

Background: Lysosome-associated membrane protein type 2a (LAMP-2A) is the receptor for chaperone-mediated autophagy (CMA).
Results: The transmembrane of LAMP-2A forms a coiled coil helix trimer in n-dodecylphosphocholine (DPC) micelle, and protein substrates interact with its cytosolic tail.
Conclusion: Protein substrates and chaperone recognize the same site with equal affinity.
Significance: Substrate recognition and recruitment are coupled in CMA.
Keywords: Autophagy, Chaperone, Nuclear Magnetic Resonance (NMR), Protein Structure, Transport, CMA, DPC, LAMP-2, Micelles
Abstract
Chaperone-mediated autophagy (CMA) is a highly regulated cellular process that mediates the degradation of a selective subset of cytosolic proteins in lysosomes. Increasing CMA activity is one way for a cell to respond to stress, and it leads to enhanced turnover of non-critical cytosolic proteins into sources of energy or clearance of unwanted or damaged proteins from the cytosol. The lysosome-associated membrane protein type 2a (LAMP-2A) together with a complex of chaperones and co-chaperones are key regulators of CMA. LAMP-2A is a transmembrane protein component for protein translocation to the lysosome. Here we present a study of the structure and dynamics of the transmembrane domain of human LAMP-2A in n-dodecylphosphocholine micelles by nuclear magnetic resonance (NMR). We showed that LAMP-2A exists as a homotrimer in which the membrane-spanning helices wrap around each other to form a parallel coiled coil conformation, whereas its cytosolic tail is flexible and exposed to the cytosol. This cytosolic tail of LAMP-2A interacts with chaperone Hsc70 and a CMA substrate RNase A with comparable affinity but not with Hsp40 and RNase S peptide. Because the substrates and the chaperone complex can bind at the same time, thus creating a bimodal interaction, we propose that substrate recognition by chaperones and targeting to the lysosomal membrane by LAMP-2A are coupled. This can increase substrate affinity and specificity as well as prevent substrate aggregation, assist in the unfolding of the substrate, and promote the formation of the higher order complex of LAMP-2A required for translocation.
Introduction
Autophagy is a highly regulated and essential process to maintain cellular homeostasis through lysosome-mediated degradation of damaged proteins and organelles (1–9). This process is categorized into three distinct pathways, micro-, macro-, and chaperone-mediated autophagy (CMA),2 based on how they are regulated, the type of substrates that are degraded, and their mechanisms in getting the substrates into the lysosomal compartments (10–15). Inhibition of autophagy can result in the accumulation of unwanted or damaged proteins in the cytosol and cause the pathogenesis of several human diseases (such as Parkinson disease, diabetes, Huntington disease, Danon disease, etc.) (16–32) as well as the inability of the cell to respond to starvation conditions (33, 34). Of all these classes of autophagy, CMA is quite unique (35). It can target degradation of selective cytosolic proteins (36, 37). This process is constitutively active in many cell types, but it is maximally activated during stress conditions (34–36, 38, 39) and contributes to amino acid recycling, protein quality control (40, 41), and cellular defense (18). In the CMA process, cytosolic proteins are recruited from the cytosol to the lysosomal membrane and translocated one by one across the membrane into the lumen for degradation (5, 16). The CMA process appears to be selective for a subset of cytosolic proteins containing a pentapeptide motif, known as the CMA targeting motif, that is biochemically related to Lys-Phe-Glu-Arg-Gln (KFERQ) (42, 43). In the cytosol, the chaperone Hsc70 recognizes this targeting sequence and recruits these proteins to the lysosomal outer membrane where the initiation of the CMA process takes place (2, 35, 44–46). The translocation of these proteins across the lysosomal membrane is established by a glycosylated membrane protein, LAMP-2 (47).
The protein LAMP-2 resides in the lysosome and is an integral membrane protein with two conserved luminal domains (constituting 90% of the entire protein), a single transmembrane (TM) domain (about 20 amino acids), and a short (10–12-amino acid) C-terminal cytosolic tail (48, 49). Glycosylation is only found in its luminal domains. The most studied LAMP-2A is found to be present in CMA complexes extracted from different tissues during CMA activation (50). In addition to Hsc70, other co-chaperones are involved together with LAMP-2A to mediate translocation of CMA substrates across the lysosomal membrane (46). Sequestration of these components by their specific antibodies inhibits CMA activity (47, 51). Earlier studies have also shown that LAMP-2A exists as a homomultimer, which upon CMA activation forms a higher oligomer complex with the chaperones (51). The formation of this higher oligomer is required for the translocation of substrates across the lysosomal membrane (51, 52). An increase in the CMA activity is directly related to the number of LAMP-2A proteins present in the lysosomal membrane (52). The accumulation of LAMP-2A in the lysosomal membrane is a result of reduction in its degradation rate or an increase in its transcription (53).
The CMA process is specific and regulated by tightly linked steps: recognition of CMA substrates in the cytosol, recruitment of substrates from cytosol to lysosomal outer membrane, and translocation of substrates across the membrane (52). However, it is not clear whether the specificity of this process comes entirely from the recognition step involving Hsc70 or the recruitment and translocation steps involving LAMP-2A. It has been presumed that only the cytosolic tail of LAMP-2A is accessible by the substrate and chaperones in the cytosol. In fact, it has been shown that mutations in the cytosolic tail of LAMP-2A can diminish the CMA activity by reducing substrate binding affinity (51). Additionally, studies of substrates binding to reconstituted isolated lysosomes containing LAMP-2A showed specificity in substrate recruitment (52). The above observations would suggest two tiers of selection of substrate: first at the recognition step in the cytosol involving Hsc70 and then at the recruitment step by the cytosolic tail of LAMP-2A. It is also important to note that some population of chaperones and co-chaperones can also be found in the lysosomal membrane that might assist in the substrate recruitment process (46). Inactive LAMP-2A exists as a homomultimer (51, 52). It is not clear what the conformation of this multimer is and whether it has a role in recruitment of substrates. Upon CMA activation, the number of LAMP-2A monomers in the CMA complex increases, and there is an additional presence of chaperones and co-chaperones. In this stage, substrate can no longer bind to LAMP-2A (52). The cause of the formation of this high oligomer complex, which is required for translocation, is not known. One possibility is that the interaction of substrate or chaperone with inactive LAMP-2A cytosolic tail can trigger aggregation through its transmembrane domain. Alternatively, the tail interaction may promote aggregation through its luminal domains. This presumes that information about substrate interaction on its cytosolic side can be transmitted to the luminal side of the LAMP-2A. Addressing some of these outstanding questions in molecular detail is necessary to understand this complex and important cellular process.
We addressed the above questions by studying the structure of a LAMP-2A protein construct containing its TM domain along with its cytosolic tail and a short (6-residue) luminal segment in a membrane mimic, DPC micelles, using NMR spectroscopy and other biophysical methods. Our data showed that the TM domain of LAMP-2A monomer has an extended α-helical conformation with a flexible cytosolic tail. Three TM monomers interact tightly in the micelle to form a coiled coil trimer. The flexible LAMP-2A cytosolic tail interacts with chaperone Hsc70 and a CMA substrate, RNase A, with comparable affinity but does not interact with a co-chaperone, Hsp40, or an RNase S peptide. No changes in the NMR chemical shifts of the transmembrane or luminal residues were detected upon cytosolic tail binding to either Hsc70 or RNase A. Thus, our study suggests that inactive LAMP-2A assembles across the lysosomal membrane as a trimer. The cytosolic tail of LAMP-2A in the multimer can bind both chaperone Hsc70 and substrate protein simultaneously. These findings parallel those of other cellular transport machineries (such as mitochondrial and endoplasmic reticulum transport) where chaperones are recruited together with substrates by the membrane transporter to avoid substrate aggregation that can inhibit translocation (54, 55). Binding of the cytosolic tail of LAMP-2A did not induce any conformational changes in its TM domain or its short luminal segment, which suggests that higher CMA oligomer complex formation is triggered primarily by interaction of LAMP-2A with its cytosolic CMA components independently of its TM and luminal domains.
