability to form ED and the strongly impaired replication capacity. The other J10 revertant, J10E, showed a small increase in viral RNA dimer formation and was again able to produce EDs. The small dimer increase nicely matches the improved, but still delayed, replication capacity of the virus. For the revertants that exhibit the largest gain in replication capacity, in vitro and in vivo RNA dimerization nicely match with virus replication.
A mature RNA dimer has been implicated in the complex process of reverse transcription (20–24, 64). Because our mutant and revertant viruses affect RNA maturation to various extents, we tested whether this is reflected in the reverse transcription capacity of the viruses (Fig. 7). SupT1 T cells were infected with similar amounts of virus (based on CA-p24 levels), and after 24 h the chromosomal DNA, containing the reverse transcribed and integrated HIV-1 provirus, was isolated. Real time PCR was performed on a short Gag DNA fragment. The Gag signal of mutants J8, J9, and J10 was strongly reduced compared with WT, corresponding to in vivo RNA dimerization and virus replication. All revertants partially restore reverse transcription and formation of the provirus.
FIGURE 7.
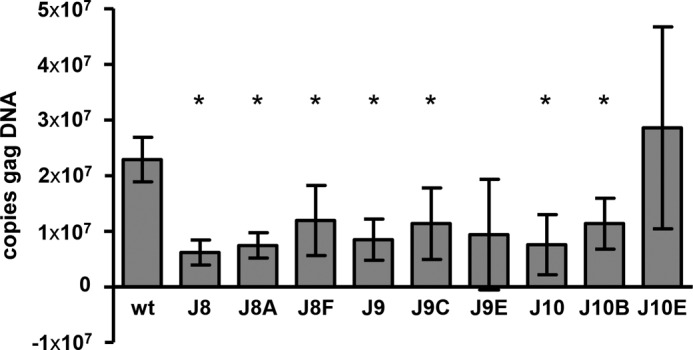
Reverse transcription and provirus formation of WT, mutant, and revertant HIV-1 variants. SupT1 T cells were infected with similar amounts of virus. After 24 h, the chromosomal DNA was isolated, and the level of integrated HIV-1 DNA was measured by real time PCR analysis amplifying a short Gag fragment. Shown are the means and S.D. (n = 3). Gag DNA levels significantly different from WT are marked with an asterisk (p < 0.05, Student's t test).
DISCUSSION
Retroviruses are unique in that they package two identical copies of the RNA genome that adopt a dimer conformation held together via noncovalent bonds. A dimeric RNA template is likely required to facilitate the complex and nonlinear reverse transcription process that generates a DNA copy that is longer than the original RNA template. The dimeric RNA genome may also allow the virus to bypass nicks or lesions in one of the RNA strands (65, 66), and it may be beneficial for virus evolution by inducing recombination events (67, 68). Therefore, it is important to learn more about the molecular structure of the RNA dimer inside virion particles. However, the exact nature of the dimer RNA genome remains somewhat mysterious. Much precise information has been collected in simplified in vitro assays with small RNA segments, but it remains unclear how these results translate into virion biology.
We tried to close this gap between in vitro and in vivo studies for HIV-1. In vitro studies suggested that early KL dimers mature into more stable RNA dimers through opening of the two DIS hairpin stems to facilitate the formation of additional intermolecular base pairs (10, 29, 69) (Fig. 1C). This ED form may relate to the RNA dimer “maturation” process described in vivo during virion maturation, yielding an RNA dimer with increased thermal stability (27, 70, 71). We previously described specific mutations near the DIS motif that stabilize this hairpin and consequently stimulate KL dimer formation in vitro. The capacity to form KL dimers did not correlate with the capacity to form ED. Instead, we observed that the three nucleotides immediately upstream of the DIS hairpin (GGA triplet) were critical for ED formation. We set out to investigate the replication capacity of these mutants to study the biological role of ED. The corresponding HIV-1 mutants exhibited rather severe replication defects. Virus evolution of these replication-impaired mutants led to revertant viruses with interesting compensatory mutations. Testing of short RNA transcripts of the mutant and revertant viruses in vitro indicated that the ability to produce a moderate level of KL and ED dimers correlates with virus replication. These results are summarized in Table 2 and reinforce the concept that retroviral RNA dimerization is a regulated process. Regulated HIV-1 RNA dimerization occurs via a riboswitch mechanism with a dimerization-incompetent (LDI) and an dimerization-competent (BMH) conformation. In general, we observed that the LDI-BMH equilibrium, which was disturbed in the DIS mutants, was, at least partially, repaired in the revertant viruses (Fig. 5 and Table 2). To our knowledge these combined results provide the first in vivo evidence for the importance of the ED structure in HIV-1 RNA dimerization and virus replication.