EXPERIMENTAL PROCEDURES
Design of Protein Constructs and Sample Preparation
The LAMP-2A gene from Homo sapiens (human, P13473) was synthesized and codon-optimized for expression in Escherichia coli (Bio Basic Inc.) (56). A LAMP-2A construct containing the TM domain and C-terminal cytosolic tail (amino acids 369–410 and referred to as TM-LAMP-2A) was designed, cloned into pMAL2px vector (New England Biolabs), and expressed as a maltose-binding protein fusion protein along with a tobacco etch virus protease cleavage site in E. coli BL21(DE3) cells. Isotopically (15N- or 13C/15N-) labeled proteins were produced in isotope-enriched M9 minimal medium. 15NH4Cl and [13C]glucose were used as the sole sources of nitrogen and carbon, respectively (Cambridge Isotopes). The E. coli cells were grown at 37 °C and incubated with 0.3 mm isopropyl 1-thio-β-d-galactopyranoside at an A600 of ∼0.8, and cell growth was continued at 18 °C for 20 h. The cells were harvested by centrifugation, resuspended in lysis buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm EDTA, 1 mm DTT, and 0.02% NaN3), and sonicated by passing thrice through a high pressure homogenizer (EmulsiFlex-C3 from Avestin, Ottawa, Canada). The lysate was centrifuged at 17,000 rpm for 30 min at 4 °C, and the supernatant was loaded to an amylose column (New England Biolabs) equilibrated with the above lysate buffer and eluted with 10 mm maltose (Sigma-Aldrich). The eluted protein fractions were cleaved with tobacco etch virus protease ( of the A280 of the protein) at room temperature overnight. The cleaved TM-LAMP-2A was further purified by HPLC (Invitrogen) using a linear gradient of acetonitrile containing 0.1% TFA. The fractions containing purified TM-LAMP-2A were lyophilized, and TFA was removed by washing with H2O several times. The purity of the TM-LAMP-2A preparation was confirmed using liquid chromatography/mass spectrometry (LC-MS) (the measured molecular mass was 4480.36 Da, whereas the theoretical molecular mass is 4480.1 Da). The Hsp40 and substrate binding domain of Hsc70 (sbd-Hsc70) were expressed in E. coli and purified using affinity chromatography. The NaCl in the buffer used for Hsp40 purification was increased to 1.5 m to minimize copurification of proteins bound to Hsp40. The protein RNase A and RNase S peptide were purchased from Amersham Biosciences and Biosynthesis, respectively.
NMR Spectroscopy
The purified TM-LAMP-2A was reconstituted in different micelles (SDS, lysopalmitoylphosphatidylglycerol, DPC, and DPC + dimyristoylphosphatidylcholine) and screened for better solubility by varying pH, salt concentrations, and temperatures using NMR by monitoring its 15N,1H HSQC spectra. Based on the spectral quality and the total number of cross-peaks in the 15N,1H HSQC, the optimal sample condition was to dissolve the TM-LAMP-2A in micelles consisting of 150 mm DPC doped with 5 mm dimyristoylphosphatidylcholine and 50 mm potassium phosphate, pH 6.5, 25 mm NaCl, 1 mm EDTA, and 0.02% NaN3. For all NMR experiments, deuterated detergents were used to prepare the micelles. All NMR experiments were carried out at 37 °C on a Bruker Avance 600 MHz spectrometer with a room temperature probe, a Bruker Avance 600 MHz, a Bruker Avance 800 MHz, or a Bruker Avance 900 MHz spectrometer with cryogenic probes. The experiments used for backbone resonances assignments were as follows: three-dimensional HNCO (57), HNCA (58), CBCA(CO)NH (59), HNCACB (60), and HBHA(CO)NH (61). Three-dimensional 15N-edited NOESY-HSQC (τmix = 80 ms) (61), four-dimensional 13C/13C-edited NOESY (τmix = 80 ms) (62), and three-dimensional 13C F1-filtered, F3-edited NOESY-HSQC (τm = 120 ms with 1024 × 128 × 56 complex points) (63) experiments were acquired for intra- as well as intermolecular NOE distance restraints. The 13C-filtered NOESY was obtained from a sample composed of a mixture of 13C/15N-labeled and unlabeled TM-LAMP-2A in a 1:1 ratio. The two proteins were purified separately, mixed after first dissolving them in acetonitrile, and lyophilized prior to finally dissolving the sample in micelles in D2O buffer. As a control, an NMR sample was prepared by first dissolving the 13C/15N-labeled and unlabeled TM-LAMP-2A in micelles in D2O buffer and then mixing them in a 1:1 ratio. Because its association in a multimer within the micelle is quite strong, no exchange between the 13C/15N-labeled and unlabeled monomers occurred; thus no cross-peaks could be observed in the 13C-filtered NOESY spectrum (data not shown).
The backbone 15N T1 measurement at 600-MHz proton resonance frequency was acquired with 256 × 1024 complex points along t1 and t2 dimensions, respectively, and inversion recovery delays of 8, 128, 384, 608, 800, 1056, 1280, and 1496 ms. The 15N T2 measurement was carried out with the same acquisition parameters using the Carr-Purcell-Meiboom-Gill pulse sequence with relaxation delays of 2, 6, 12, 20, 30, 42, 56, and 70 ms (64).
To monitor LAMP-2A and chaperone Hsc70 protein interaction, a set of 15N,1H HSQC spectra were recorded for 15N-labeled (TM-LAMP-2A) protein as a function of increasing concentrations of unlabeled sbd-Hsc70. At each titration, 15N-labeled TM-LAMP-2A and unlabeled sbd-Hsc70 were slowly mixed and concentrated (at 2,000 rpm using an Amicon ultracentrifuge) to keep the relative micelles and TM-LAMP-2A concentrations the same. Similarly, interactions of TM-LAMP-2A with co-chaperone Hsp40; one of the well studied CMA substrates, RNase A; and RNase S peptide (with KFERQ motif) were also studied. The residue-specific HN chemical shift perturbation was calculated by using the following equation: chemical shift perturbation (CSP) = [((ΔH)2 + (ΔN/10)2)]1/2 where ΔH and ΔN are the differences in the 1H and 15N chemical shifts of the bound and unbound TM-LAMP-2A. To obtain the binding affinity, the chemical shift changes were fit to a two-state model: (P + L ⇒ PL), with species “L” being titrated at concentration “l” into solution with species “P” held at constant concentration “p” using the following equation: Δl = (Δmax/2p) (l + Kd + p − ((l + Kd + p)2 − 4lp)1/2 where Δl is the change in the observed shift of species L from the free state, Δmax is the maximum shift change on saturation, and Kd is the dissociation constant. The two parameters extracted from the fit are Kd and Δmax. To simultaneously fit several residues, the scale of their chemical shift changes must be normalized. One residue was arbitrarily chosen as a reference, and the remainder were scaled by a factor, fi = ∑Δ0(l)Δi(l)/∑Δi2(l) where Δ0(l) is the perturbation at concentration l of the reference residue and Δi(l) is the perturbation of the residue to be scaled, and the sums were carried out over all concentrations.
All of the NMR data were processed using NMRPipe and analyzed with nmrDraw, PIPP, and CARA (65–67). NMR experimental errors were estimated based on the spectral noise as described previously (68).
Paramagnetic Relaxation Enhancement
An extended construct of TM-LAMP-2A (amino acids 360–410) containing a cysteine in the N terminus was expressed similarly as described above. After elution from the maltose column, the maltose-binding protein-fused cysteine construct of TM-LAMP-2A was soaked with 10 mm tris(2-carboxyethyl)phosphine to reduce any possible cysteine disulfide bond formation. The protein buffer was then exchanged into a reaction buffer (100 mm Tris-HCl, pH 8.0, 100 mm NaCl, and 1 mm EDTA) using a PD-10 desalting column. A spin probe labeling reaction was carried out by mixing the protein sample with 20× molar excess of the paramagnetic probe PROXYL ((3-(3-(2-iodoacetamido)propylcarbamoyl)proxyl); Toronto Research Chemicals Inc.) at room temperature overnight. After this reaction, the excess spin probe was removed using a PD-10 column, and the protein was cleaved with tobacco etch virus and purified using HPLC as described above. The spin probe labeling was confirmed by LC/MS. The two-dimensional 15N,1H HSQC spectrum of the spin-labeled and reference sample was recorded, and no obvious chemical shift changes were observed, indicating that spin labeling did not disrupt the TM-LAMP-2A structure.
The solvent PRE experiments were performed by stepwise addition of the water-soluble spin label reagent gadodiamide (aqua[5,8-bis(carboxymethyl)-11-[2-(methylamino)-2-oxoethyl]-3-oxo-2,5,8,11-tetraazatridecan-13-oato (3-)-N5,N8,N11,O3,O5,O8,O11,O13]gadolinium hydrate; trade name Omniscan, GE Healthcare) into the NMR sample to final concentrations of 0.25, 0.5, and 1.0 mm. A reference spectrum was recorded prior to the addition of spin label reagent. In both PRE studies, the PRE 1HN-Γ2 rates were determined from a two-time point (0.04 and 16 ms) interleaved measurement using a transverse relaxation-optimized spectroscopy-based experiment (69).
Analytical Ultracentrifugation
Sedimentation equilibrium (density matching protocol) experiments were performed at 37 °C on TM-LAMP-2A reconstituted in micelles using a Beckman XL-I analytical ultracentrifuge. To eliminate the contribution of the micelle to the buoyant molecular mass of the TM-LAMP-2A·micelle complex, experiments were carried out at a solvent density that was adjusted to equal that of the micelles (70, 71). This density adjustment was carried out by centrifugation of micelles in a buffer containing different percentages of deuterium oxide (D2O) at three different centrifugal speeds (17,000, 22,000, and 32,000 rpm). The density match was calculated from a plot of the radial gradient of fringe displacement versus buffer density (71). The density matching protocol of sedimentation equilibrium analytical ultracentrifugation experiments to determine the oligomeric state was carried out at three different centrifugation speeds (17,000, 22,000, and 32,000 rpm) and at two sample concentrations (140 and 365 μm) to produce consistency in the results and allow for a global fit.
Chemical Cross-linking
Cross-linkers are either homo- or heterobifunctional reagents with reactive groups (identical or non-identical), allowing the establishment of intra- as well as intermolecular cross-linkages with the formation of covalent bonds (72). Intersubunit cross-link has been used for determination of quaternary structure and stoichiometry of the subunits within homo-oligomeric protein complexes. The chemical cross-linking experiment on TM-LAMP-2A (amino acids 360–410 having two primary amines, “Lys”) reconstituted in micelles was carried out with an amine-reactive imido ester cross-linker, dimethyl suberimidate (Thermo Scientific Inc.). In this reaction, 80 μm protein was treated with dimethyl suberimidate (20× molar excess) for 1 h at room temperature. The reaction was stopped by adding primary amine (50 mm Tris-HCl, pH 8.0) and incubating for 15 min at room temperature followed by adding 10 mm DTT to reduce the cysteine. The reaction mixture was analyzed using SDS-PAGE (silver staining protocol). The protocol used for this reaction was carried out according to the manufacturer (Thermo Scientific Inc.).
NMR Structure Calculation
The cross-peaks of NOESY spectra were identified and assigned, and the corresponding peak intensities were translated into a continuous distribution of 1H-1H distances. The dihedral angles φ and ψ were calculated from the assigned backbone chemical shifts using the program TALOS. Generic hydrogen bond distance restraints were imposed for residues located at well defined α-helical regions. With the above restraints, the structure of TM-LAMP-2A was calculated using a simulated annealing protocol in which the bath temperature was cooled slowly from 3,500 to 298 K in the program Xplor-NIH (73). The final monomer structure calculation used 174 inter- and 134 intraresidual distance restraints, 34 hydrogen bond distance restraints, and 58 φ and 58 ψ dihedral angle restraints. For the oligomer structure calculation, a starting template of oligomer structure was generated using the lowest energy monomer structure and duplicated into respective subunits using Xplor-NIH (73). All the constraints used for monomer structure calculation were applied separately to the individual subunits to make them identical. Similarly the 15 intermolecular NOE restraints derived from the combination of 13C-filtered NOESY and four-dimensional HCCH NOESY experiments were applied in a pairwise manner to all of the monomers to impose symmetry in calculating the structure of the oligomer. The non-crystallographic symmetry restraint was not used in the calculation.
RESULTS
Secondary Structure of Transmembrane Domain of LAMP-2A in DPC Micelles
The two-dimensional 15N,1H HSQC spectrum of TM-LAMP-2A in the optimized NMR buffer showed varying peak intensities, suggesting that TM-LAMP-2A residues might experience different effective rotational correlation times likely due to their different states of association with the micelles. Analysis of the NMR data sets yielded near complete backbone and side chain resonance assignments of TM-LAMP-2A. Close to 90% of the backbone amides were assigned (Biological Magnetic Resonance Bank accession number 19941). The resonances of amides that were not assigned were either because of resonance overlap or they were too broad to be detected (residues Ser369, Ala370, Val377, Pro378, Val381, His403, and His404). The 1HN NMR resonances showed a small dispersion (∼1.2 ppm), indicating the presence of mostly helical and random coil conformations (Fig. 1A). The calculated chemical shift index from the assigned Cα and Cβ secondary chemical shifts indicated the presence of an extended α-helical transmembrane domain (residues Ala380–Leu400), which is further corroborated by the interproton NOE connectivity data (Fig. 1B). The solvent PREs measured on the TM-LAMP-2A show that there is a significant increase in 1HN-Γ2 for the residues corresponding to the cytosolic tail and luminal domain, whereas the residues belonging to the transmembrane domain show less significant solvent PRE. The solvent PRE is a manifestation of the closest distance that the solvent molecule can get to an amide proton. Therefore, the lesser PRE observed for the transmembrane residues signifies that they are further away from the water interface and must be membrane-associated (Fig. 1C). Respondek et al. (74) have shown that for a helix in a micelle the PRE follows the periodicity of residues in the helix, going from a maximum PRE in the helix to a minimum every three residues. In our study, the PRE behaviors for those transmembrane residues (Ala380–Leu400) are also not uniform. Upon closer inspection, one could identify residues Ala383, Gly387, Leu393, and Ile398 to have higher PRE values than the rest. However, their periodicity is not the same as found in Respondek et al. (74), suggesting non-equal solvent environment on one face of the helix relative to the other. Although this inspection is qualitative, it does suggest the possibility that TM-LAMP-2A is not a monomer in the micelle.
FIGURE 1.

A, the two-dimensional 15N,1H HSQC spectrum of TM domain of human LAMP-2A in DPC micelles. The assignments are indicated by the one-letter amino acid code followed by the corresponding number along the protein primary sequence. The resonances connected with horizontal lines are correlations from side chain NH2 spin pairs belonging to Asn and Gln residues. B, secondary structures of TM domain of LAMP-2A in DPC micelles. The secondary chemical shift index ΔCα − ΔCβ defines the presence of one TM helix. The values of ΔCα and ΔCβ were obtained as the differences between the experimentally observed 13Cα and 13Cβ chemical shifts and the corresponding random coil chemical shifts. The consecutive positive bars in the chemical shift index plot indicate the presence of α-helical conformation for the TM domain, whereas the cytosolic and N-terminal tails do not adopt any specific secondary conformation. The defined TM helix is also identified by the characteristic medium range interproton NOE connectivity of Hαi to Hβi + 3 and strong HNi to HNi + 1 NOE. The secondary structural elements are shown at the bottom of panel B. C, impact of solvent PRE on amide proton transverse relaxation rate (1HN-Γ2) of TM domain of LAMP-2A. No significant effect could be observed in 1HN-Γ2 for the TM domain, confirming its insertion in the DPC micelles. Residue numbering is kept the same as the full-length LAMP-2A.