The GGA triplet immediately upstream of the DIS hairpin seems of critical importance for ED formation and viral replication. This is demonstrated by comparison of the J8 and J10 mutants and four revertants thereof: J8A, J8F, J10B, and J10E. In both J8 and J10, the GGA triplet was mutated, and in vitro ED formation, viral genomic RNA dimerization, and viral replication are strongly affected for both viruses (in Table 2: ED formation −, monomer ∼40%, replication −). The revertants J8A, J8F, and J10E restore all of these processes. J8A with a single substitution in the GGG triplet downstream of the DIS hairpin is modestly improved (in Table 2: ED formation +, monomer 34%, replication ++), but J8F with two substitutions in the upstream GGA triplet has a near WT-like phenotype (in Table 2: ED formation +, monomer 18%, replication +++). Both J8A and J8F destabilize the extended DIS hairpin. The additional positive effect of J8F compared with J8A therefore indicates the greater importance of the GGA triplet compared with the GGG triplet. Both J10 revertants acquired a single substitution at the same position in the upstream triplet (J10B: C to U, J10E; C to A), but with different phenotypic effects. J10B reversion does not repair the purine content and has only limited effects (in Table 2: ED formation −, monomer 35%, replication +). J10E introduces a purine and improves both the in vitro ED forming ability and virus replication capacity to a large extent (ED formation +, replication ++). Only a slight effect on viral RNA dimerization was measured for J10E (monomer 31%, versus 36% for J10). The two substitutions in the GGA triplet in J8F have a more profound effect than the single substitution in the same triplet of J10B and J10E. These combined results strengthen the notion of an important role of the GGA triplet in ED formation and virus replication and the idea that ED formation is required for efficient virus replication.
Further experimentation is needed to explain in mechanistic detail how the upstream GGA triplet affects HIV-1 RNA dimerization, ED formation in particular. There could be an unexpected stacking effect of these single-stranded nucleotides on DIS folding or melting during the KL to ED conversion (72). The combined experimental results indicate that the actual sequence of this motif is important, which may suggest the sequence specific binding of a protein co-factor. The most likely candidate is the viral nucleocapsid/Gag protein that is known to occupy multiple sites within the 5′ leader to support several RNA functions (34, 73, 74).
Because the ED-deficient HIV-1 mutants J8 and J10 package normal genomic RNA levels, the results suggest that the early KL dimer form is sufficient to trigger efficient genome packaging, which is consistent with previous reports (23, 24, 75, 76). The inability of these mutant HIV-1 RNA genomes to mature into more stable ED dimers may trigger subsequent defects, e.g. genome disintegration over time or incomplete reverse transcription as suggested previously (64). Our results indeed suggest a link between RNA dimer maturation and reverse transcription.
We recently performed an in vivo SELEX experiment in which sequence elements flanking the DIS and SD hairpins (nt 237–242, 278–281, and 301–305) were randomized, followed by the selection of replication-competent virus variants (59). These experiments demonstrated an extremely high selection pressure for the WT sequence in the 5′ DIS flank, which includes the GGA triplet that was identified in this study as an important motif for ED formation. A strong but less extreme selection pressure was scored for the 3′ DIS flank that encompasses the GGG triplet. The current virus evolution data confirm the in vivo SELEX data in pointing to the critical importance of the GGA triplet and to a lesser extent the GGG motif. The SELEX experiment showed strong selection pressure on the 237GCAGGA242 motif that encompasses the critical GGA motif. By sharp contrast, no selection pressure for a particular sequence was observed in the 3′ flank of the SD hairpin (nt 301–305). A phylogenetic survey indicates that the upstream GGA triplet is absolutely conserved among HIV-1 subtypes, except for the outlier (O) HIV-1 group (Fig. 8). The downstream GGG triplet is less well conserved. The HIV-2 DIS hairpin is also flanked by conserved purine motifs, GGAG, at the 5′ and 3′ side and an in vivo selection experiment selected purines in the 5′ flank (77). Disruption of the 5′ GGAG motif of HIV-2 affected RNA genome dimerization and virus replication (78). In general, all HIV and SIV variants are likely to use similar molecular strategies to regulate RNA dimerization and packaging (79).
FIGURE 8.

Phylogenetic analysis of DIS sequences in HIV-SIV isolates. The consensus sequence of the DIS hairpin and flanking sequences (nt 392–448) is shown for different HIV-1 and HIV-2 subtypes. For HIV-1 strains, nucleotides differing from the subtype B consensus are in bold type. For HIV-2 and SIV strains, nucleotides differing between the depicted strains are in bold type. The 6-nucleotide DIS palindrome is underlined, and the two purine stretches flanking the DIS hairpin are boxed. Sequences are from the HIV-1 sequence compendium 2012 (80).
Acknowledgment
We thank Stephan Heynen for performing CA-p24 ELISA.
This work was supported by Netherlands Organization for Scientific Research, Chemical Sciences Division Grant 700.59.301.
- DIS
- dimerization initiation signal
- KL
- kissing loop
- ED
- extended dimer
- LDI
- long distance interaction
- nt
- nucleotide
- BMH
- branched multiple-hairpins.
REFERENCES
- 1. Darlix J. L., Gabus C., Nugeyre M. T., Clavel F., Barré-Sinoussi F. (1990) cis elements and trans-acting factors involved in the RNA dimerization of the human immunodeficiency virus HIV-1. J. Mol. Biol. 216, 689–699 [DOI] [PubMed] [Google Scholar]
- 2. Marquet R., Baudin F., Gabus C., Darlix J. L., Mougel M., Ehresmann C., Ehresmann B. (1991) Dimerization of human immunodeficiency virus (type 1) RNA: stimulation by cations and possible mechanism. Nucleic Acids Res. 19, 2349–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paillart J. C., Marquet R., Skripkin E., Ehresmann B., Ehresmann C. (1994) Mutational analysis of the bipartite dimer linkage structure of human immunodeficiency virus type 1 genomic RNA. J. Biol. Chem. 269, 27486–27493 [PubMed] [Google Scholar]
- 4. Paillart J. C., Skripkin E., Ehresmann B., Ehresmann C., Marquet R. (1996) A loop-loop “kissing” complex is the essential part of the dimer linkage of genomic HIV-1 RNA. Proc. Natl. Acad. Sci. U.S.A. 93, 5572–5577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Skripkin E., Paillart J. C., Marquet R., Ehresmann B., Ehresmann C. (1994) Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc. Natl. Acad. Sci. U.S.A. 91, 4945–4949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berkhout B., van Wamel J. L. (1996) Role of the DIS hairpin in replication of human immunodeficiency virus type 1. J. Virol. 70, 6723–6732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ennifar E., Walter P., Ehresmann B., Ehresmann C., Dumas P. (2001) Crystal structures of coaxially stacked kissing complexes of the HIV-1 RNA dimerization initiation site. Nat. Struct. Biol. 8, 1064–1068 [DOI] [PubMed] [Google Scholar]
- 8. St Louis D. C., Gotte D., Sanders-Buell E., Ritchey D. W., Salminen M. O., Carr J. K., McCutchan F. E. (1998) Infectious molecular clones with the nonhomologous dimer initiation sequences found in different subtypes of human immunodeficiency virus type 1 can recombine and initiate a spreading infection in vitro. J. Virol. 72, 3991–3998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feng Y. X., Copeland T. D., Henderson L. E., Gorelick R. J., Bosche W. J., Levin J. G., Rein A. (1996) HIV-1 nucleocapsid protein induces “maturation” of dimeric retroviral RNA in vitro. Proc. Natl. Acad. Sci. U.S.A. 93, 7577–7581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Muriaux D., Fossé P., Paoletti J. (1996) A kissing complex together with a stable dimer is involved in the HIV-1Lai RNA dimerization process in vitro. Biochemistry 35, 5075–5082 [DOI] [PubMed] [Google Scholar]
- 11. Theilleux-Delalande V., Girard F., Huynh-Dinh T., Lancelot G., Paoletti J. (2000) The HIV-1(Lai) RNA dimerization: thermodynamic parameters associated with the transition from the kissing complex to the extended dimer. Eur. J. Biochem. 267, 2711–2719 [DOI] [PubMed] [Google Scholar]
- 12. Abbink T. E., Berkhout B. (2003) A novel long distance base-pairing interaction in human immunodeficiency virus type 1 RNA occludes the Gag start codon. J. Biol. Chem. 278, 11601–11611 [DOI] [PubMed] [Google Scholar]