Backbone Dynamics of the Transmembrane Domain of LAMP-2A in DPC Micelles
The backbone 15N T1 and T2 relaxations were measured for TM-LAMP-2A at 600-MHz proton resonance frequencies, and the results are shown in Fig. 2. The 15N T2 relaxation data indicated that the helical transmembrane domain of LAMP-2A is rigid compared with its non-helical regions (Fig. 2B). This shows that the rigid helical transmembrane domain tumbles together with the micelles. In contrast, the cytosolic tail is highly flexible with high T2 and low T1 values (Fig. 2, A and B). The average T1 and T2 relaxation times for the rigid helical transmembrane domain are 1153.5 ± 20.1 and 55.1 ± 2.9 ms, respectively. The variations in its T1/T2 ratios (Fig. 2C) indicated that the molecule is somewhat anisotropic in DPC micelles. The calculated optimal τc from 15N T1 and T2 corresponding to the transmembrane domain using model-free spectral densities is 14.4 ns, which corresponds to a molecular mass of ∼29 kDa if one assumes a globular protein. This calculated molecular size is not in an agreement with a TM-LAMP-2A monomer in a micelle considering a micelle size of 18.5 kDa (75). Note that the micelle size will vary depending on the composition of the detergent used in its preparation.
FIGURE 2.
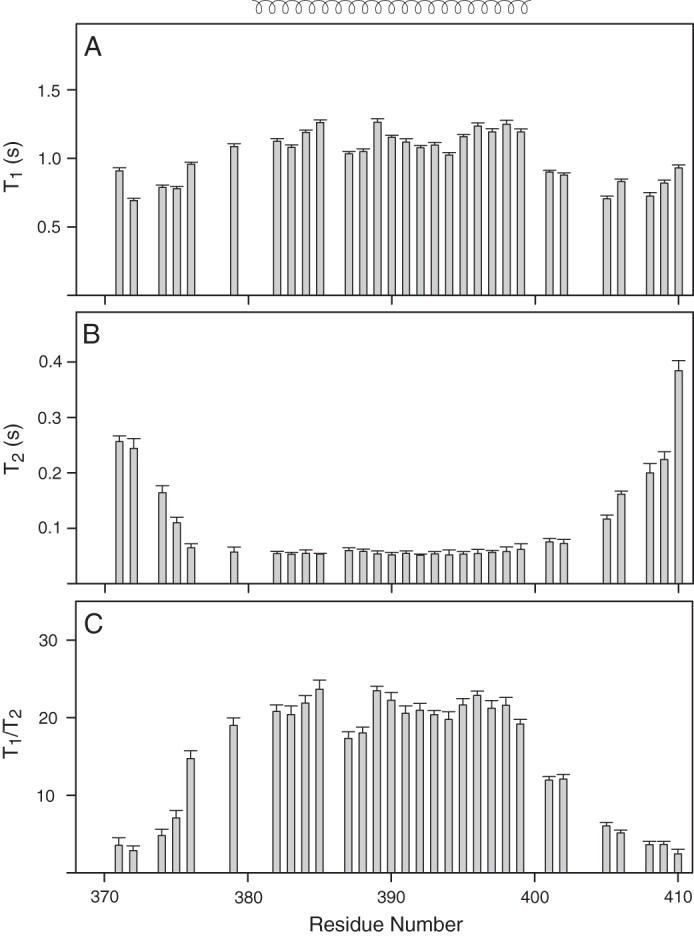
Backbone 15N relaxation dynamics of TM domain of human LAMP-2A in DPC micelles. The 15N T1 (A) and T2 (B) relaxation data of TM-LAMP-2A and their ratio (C) are plotted as a function of residue number. Relaxation values could not be estimated for residues Asp373, Ala380, Val381, Ala386, Leu400, and Tyr407 due to spectral overlap and weak intensity resulting in improper fit. The residues Ser369, Ala370, Val377, Pro378, His403, and His404 are not assigned. The mean value of T1 and T2 for the structured region is 1153.5 ± 20.1 and 55.1 ± 2.9 ms, respectively, corresponding to an estimated rotational correlation time (τc) of 14.4 ns. The secondary structural elements are shown at the top of A. Error bars represent fitting errors in the relaxation times.
Characterization of the Oligomeric State of TM-LAMP-2A
To understand the function of membrane proteins such as LAMP-2A, it is very important to know the stoichiometry of their membrane-bound form over wide ranges of concentrations. To confirm our correlation time analysis using the NMR relaxation data, a dynamic light scattering measurement was carried out at 37 °C on the TM-LAMP-2A sample. An effective hydrodynamic radius of 2.4 nm was derived from the dynamic light scattering measurement. Using a Debye-Einstein relationship, a correlation time of 14.2 ns was calculated that corresponds to the above hydrodynamic radius. This is consistent with the NMR relaxation-derived correlation time. To determine how many monomers of TM-LAMP-2A are in a micelle, the effective size of the micelle alone was determined by dynamic light scattering to be about 16 kDa. This leads to an estimate of the total effective weight of the TM-LAMP-2A in a micelle of about 12.6 kDa, which corresponds to roughly three monomers.
Another biophysical measurement that can corroborate our estimation of the oligomeric state of TM-LAMP-2A is sedimentation equilibrium analytical centrifugation. To measure the effective size of TM-LAMP-2A alone, the contribution from the micelle to the overall size of the particle has to be taken into account. One way to achieve this is by creating a solvent condition that matches the density of the micelle so that it does not migrate. The density matching condition in our case was met when we mixed our buffer to consist of 43% D2O at 37 °C. The sedimentation equilibrium curve (Fig. 3A) obtained using the density matching protocol was best fit to an equilibrium between a monomer and a trimer (global χ2 = 0.34; Fig. 3B). The result of this fitting suggests a trimer (13.4 kDa) population of 67%, a monomer population of 32%, and a higher oligomer species (75.2 kDa) with a population of 1%. This result was found to be consistent over two different concentrations (140 and 365 μm) and at three different centrifugal speeds (17,000, 22,000, and 32,000 rpm).
FIGURE 3.

Sedimentation equilibrium of human TM domain of LAMP-2A in DPC micelles. A, the sedimentation curves obtained at centrifugation speeds of 17,000 (red), 22,000 (blue), and 32,000 (black) rpm (solid circles). These curves were collected with a protein concentration of 365 μm. The sedimentation equilibrium curves acquired with the density matching protocol were best fit to a monomer-trimer equilibrium (global χ2 = 0.34), and the fitting result is shown as a solid line. B, the residuals of the above fitting routine. C, chemical cross-linking of TM-LAMP-2A characterized by SDS-PAGE using a silver staining protocol. The silver-stained SDS-polyacrylamide gel of the reference sample (left panel) and that of TM-LAMP-2A in the presence of chemical cross-linker dimethyl suberimidate (DMS) (right panel) are shown. Lane labeled M contains molecular mass markers.
The experimental results we have reported thus far only show that more than one TM-LAMP-2A monomer exist in a micelle. It does not prove that these monomers are tightly interacting with each other. To show that these TM-LAMP-2A molecules are not far apart from each other, we carried out a cross-linking experiment. SDS-PAGE of TM-LAMP-2A in a micelle following a cross-linking reaction using dimethyl suberimidate is shown in Fig. 3C. This SDS gel revealed a band corresponding to a trimer of the molecule. The same gel also showed bands corresponding to a monomer as well as a dimer of TM-LAMP-2A. These bands appear to be due to the potential of different types of cross-linking to occur and efficiency of the cross-linker to link the different monomers.