- 13. Abbink T. E., Ooms M., Haasnoot P. C., Berkhout B. (2005) The HIV-1 leader RNA conformational switch regulates RNA dimerization but does not regulate mRNA translation. Biochemistry 44, 9058–9066 [DOI] [PubMed] [Google Scholar]
- 14. Huthoff H., Berkhout B. (2002) Multiple secondary structure rearrangements during HIV-1 RNA dimerization. Biochemistry 41, 10439–10445 [DOI] [PubMed] [Google Scholar]
- 15. Lu K., Heng X., Garyu L., Monti S., Garcia E. L., Kharytonchyk S., Dorjsuren B., Kulandaivel G., Jones S., Hiremath A., Divakaruni S. S., LaCotti C., Barton S., Tummillo D., Hosic A., Edme K., Albrecht S., Telesnitsky A., Summers M. F. (2011) NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science 334, 242–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sakuragi J., Ode H., Sakuragi S., Shioda T., Sato H. (2012) A proposal for a new HIV-1 DLS structural model. Nucleic Acids Res. 40, 5012–5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watts J. M., Dang K. K., Gorelick R. J., Leonard C. W., Bess J. W., Jr., Swanstrom R., Burch C. L., Weeks K. M. (2009) Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460, 711–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huthoff H., Berkhout B. (2001) Two alternating structures of the HIV-1 leader RNA. RNA 7, 143–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Höglund S., Ohagen A., Goncalves J., Panganiban A. T., Gabuzda D. (1997) Ultrastructure of HIV-1 genomic RNA. Virology 233, 271–279 [DOI] [PubMed] [Google Scholar]
- 20. van Wamel J. L., Berkhout B. (1998) The first strand transfer during HIV-1 reverse transcription can occur either intramolecularly or intermolecularly. Virology 244, 245–251 [DOI] [PubMed] [Google Scholar]
- 21. Moore M. D., Hu W. S. (2009) HIV-1 RNA dimerization: it takes two to tango. AIDS Rev. 11, 91–102 [PMC free article] [PubMed] [Google Scholar]
- 22. Nikolaitchik O. A., Galli A., Moore M. D., Pathak V. K., Hu W. S. (2011) Multiple barriers to recombination between divergent HIV-1 variants revealed by a dual-marker recombination assay. J. Mol. Biol. 407, 521–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen J., Nikolaitchik O., Singh J., Wright A., Bencsics C. E., Coffin J. M., Ni N., Lockett S., Pathak V. K., Hu W. S. (2009) High efficiency of HIV-1 genomic RNA packaging and heterozygote formation revealed by single virion analysis. Proc. Natl. Acad. Sci. U.S.A. 106, 13535–13540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moore M. D., Fu W., Nikolaitchik O., Chen J., Ptak R. G., Hu W. S. (2007) Dimer initiation signal of human immunodeficiency virus type 1: its role in partner selection during RNA copackaging and its effects on recombination. J. Virol. 81, 4002–4011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bender W., Davidson N. (1976) Mapping of poly(A) sequences in the electron microscope reveals unusual structure of type C oncornavirus RNA molecules. Cell 7, 595–607 [DOI] [PubMed] [Google Scholar]
- 26. Fu W., Rein A. (1993) Maturation of dimeric viral RNA of Moloney murine leukemia virus. J. Virol. 67, 5443–5449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Song R., Kafaie J., Yang L., Laughrea M. (2007) HIV-1 viral RNA is selected in the form of monomers that dimerize in a three-step protease-dependent process; the DIS of stem-loop 1 initiates viral RNA dimerization. J. Mol. Biol. 371, 1084–1098 [DOI] [PubMed] [Google Scholar]
- 28. Laughrea M., Jetté L. (1994) A 19-nucleotide sequence upstream of the 5′ major splice donor is part of the dimerization domain of human immunodeficiency virus 1 genomic RNA. Biochemistry 33, 13464–13474 [DOI] [PubMed] [Google Scholar]
- 29. Laughrea M., Jetté L. (1996) Kissing-loop model of HIV-1 genome dimerization: HIV-1 RNAs can assume alternative dimeric forms, and all sequences upstream or downstream of hairpin 248–271 are dispensable for dimer formation. Biochemistry 35, 1589–1598 [DOI] [PubMed] [Google Scholar]