Structure of Transmembrane Domain of LAMP-2A in DPC Micelles
Structures of TM-LAMP-2A were calculated using the following restraints: NOE-derived distances, TALOS-derived dihedral angles, and generic hydrogen bonds. The calculated 20 lowest energy TM-LAMP-2A structures (Protein Data Bank code 2MOF) do not have any distance or dihedral angle violations greater than 0.5 Å and 5°, respectively. These structures show an extended transmembrane helix (residues Ala380–Leu400) with backbone root mean square deviation of 0.52 ± 0.14 Å, whereas its N terminus (residues Ser369–Ile379) and cytosolic tail (residues Lys401–Phe410) are flexible and solvent-exposed (Fig. 4A). Because we only observed one set of resonances from the oligomer, we have to assume and therefore impose symmetry in the structure calculation. Intermolecular distance restraints between LAMP-2A monomers were measured using a 13C-filtered NOESY experiment. Two representative strips of the 13C-filtered spectrum of LAMP-2A are shown in Fig. 4B. The oligomer structure of TM-LAMP-2A is composed of three molecules wrapped around each other to form a parallel coiled coil trimer. The calculated 20 lowest energy TM-LAMP-2A trimer structures (Protein Data Bank code 2MOM; Fig. 4, C and D) show a good convergence with a backbone root mean square deviation for the helical segment (residues Ala380–Leu400) of 0.67 ± 0.16 Å. No NOE contacts were observed for segments other than the transmembrane helix; therefore as expected the N-terminal and the cytosolic tails are still disordered in the trimer structure. The full structural statistics for the trimer describing the potential energies used in the calculation are shown in Table 1. In the oligomer structure, the individual subunits are closely packed (Fig. 4E) to form a conformation where the helices are oriented at 17° relative to each other. In addition, the N-terminal opening (diameter of 10.8 Å) in this three-helix bundle is slightly different from the cytosolic side (diameter of 7.4 Å). Interactions among residues Val381, Leu385, Val388, Leu389, Val392, Ala395, and Tyr396 stabilize the three-helix bundle conformation (Fig. 4D).
FIGURE 4.

NMR-derived solution structure of TM domain of human LAMP-2A in DPC micelles. A, a superposition of an ensemble of 20 lowest energy structures of TM-LAMP-2A (backbone root mean square deviation, 0.52 ± 0.14 Å). The calculated structures show an extended α-helical transmembrane domain and a flexible cytosolic tail at the C terminus. B, selected strips of 13C-filtered NOESY at the Val388-Hα and Tyr396-Hα chemical shifts obtained from a sample composed of a mixture of 13C/15N-labeled and unlabeled TM-LAMP-2A in a 1:1 ratio. C, ensemble of 20 superimposed lowest energy structures of TM-LAMP-2A trimer (backbone root mean square deviation, 0.67 ± 0.16 Å). In the calculated structure of the trimer, three TM helices wrap around each other to form a symmetric coiled coil conformation. For clarity, only the structured region of TM domain is shown here. D, lowest energy structure of the trimer showing residues Val381, Leu385, Val388, Leu389, Val392, Ala395, and Tyr396 forming the core of the trimer. In this figure, A, B, and C represents three individual subunits. E, the top view (N terminus) of the TM-LAMP-2A showing some of the hydrophobic interactions that stabilize the core of the trimer.
TABLE 1.
Structural statistics of an ensemble of 20 lowest energy structures of TM domain of human LAMP-2A trimer in DPC micelles (Protein Data Bank code 2MOM) derived from Xplor-NIH and PSVS 1.5
RMSD, root mean square deviation.

a Total number of restraints.
b Residues in regular secondary structure: 380–400.
Finally, to validate our TM-LAMP-2A three-helix bundle structure, we carried out PRE measurement. This was performed on a sample where isotopically labeled 15N-labeled TM-LAMP-2A was mixed with unlabeled TM-LAMP-2A that contains a spin label attached to a cysteine at the N terminus. We measured close distances between the spin label on one TM-LAMP-2A monomer to specific residues in the other monomers by observing significant transverse relaxation enhancement of their amide proton resonances (1HN-Γ2). The PRE result is shown in Fig. 5A. Qualitatively, residues close to the N terminus show larger PRE. However, if the mixture of monomers was inhomogeneous or the conformation of our trimer structure is not the correct representative, such as the presence of a mixture of “head-head” and “head-tail” arrangement of the trimer, then a detailed spin label to proton distance versus PRE comparison will not be in good agreement. Therefore our PRE result was compared with the intermolecular distances from the structure of TM-LAMP-2A trimer that we determined. The PRE from a single spin label in the trimer will affect the other two monomers, and the measured PRE value will be the sum of effect experienced by the two monomers. The PRE was plotted against the sum of the inversed sixth power of the distances and is shown in Fig. 5B. The average distances were calculated from the 20 lowest energy structures to represent an ensemble. The observed good agreement is an independent cross-validation of our calculated structures of TM-LAMP-2A, and we could rule out the possibility of mixed symmetry in LAMP-2A. Interestingly, we also compared our PRE data against a calculated symmetric dimer or tetramer structure of LAMP-2A, and their agreements are much worse than the trimer structure (data not shown).
FIGURE 5.
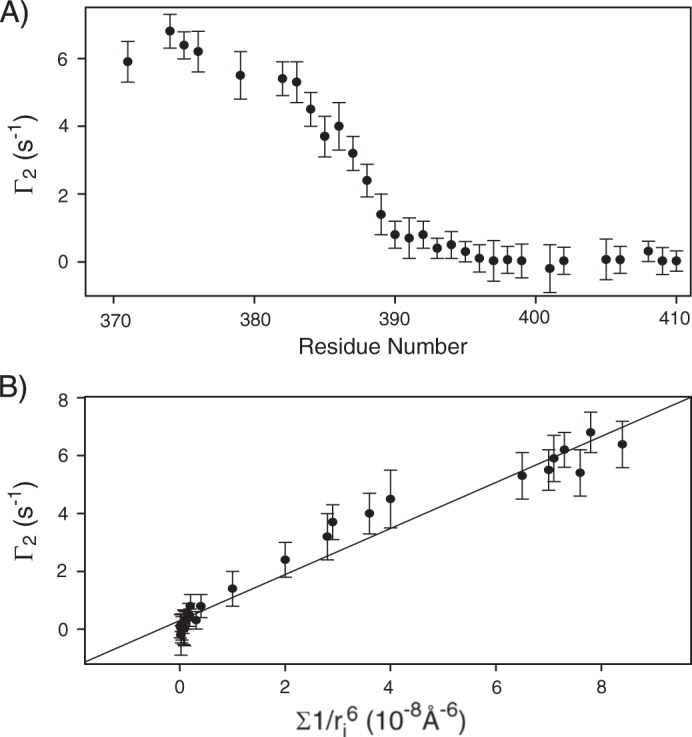
A, the PRE from mixing the spin probe attached to unlabeled TM-LAMP-2A with 15N-labeled TM-LAMP-2A was determined by measuring the change in relaxation rates, 1HN-Γ2, relative to a sample not containing the spin label. The enhanced 1HN-Γ2 indicates the tight association of the TM-LAMP-2A monomers in the micelles. B, correlation between 1HN-Γ2 and the calculated sum of inversed sixth power distances from individual backbone amide proton from each monomer to the spin label based on the NMR-derived trimer structure of TM-LAMP-2A. Error bars correspond to reproducibilities from two different data sets.
TM-LAMP-2A Interacts with Hsc70 and RNase A but Not with Hsp40 and RNase S Peptide
The interactions between TM-LAMP-2A and other proteins were monitored by changes in its 1H and 15N NMR chemical shifts upon formation of a complex. In the presence of the substrate binding domain of Hsc70, only residues corresponding to the cytosolic tail of TM-LAMP-2A show significant CSPs (Fig. 6A). The CSPs are plotted as a function of residue numbers and are shown in Fig. 6B. The relative positions of those residues with significant CSP in TM-LAMP-2A structure are shown in Fig. 6C. A similar pattern of chemical shift changes was also observed when a CMA substrate, RNase A, was added to TM-LAMP-2A (data not shown). In contrast, however, no CSPs in TM-LAMP-2A were observed upon addition of Hsp40 and RNase S peptide into the sample (data not shown). The amounts of chemical shift changes of different cytosolic residues of TM-LAMP-2A were plotted as a function of concentration of Hsc70 and RNase A in Fig. 6, D and E, respectively. These titration curves were fit to a two-state binding model (see “Experimental Procedures”), resulting in Kd values of 49.9 ± 12.7 μm for Hsc70 and 87.5 ± 19.5 μm for RNase A interaction with TM-LAMP-2A. The dissociation constant found for RNase A is consistent with the value measured using isolated lysosome published previously (43).
FIGURE 6.

A, overlay of the contour plot of 1H-15N backbone resonances of those residues of TM-LAMP-2A that undergo significant chemical shift changes during chaperone Hsc70 titration (black and red correspond to unbound and bound TM-LAMP-2A, respectively). B, residue-specific CSP of TM domain of LAMP-2A upon addition of the substrate binding domain of Hsc70. The secondary structural elements are shown at the top of B. Residues experiencing significant perturbations are those with CSP values above the standard of deviation (0.016) cutoff shown in a dotted line. These residues belong to the cytosolic tail of LAMP-2A. C, a ribbon representation of TM-LAMP-2A with residues having significant chemical shift perturbation shown as stick models. D and E, fitting of the experimental chemical shift titration data (circle, Lys401; diamond, His402; triangle, Ala405; star, Gly406) to determine Kd for LAMP-2A binding to Hsc70 and RNase A, respectively. The results from the fit are shown as solid lines.
DISCUSSION
The underlying molecular mechanism for substrate specificity and targeting to lysosomes in the CMA process is not clearly understood. To investigate the mechanism, we solved the NMR structure of the TM domain of LAMP-2A and characterized its interaction with chaperones and CMA substrates in DPC micelles. Our findings revealed the existence of LAMP-2A as a symmetric trimer. A number of key hydrophobic interactions stabilize this conformation. We did not observe any exchange between them in the micelles over a long period of time. In fact, we had to modify our sample preparation to create a mixed isotopically labeled TM-LAMP-2A (see “Experimental Procedures”). We believe this conformation is the stable, inactive conformation of TM-LAMP-2A. This oligomeric state is not altered in the presence of chaperones and CMA substrates because we did not observe any change in the NMR resonance line widths compared with the free TM-LAMP-2A.
The existence of inactive LAMP-2A as an oligomer reported previously (51, 52, 76) is in agreement with our structure of the TM-LAMP-2A trimer. Further formation of higher order oligomer of LAMP-2A upon CMA activation is one of the important steps to translocate substrates into lysosomes for degradation (51, 52). Mutation of two glycine residues into alanines in the TM region of LAMP-2A (GG/AA) alters the oligomerization state, and thus the cells expressing the GG/AA LAMP-2A mutant showed significantly lower ability to translocate and degrade the substrates via CMA (52, 77). In our structure of TM-LAMP-2A, the glycine residues in the TM region are outside of the core of the trimer. Therefore the reported result of glycine to alanine mutation in the TM region would suggest that higher order oligomers of LAMP-2A are formed by bringing together multiple trimer building blocks through their lipid-interacting surface to form a membrane-translocating pore.
The cytosolic tail of LAMP-2A is the only part of the protein that is initially accessible to CMA substrates. Therefore, substrate affinity and specificity must be defined through this LAMP-2A tail. Indeed, the observed NMR chemical shift changes in TM-LAMP-2A upon addition of Hsc70 and RNase A were confined to residues in its short cytosolic tail. Our data also indicated that the chaperone Hsc70 and CMA substrate RNase A bind to the cytosolic tail of LAMP-2A with similar affinity, whereas the co-chaperone Hsp40 does not interact with LAMP-2A. Therefore, the LAMP-2A cytosolic tail provides a measure of specificity toward CMA substrate. This step was shown to be a rate-limiting step in the CMA process (53).
At first glance, it appears somewhat contradictory that CMA, a specific biological transport system, utilizes Hsc70, a chaperone known to bind a large number of cytosolic proteins, as a substrate recruiter, whereas the transport receptor, LAMP-2A, has similar affinity for the CMA substrate and Hsc70. This paradox is also compounded by the fact that the chaperone Hsc70 is also a CMA substrate. A clear argument, however, can be made why the above observations make sense based on our TM-LAMP-2A structure, which revealed three cytosolic tails of LAMP-2A that are dynamic, exposed, and in close proximity to each other. The chaperone Hsc70 binds cytosolic proteins that potentially can be CMA substrates by recognizing a KFERQ motif sequence of residues. This recognition sequence is not highly specific, and in fact, any sequence that is chemically similar will be recognized (42). This interaction provides some specificity but is not the sole determinant of CMA substrate recognition. If the Hsc70·substrate complex binds to the LAMP-2A trimer with each component interacting with one LAMP-2A cytosolic tail, this would result in a higher effective affinity than each binding individually. This type of bimodal interaction would also result in a much higher specificity for CMA substrate. Only those proteins that have binding affinity for both Hsc70 (through a KFERQ-like motif) and the cytosolic tail of LAMP-2A will be selected for CMA degradation. For this to occur, the interaction sites of Hsc70 and LAMP-2A on the substrate must be distinct.
Previous work has shown that RNase S peptide having the KFERQ motif inhibits the CMA activity (46, 78–80), and our result shows that RNase S peptide does not bind to the cytosolic tail of LAMP-2A. Taken together, these results imply that the binding sites for Hsc70 and LAMP-2A on RNase A do not overlap, and therefore RNase S peptide inhibition of CMA must be established through Hsc70 sequestration from recruiting proper substrates. We therefore propose a hypothesis where substrate recognition in the cytosol by Hsc70 and lysosomal membrane targeting, the two initial steps in the CMA process, must be coupled. This coupling between Hsc70 binding of substrate and their simultaneous interaction with the cytosolic tail of LAMP-2A is key in providing specificity in the CMA process. In addition to the above role, Hsc70 binding to the LAMP-2A cytosolic tail ensures there is a population of this chaperone around the receptor to prevent aggregation as well as to promote the unfolding of protein substrates that is required for translocation (46, 52, 81).
The active form of LAMP-2A was associated with the formation of a higher ordered oligomer complex involving multiple LAMP-2A and other regulator proteins, such as Hsp70-interacting protein, Hsp70-Hsp90 organizing protein, BAG-1, Hsp40, and Hsp90 (46). It is still not clear what the key molecular trigger might be that activates the CMA process. The present study only used the substrate binding domain of Hsc70, and it is possible the Hsc70 nucleotide binding domain could play some role in CMA. It is also not very well understood what roles the luminal domains of LAMP-2A play in the translocation of substrates through the lysosomal membrane. For the luminal domain of LAMP-2A to be actively involved in the substrate translocation, information about substrate binding on its cytosolic side must be transferred to the luminal side. This process must involve the transmembrane domain. However, we did not observe any change in the chemical shifts of residues in the transmembrane domain when Hsc70 or RNase A binds the cytosolic tail of LAMP-2A. This showed that there was no conformational rearrangement of the TM-LAMP-2A associated with substrate binding. Therefore the rearrangement must occur in the higher oligomeric form of LAMP-2A and not within its inactive trimer form. Alternatively, the other regulatory proteins involved in the CMA complex might drive this process.
In summary, our data collectively suggest that substrate specificity and affinity in the CMA process are due to the coupling between chaperone Hsc70 recognition of the substrate and their simultaneous interaction with the LAMP-2A cytosolic tail. The presence of Hsc70 molecules at the same site as substrate on LAMP-2A also serves to prevent aggregation and to promote unfolding of the substrate prior to translocation across the membrane. This coupled interaction mechanism could be a general mechanism in other transport machinery (such as mitochondrial or endoplasmic reticulum) because they share the same chaperone and co-chaperone proteins as regulatory factors.
Acknowledgments
We thank Motoshi Suzuki for help with critical scientific discussions, Charles Schwieters for professional help with Xplor-NIH, Duck-Yeon Lee of the National Heart, Lung, and Blood Institute Biochemical Facility for expertise regarding mass spectrometry. The protein Hsp40 was a kind gift from Dr. James Gruschus.