- 30. Nikolaitchik O. A., Dilley K. A., Fu W., Gorelick R. J., Tai S. H., Soheilian F., Ptak R. G., Nagashima K., Pathak V. K., Hu W. S. (2013) Dimeric RNA recognition regulates HIV-1 genome packaging. PLoS Pathog. 9, e1003249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hill M. K., Shehu-Xhilaga M., Campbell S. M., Poumbourios P., Crowe S. M., Mak J. (2003) The dimer initiation sequence stem-loop of human immunodeficiency virus type 1 is dispensable for viral replication in peripheral blood mononuclear cells. J. Virol. 77, 8329–8335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jones K. L., Sonza S., Mak J. (2008) Primary T-lymphocytes rescue the replication of HIV-1 DIS RNA mutants in part by facilitating reverse transcription. Nucleic Acids Res. 36, 1578–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Andersen E. S., Contera S. A., Knudsen B., Damgaard C. K., Besenbacher F., Kjems J. (2004) Role of the trans-activation response element in dimerization of HIV-1 RNA. J. Biol. Chem. 279, 22243–22249 [DOI] [PubMed] [Google Scholar]
- 34. Heng X., Kharytonchyk S., Garcia E. L., Lu K., Divakaruni S. S., LaCotti C., Edme K., Telesnitsky A., Summers M. F. (2012) Identification of a minimal region of the HIV-1 5′-leader required for RNA dimerization, NC binding, and packaging. J. Mol. Biol. 417, 224–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huthoff H., Berkhout B. (2001) Mutations in the TAR hairpin affect the equilibrium between alternative conformations of the HIV-1 leader RNA. Nucleic Acids Res. 29, 2594–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Song R., Kafaie J., Laughrea M. (2008) Role of the 5′ TAR stem-loop and the U5-AUG duplex in dimerization of HIV-1 genomic RNA. Biochemistry 47, 3283–3293 [DOI] [PubMed] [Google Scholar]
- 37. De Guzman R. N., Wu Z. R., Stalling C. C., Pappalardo L., Borer P. N., Summers M. F. (1998) Structure of the HIV-1 nucleocapsid protein bound to the SL3 psi-RNA recognition element. Science 279, 384–388 [DOI] [PubMed] [Google Scholar]
- 38. Das A. T., Vrolijk M. M., Harwig A., Berkhout B. (2012) Opening of the TAR hairpin in the HIV-1 genome causes aberrant RNA dimerization and packaging. Retrovirology 9, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vrolijk M. M., Ooms M., Harwig A., Das A. T., Berkhout B. (2008) Destabilization of the TAR hairpin affects the structure and function of the HIV-1 leader RNA. Nucleic Acids Res. 36, 4352–4363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ohishi M., Nakano T., Sakuragi S., Shioda T., Sano K., Sakuragi J. (2011) The relationship between HIV-1 genome RNA dimerization, virion maturation and infectivity. Nucleic Acids Res. 39, 3404–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Russell R. S., Liang C., Wainberg M. A. (2004) Is HIV-1 RNA dimerization a prerequisite for packaging? Yes, no, probably? Retrovirology 1, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sakuragi J., Ueda S., Iwamoto A., Shioda T. (2003) Possible role of dimerization in human immunodeficiency virus type 1 genome RNA packaging. J. Virol. 77, 4060–4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jalalirad M., Laughrea M. (2010) Formation of immature and mature genomic RNA dimers in wild-type and protease-inactive HIV-1: differential roles of the Gag polyprotein, nucleocapsid proteins NCp15, NCp9, NCp7, and the dimerization initiation site. Virology 407, 225–236 [DOI] [PubMed] [Google Scholar]
- 44. Jouvenet N., Simon S. M., Bieniasz P. D. (2009) Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. U.S.A. 106, 19114–19119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kutluay S. B., Bieniasz P. D. (2010) Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathog. 6, e1001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peden K., Emerman M., Montagnier L. (1991) Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virology 185, 661–672 [DOI] [PubMed] [Google Scholar]