This work was supported by the Intramural Research Program of the National Institutes of Health, NHLBI.
The atomic coordinates and structure factors (codes 2MOF and 2MOM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The chemical shifts can be accessed through the Biological Magnetic Resonance Bank (BMRB) under BMRB accession number 19941.
- CMA
- chaperone-mediated autophagy
- LAMP-2
- lysosome-associated membrane protein type 2
- TM
- transmembrane
- DPC
- n-dodecylphosphocholine
- HSQC
- heteronuclear single quantum correlation
- sbd
- substrate binding domain
- CSP
- chemical shift perturbation
- PRE
- paramagnetic relaxation enhancement.
REFERENCES
- 1. De Duve C., Wattiaux R. (1966) Functions of lysosomes. Annu. Rev. Physiol. 28, 435–492 [DOI] [PubMed] [Google Scholar]
- 2. Cuervo A. M., Dice J. F. (1998) Lysosomes, a meeting point of proteins, chaperones, and proteases. J. Mol. Med. 76, 6–12 [DOI] [PubMed] [Google Scholar]
- 3. Goldberg A. L. (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899 [DOI] [PubMed] [Google Scholar]
- 4. Cuervo A. M. (2004) Autophagy: many paths to the same end. Mol. Cell. Biochem. 263, 55–72 [DOI] [PubMed] [Google Scholar]
- 5. Dice J. F. (2000) Lysosomal Pathways of Protein Degradation. Landes Bioscience, Austin, TX [Google Scholar]
- 6. Agarraberes F. A., Dice J. F. (2001) Protein translocation across membranes. Biochim. Biophys. Acta 1513, 1–24 [DOI] [PubMed] [Google Scholar]
- 7. Singh R., Cuervo A. M. (2011) Autophagy in the cellular energetic balance. Cell Metab. 13, 495–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mizushima N., Klionsky D. J. (2007) Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 27, 19–40 [DOI] [PubMed] [Google Scholar]
- 9. Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A. M., Czaja M. J. (2009) Autophagy regulates lipid metabolism. Nature 458, 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klionsky D. J., Cuervo A. M., Seglen P. O. (2007) Methods for monitoring autophagy from yeast to human. Autophagy 3, 181–206 [DOI] [PubMed] [Google Scholar]
- 11. Dice J. F. (2010) Artophagy: the art of autophagy—macroautophagy. Interview by Daniel J. Klionsky. Autophagy 6, 320–321 [DOI] [PubMed] [Google Scholar]
- 12. Yang Z., Klionsky D. J. (2010) Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12, 814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sahu R., Kaushik S., Clement C. C., Cannizzo E. S., Scharf B., Follenzi A., Potolicchio I., Nieves E., Cuervo A. M., Santambrogio L. (2011) Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 20, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dice J. F. (2009) Chaperone-mediated autophagy: the heretofore untold story of J. Fred “Paulo” Dice. Interview by Daniel J. Klionsky. Autophagy 5, 1079–1084 [DOI] [PubMed] [Google Scholar]
- 15. Dice J. F. (2007) Chaperone-mediated autophagy. Autophagy 3, 295–299 [DOI] [PubMed] [Google Scholar]
- 16. Massey A. C., Zhang C., Cuervo A. M. (2006) Chaperone-mediated autophagy in aging and disease. Curr. Top. Dev. Biol. 73, 205–235 [DOI] [PubMed] [Google Scholar]
- 17. Cuervo A. M. (2004) Autophagy: in sickness and in health. Trends Cell Biol. 14, 70–77 [DOI] [PubMed] [Google Scholar]
- 18. Mizushima N., Levine B., Cuervo A. M., Klionsky D. J. (2008) Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chu C. T. (2006) Autophagic stress in neuronal injury and disease. J. Neuropathol. Exp. Neurol. 65, 423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sridhar S., Botbol Y., Macian F., Cuervo A. M. (2012) Autophagy and disease: always two sides to a problem. J. Pathol. 226, 255–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koga H., Cuervo A. M. (2011) Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol. Dis. 43, 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wong E., Cuervo A. M. (2010) Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 13, 805–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Massey A., Kiffin R., Cuervo A. M. (2004) Pathophysiology of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 36, 2420–2434 [DOI] [PubMed] [Google Scholar]
- 24. Xilouri M., Vogiatzi T., Vekrellis K., Stefanis L. (2008) α-Synuclein degradation by autophagic pathways: a potential key to Parkinson's disease pathogenesis. Autophagy 4, 917–919 [DOI] [PubMed] [Google Scholar]
- 25. Vogiatzi T., Xilouri M., Vekrellis K., Stefanis L. (2008) Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 283, 23542–23556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kabuta T., Setsuie R., Mitsui T., Kinugawa A., Sakurai M., Aoki S., Uchida K., Wada K. (2008) Aberrant molecular properties shared by familial Parkinson's disease-associated mutant UCH-L1 and carbonyl-modified UCH-L1. Hum. Mol. Genet. 17, 1482–1496 [DOI] [PubMed] [Google Scholar]
- 27. Kabuta T., Furuta A., Aoki S., Furuta K., Wada K. (2008) Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J. Biol. Chem. 283, 23731–23738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shintani T., Klionsky D. J. (2004) Autophagy in health and disease: a double-edged sword. Science 306, 990–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu W. H., Kumar A., Peterhoff C., Shapiro Kulnane L., Uchiyama Y., Lamb B. T., Cuervo A. M., Nixon R. A. (2004) Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for β-amyloid peptide over-production and localization in Alzheimer's disease. Int. J. Biochem. Cell Biol. 36, 2531–2540 [DOI] [PubMed] [Google Scholar]
- 30. Cuervo A. M., Stefanis L., Fredenburg R., Lansbury P. T., Sulzer D. (2004) Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295 [DOI] [PubMed] [Google Scholar]
- 31. Proenca C. C., Stoehr N., Bernhard M., Seger S., Genoud C., Roscic A., Paganetti P., Liu S., Murphy L. O., Kuhn R., Bouwmeester T., Galimberti I. (2013) Atg4b-dependent autophagic flux alleviates Huntington's disease progression. PLoS One 8, e68357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martinez-Vicente M., Talloczy Z., Wong E., Tang G., Koga H., Kaushik S., de Vries R., Arias E., Harris S., Sulzer D., Cuervo A. M. (2010) Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 13, 567–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Massey A. C., Kaushik S., Sovak G., Kiffin R., Cuervo A. M. (2006) Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. U.S.A. 103, 5805–5810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cuervo A. M., Knecht E., Terlecky S. R., Dice J. F. (1995) Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. Cell Physiol. 269, C1200–C1208 [DOI] [PubMed] [Google Scholar]
- 35. Kaushik S., Cuervo A. M. (2012) Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 22, 407–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cuervo A. M. (2010) Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol. Metab. 21, 142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dice J. F., Terlecky S. R. (1990) Targeting of cytosolic proteins to lysosomes for degradation. Crit. Rev. Ther. Drug Carrier Syst. 7, 211–233 [PubMed] [Google Scholar]
- 38. Kaushik S., Massey A. C., Mizushima N., Cuervo A. M. (2008) Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell 19, 2179–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kiffin R., Christian C., Knecht E., Cuervo A. M. (2004) Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 15, 4829–4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roth D. M., Balch W. E. (2011) Modeling general proteostasis: proteome balance in health and disease. Curr. Opin. Cell Biol. 23, 126–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cuervo A. M., Wong E. (2014) Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 24, 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dice J. F. (1990) Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 15, 305–309 [DOI] [PubMed] [Google Scholar]
- 43. Cuervo A. M., Terlecky S. R., Dice J. F., Knecht E. (1994) Selective binding and uptake of ribonuclease A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosomes. J. Biol. Chem. 269, 26374–26380 [PubMed] [Google Scholar]
- 44. Kaushik S., Cuervo A. M. (2012) Chaperones in autophagy. Pharmacol. Res. 66, 484–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chiang H. L., Terlecky S. R., Plant C. P., Dice J. F. (1989) A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246, 382–385 [DOI] [PubMed] [Google Scholar]
- 46. Agarraberes F. A., Dice J. F. (2001) A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 114, 2491–2499 [DOI] [PubMed] [Google Scholar]
- 47. Cuervo A. M., Dice J. F. (1996) A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501–503 [DOI] [PubMed] [Google Scholar]
- 48. Gough N. R., Hatem C. L., Fambrough D. M. (1995) The family of LAMP-2 proteins arises by alternative splicing from a single gene: characterization of the avian LAMP-2 gene and identification of mammalian homologs of LAMP-2b and LAMP-2c. DNA Cell Biol. 14, 863–867 [DOI] [PubMed] [Google Scholar]
- 49. Hatem C. L., Gough N. R., Fambrough D. M. (1995) Multiple mRNAs encode the avian lysosomal membrane protein LAMP-2, resulting in alternative transmembrane and cytoplasmic domains. J. Cell Sci. 108, 2093–2100 [DOI] [PubMed] [Google Scholar]
- 50. Konecki D. S., Foetisch K., Zimmer K. P., Schlotter M., Lichter-Konecki U. (1995) An alternatively spliced form of the human lysosome-associated membrane protein-2 gene is expressed in a tissue-specific manner. Biochem. Biophys. Res. Commun. 215, 757–767 [DOI] [PubMed] [Google Scholar]
- 51. Cuervo A. M., Dice J. F. (2000) Unique properties of lamp2a compared to other lamp2 isoforms. J. Cell Sci. 113, 4441–4450 [DOI] [PubMed] [Google Scholar]
- 52. 52 Bandyopadhyay U., Kaushik S., Varticovski L., Cuervo A. M. (2008) The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 28, 5747–5763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cuervo A. M., Dice J. F. (2000) Regulation of lamp2a levels in the lysosomal membrane. Traffic 1, 570–583 [DOI] [PubMed] [Google Scholar]
- 54. Young J. C., Agashe V. R., Siegers K., Hartl F. U. (2004) Pathways of chaperone-mediated protein folding in the cytosol. Nat. Rev. Mol. Cell Biol. 5, 781–791 [DOI] [PubMed] [Google Scholar]
- 55. Ellis R. J. (2003) Molecular chaperones: Plugging the transport gap. Nature 421, 801–802 [DOI] [PubMed] [Google Scholar]
- 56. Konecki D. S., Foetisch K., Schlotter M., Lichter-Konecki U. (1994) Complete cDNA sequence of human lysosome-associated membrane protein-2. Biochem. Biophys. Res. Commun. 205, 1–5 [DOI] [PubMed] [Google Scholar]
- 57. Kay L. E., Xu G. Y., Yamazaki T. (1994) Enhanced-sensitivity triple-resonance spectroscopy with minimal H2O saturation. J. Magn. Reson. A 109, 129–133 [Google Scholar]
- 58. Grzesiek S., Bax A. (1992) Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J. Magn. Reson. 96, 432–440 [Google Scholar]
- 59. Grzesiek S., Bax A. (1992) Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 114, 6291–6293 [Google Scholar]
- 60. Wittekind M., Mueller L. (1993) HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the α- and β-carbon resonances in proteins. J. Magn. Reson. B 101, 201–205 [Google Scholar]
- 61. Bax A., Grzesiek S. (1993) Methodological advances in protein NMR. Acc. Chem. Res. 26, 131–138 [Google Scholar]
- 62. Clore G. M., Kay L. E., Bax A., Gronenborn A. M. (1991) Four-dimensional 13C/13C-edited nuclear Overhauser enhancement spectroscopy of a protein in solution: application to interleukin 1β. Biochemistry 30, 12–18 [DOI] [PubMed] [Google Scholar]
- 63. Zwahlen C. L. P., Vincent S. J. F., Greenblatt J., Konrat R., Kay L. E. (1997) Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: application to a bacteriophage N-peptide/boxB RNA complex. J. Am. Chem. Soc. 119, 6711–6721 [Google Scholar]
- 64. Barbato G., Ikura M., Kay L. E., Pastor R. W., Bax A. (1992) Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry 31, 5269–5278 [DOI] [PubMed] [Google Scholar]
- 65. Keller R. (2004) The Computer Aided NMR Resonance Assignment Tutorial. CANTINA Verlag, Goldau, Germany [Google Scholar]
- 66. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 67. Garrett D. S., Powers R., Gronenborn A. M., Clore G. M. (1991) A common sense approach to peak picking two-, three-and four-dimensional spectra using automatic computer analysis of contour diagrams. J. Magn. Reson. 95, 214–220 [DOI] [PubMed] [Google Scholar]
- 68. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., Kay L. E. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 [DOI] [PubMed] [Google Scholar]
- 69. Iwahara J., Schwieters C. D., Clore G. M. (2004) Ensemble approach for NMR structure refinement against 1H paramagnetic relaxation enhancement data arising from a flexible paramagnetic group attached to a macromolecule. J. Am. Chem. Soc. 126, 5879–5896 [DOI] [PubMed] [Google Scholar]
- 70. Lustig A., Engel A., Tsiotis G., Landau E. M., Baschong W. (2000) Molecular weight determination of membrane proteins by sedimentation equilibrium at the sucrose or Nycodenz-adjusted density of the hydrated detergent micelle. Biochim. Biophys. Acta 1464, 199–206 [DOI] [PubMed] [Google Scholar]
- 71. Fleming K. G. (2008) Determination of membrane protein molecular weight using sedimentation equilibrium analytical ultracentrifugation. Curr. Protoc. Protein Sci. Chapter 7, Unit 7.12.1–7.12.13 [DOI] [PubMed] [Google Scholar]
- 72. Kluger R., Alagic A. (2004) Chemical cross-linking and protein-protein interactions—a review with illustrative protocols. Bioorg. Chem 32, 451–472 [DOI] [PubMed] [Google Scholar]
- 73. Schwieters C. D., Kuszewski J. J., Tjandra N., Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 74. Respondek M., Madl T., Göbl C., Golser R., Zangger K. (2007) Mapping the orientation of helices in micelle-bound peptides by paramagnetic relaxation waves. J. Am. Chem. Soc. 129, 5228–5234 [DOI] [PubMed] [Google Scholar]
- 75. Roche J., Louis J. M., Grishaev A., Ying J., Bax A. (2014) Dissociation of the trimeric gp41 ectodomain at the lipid-water interface suggests an active role in HIV-1 Env-mediated membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 111, 3425–3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Akasaki K., Tsuji H. (1998) Purification and characterization of a soluble form of lysosome-associated membrane glycoprotein-2 (lamp-2) from rat liver lysosomal contents. Biochem. Mol. Biol. Int. 46, 197–206 [DOI] [PubMed] [Google Scholar]
- 77. Russ W. P., Engelman D. M. (2000) The GxxxG motif: a framework for transmembrane helix-helix association. J. Mol. Biol. 296, 911–919 [DOI] [PubMed] [Google Scholar]
- 78. Terlecky S. R., Dice J. F. (1993) Polypeptide import and degradation by isolated lysosomes. J. Biol. Chem. 268, 23490–23495 [PubMed] [Google Scholar]
- 79. Terlecky S. R., Chiang H. L., Olson T. S., Dice J. F. (1992) Protein and peptide binding and stimulation of in vitro lysosomal proteolysis by the 73-kDa heat shock cognate protein. J. Biol. Chem. 267, 9202–9209 [PubMed] [Google Scholar]
- 80. Backer J. M., Bourret L., Dice J. F. (1983) Regulation of catabolism of microinjected ribonuclease A requires the amino-terminal 20 amino acids. Proc. Natl. Acad. Sci. U.S.A. 80, 2166–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kaushik S., Bandyopadhyay U., Sridhar S., Kiffin R., Martinez-Vicente M., Kon M., Orenstein S. J., Wong E., Cuervo A. M. (2011) Chaperone-mediated autophagy at a glance. J. Cell Sci. 124, 495–499 [DOI] [PMC free article] [PubMed] [Google Scholar]