- 47. Berkhout B., van Wamel J., Klaver B. (1995) Requirements for DNA strand transfer during reverse transcription in mutant HIV-1 virions. J. Mol. Biol. 252, 59–69 [DOI] [PubMed] [Google Scholar]
- 48. Das A. T., Zhou X., Vink M., Klaver B., Verhoef K., Marzio G., Berkhout B. (2004) Viral evolution as a tool to improve the tetracycline-regulated gene expression system. J. Biol. Chem. 279, 18776–18782 [DOI] [PubMed] [Google Scholar]
- 49. Jeeninga R. E., Jan B., van den Berg H., Berkhout B. (2006) Construction of doxycyline-dependent mini-HIV-1 variants for the development of a virotherapy against leukemias. Retrovirology 3, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Berkhout B., Das A. T. (2009) Virus evolution as a tool to study HIV-1 biology. Methods Mol. Biol. 485, 436–451 [DOI] [PubMed] [Google Scholar]
- 51. Das A. T., Klaver B., Klasens B. I., van Wamel J. L., Berkhout B. (1997) A conserved hairpin motif in the R-U5 region of the human immunodeficiency virus type 1 RNA genome is essential for replication. J. Virol. 71, 2346–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Klaver B., Berkhout B. (1994) Evolution of a disrupted TAR RNA hairpin structure in the HIV-1 virus. EMBO J. 13, 2650–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wei X., Decker J. M., Liu H., Zhang Z., Arani R. B., Kilby J. M., Saag M. S., Wu X., Shaw G. M., Kappes J. C. (2002) Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46, 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wei X., Decker J. M., Wang S., Hui H., Kappes J. C., Wu X., Salazar-Gonzalez J. F., Salazar M. G., Kilby J. M., Saag M. S., Komarova N. L., Nowak M. A., Hahn B. H., Kwong P. D., Shaw G. M. (2003) Antibody neutralization and escape by HIV-1. Nature 422, 307–312 [DOI] [PubMed] [Google Scholar]
- 55. Reuter J. S., Mathews D. H. (2010) RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pasternak A. O., Adema K. W., Bakker M., Jurriaans S., Berkhout B., Cornelissen M., Lukashov V. V. (2008) Highly sensitive methods based on seminested real-time reverse transcription-PCR for quantitation of human immunodeficiency virus type 1 unspliced and multiply spliced RNA and proviral DNA. J. Clin. Microbiol. 46, 2206–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Das A. T., Klaver B., Berkhout B. (1995) Reduced replication of human immunodeficiency virus type 1 mutants that use reverse transcription primers other than the natural tRNA(3Lys). J. Virol. 69, 3090–3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Das A. T., Berkhout B. (2010) HIV-1 evolution: frustrating therapies, but disclosing molecular mechanisms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 1965–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. van Bel N., Das A. T., Berkhout B. (2014) In vivo SELEX of single-stranded domains in the HIV-1 leader RNA. J. Virol. 88, 1870–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Clever J. L., Parslow T. G. (1997) Mutant human immunodeficiency virus type 1 genomes with defects in RNA dimerization or encapsidation. J. Virol. 71, 3407–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Haddrick M., Lear A. L., Cann A. J., Heaphy S. (1996) Evidence that a kissing loop structure facilitates genomic RNA dimerisation in HIV-1. J. Mol. Biol. 259, 58–68 [DOI] [PubMed] [Google Scholar]
- 62. Laughrea M., Jetté L., Mak J., Kleiman L., Liang C., Wainberg M. A. (1997) Mutations in the kissing-loop hairpin of human immunodeficiency virus type 1 reduce viral infectivity as well as genomic RNA packaging and dimerization. J. Virol. 71, 3397–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. L'Hernault A., Weiss E. U., Greatorex J. S., Lever A. M. (2012) HIV-2 genome dimerization is required for the correct processing of Gag: a second-site reversion in matrix can restore both processes in dimerization-impaired mutant viruses. J. Virol. 86, 5867–5876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Berkhout B., Das A. T., van Wamel J. L. (1998) The native structure of the human immunodeficiency virus type 1 RNA genome is required for the first strand transfer of reverse transcription. Virology 249, 211–218 [DOI] [PubMed] [Google Scholar]
- 65. DeStefano J. J. (1994) Kinetic analysis of the catalysis of strand transfer from internal regions of heteropolymeric RNA templates by human immunodeficiency virus reverse transcriptase. J. Mol. Biol. 243, 558–567 [DOI] [PubMed] [Google Scholar]
- 66. King S. R., Duggal N. K., Ndongmo C. B., Pacut C., Telesnitsky A. (2008) Pseudodiploid genome organization AIDS full-length human immunodeficiency virus type 1 DNA synthesis. J. Virol. 82, 2376–2384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hu W. S., Temin H. M. (1990) Retroviral recombination and reverse transcription. Science 250, 1227–1233 [DOI] [PubMed] [Google Scholar]
- 68. Hu W. S., Temin H. M. (1990) Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. U.S.A. 87, 1556–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Greatorex J., Lever A. (1998) Retroviral RNA dimer linkage. J. Gen. Virol. 79, 2877–2882 [DOI] [PubMed] [Google Scholar]
- 70. Fu W., Gorelick R. J., Rein A. (1994) Characterization of human immunodeficiency virus type 1 dimeric RNA from wild-type and protease-defective virions. J. Virol. 68, 5013–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sundquist W. I., Kräusslich H. G. (2012) HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2, a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Walter A. E., Turner D. H., Kim J., Lyttle M. H., Müller P., Mathews D. H., Zuker M. (1994) Coaxial stacking of helixes enhances binding of oligoribonucleotides and improves predictions of RNA folding. Proc. Natl. Acad. Sci. U.S.A. 91, 9218–9222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wilkinson K. A., Gorelick R. J., Vasa S. M., Guex N., Rein A., Mathews D. H., Giddings M. C., Weeks K. M. (2008) High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. 6, e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Abd El-Wahab E. W., Smyth R. P., Mailler E., Bernacchi S., Vivet-Boudou V., Hijnen M., Jossinet F., Mak J., Paillart J. C., Marquet R. (2014) Specific recognition of the HIV-1 genomic RNA by the Gag precursor. Nat. Commun. 5, 4304. [DOI] [PubMed] [Google Scholar]
- 75. Chin M. P., Rhodes T. D., Chen J., Fu W., Hu W. S. (2005) Identification of a major restriction in HIV-1 intersubtype recombination. Proc. Natl. Acad. Sci. U.S.A. 102, 9002–9007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Moore M. D., Nikolaitchik O. A., Chen J., Hammarskjöld M. L., Rekosh D., Hu W. S. (2009) Probing the HIV-1 genomic RNA trafficking pathway and dimerization by genetic recombination and single virion analyses. PLoS Pathog. 5, e1000627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Baig T. T., Lanchy J. M., Lodmell J. S. (2009) Randomization and in vivo selection reveal a GGRG motif essential for packaging human immunodeficiency virus type 2 RNA. J. Virol. 83, 802–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. L'Hernault A., Greatorex J. S., Crowther R. A., Lever A. M. (2007) Dimerisation of HIV-2 genomic RNA is linked to efficient RNA packaging, normal particle maturation and viral infectivity. Retrovirology 4, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Baig T. T., Strong C. L., Lodmell J. S., Lanchy J. M. (2008) Regulation of primate lentiviral RNA dimerization by structural entrapment. Retrovirology 5, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kuiken C., Foley B., Leitner T., Apetrei C., Hahn B. H., Mizrachi I., Mullins J., Rambaut A., Wolinsky S., Korber B. (2013) HIV Sequence Compendium 2012, LA-UR 13-26007, Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, NM [Google Scholar]