

Background: The arabinosyltransferase EmbC plays an essential role in the synthesis of the cell wall component lipoarabinomannan.
Results: We identified a key motif for EmbC catalytic activity and determined that its structural features are unique over other arabinosyltransferases.
Conclusion: The proline-rich motif is required for the addition of branched arabinose chains to lipoarabinomannan.
Significance: This extends our understanding of lipoarabinomannan synthesis.
Keywords: Cell Wall, Glycolipid, Infectious Disease, Mycobacteria, Mycobacterium tuberculosis, Arabinosyltransferase, Essential Gene, Ethambutol
Abstract
The Mycobacterium tuberculosis cell wall is a complex structure essential for the viability of the organism and its interaction with the host. The glycolipid lipoarabinomannan (LAM) plays an important role in mediating host-bacteria interactions and is involved in modulation of the immune response. The arabinosyltransferase EmbC required for LAM biosynthesis is essential. We constructed recombinant strains of M. tuberculosis expressing a variety of alleles of EmbC. We demonstrated that EmbC has a functional signal peptide in M. tuberculosis. Over- or underexpression of EmbC resulted in reduced or increased sensitivity to ethambutol, respectively. The C-terminal domain of EmbC was essential for activity because truncated alleles were unable to mediate LAM production in Mycobacterium smegmatis and were unable to complement an embC deletion in M. tuberculosis. The C-terminal domain of the closely related arabinosyltransferase EmbB was unable to complement the function of the EmbC C-terminal domain. Two functional motifs were identified. The GT-C motif contains two aspartate residues essential for function in the DDX motif. The proline-rich region contains two highly conserved asparagines (Asn-638 and Asn-652). Mutation of these residues was tolerated, but loss of Asn-638 resulted in the synthesis of truncated LAM, which appeared to lack arabinose branching. All embC alleles that were incapable of complementing LAM production in M. smegmatis were not viable in M. tuberculosis, supporting the hypothesis that LAM itself is essential in M. tuberculosis.
Introduction
Tuberculosis is the leading cause of death from bacterial infection (1). Improved diagnostics, therapeutics, and prophylactics are all required urgently because the threat of drug resistance is growing. It is imperative to understand the biology, physiology, and pathogenicity of its causative agent, Mycobacterium tuberculosis, to improve our chance of eradicating the disease. The cell wall of M. tuberculosis is a complex structure essential for the viability of the organism and is the target of several therapeutic agents (2, 3). Current models incorporate the presence of a polysaccharide capsule at the very outer surface, mainly composed of glucan, arabinomannan, and mannan, as well as some proteins and lipids (4). Inside of this is an asymmetric lipid bilayer composed of the long chain fatty acids (mycolic acids) and other lipids and glycolipids. Mycolic acids are esterified to arabinogalactan, which is linked, in turn, to peptidoglycan, forming the mycolate-arabinogalactan-peptidoglycan complex (2).
In addition to the core structural mycolate-arabinogalactan-peptidoglycan complex, the cell wall contains complex lipoglycans, glycolipids, sulfolipids, polysaccharides, and proteins (2). The lipoglycans include a number of glycosylated derivatives of phosphatidyl inositol, the most abundant of which are lipoarabinomannan (LAM)2 and its proposed precursors: lipomannan (LM) and phosphatidyl inositol mannosides. LAM is a complex molecule composed of three main parts, the phosphatidyl inositol anchor, the mannan backbone, and the arabinan chain. The arabinan chain is composed of, on average, 55–70 arabinofuranose (Araf) residues that comprise a linear α(1→5)-Araf backbone branched by α(3→5)-Araf residues that are further elaborated with either linear tetra-arabinoside (Ara4) or branched hexa-arabinoside regions terminating in β(1→2)-Araf residues (5, 6). In pathogenic mycobacteria, including M. tuberculosis, these terminating β(1→2)-Araf residues are capped with α(1 → 2) mannosides (6–8). LAM isolated from mycobacteria exists as a heterogeneous population of molecules in which the degrees of acylation, branching, and mannose capping can all vary (7, 8). Although it is evident that LAM plays a major role in mediating host-bacterium interactions and pathogenicity through its interaction with both Toll-like receptor 2 and the macrophage mannose receptor (9–13), the physiological role of LAM in the bacterial cell is unknown.
M. tuberculosis strains expressing variant and truncated forms of LAM have been isolated (14), but there are no strains that completely lack LAM. In the fast-growing, non-pathogenic Mycobacterium smegmatis, LAM is non-essential, and LAM synthesis can be abrogated by the deletion or catalytic mutation of the arabinosyltransferase EmbC, leaving the synthesis of closely related lipomannan and arabinogalactan intact (20). In M. tuberculosis, the essentiality of the arabinosyltransferase EmbC, the only known function of which is biosynthesis of the arabinan portion of LAM, also points to the importance of LAM (15). M. tuberculosis has three arabinosyltransferases with different functions. EmbA and EmbB direct the addition of arabinan into arabinogalactan (16–18), whereas EmbC is involved in the arabinosylation of LAM by extension of an arabinan-primed LM with α(1→5)-Araf residues (14, 15, 19). The Emb proteins are targets of the frontline antitubercular agent ethambutol, which inhibits the activity of all three, resulting in cessation of both arabinogalactan and LAM synthesis and, ultimately, cell death (17, 20–24). embA, embB, and embC are all individually dispensable in M. smegmatis (18, 19). In contrast, embA and embC are essential in M. tuberculosis, whereas embB is predicted to be essential (15, 16, 25).
EmbC is an integral membrane protein member of the glycosyltransferase C superfamily (26) which utilizes a lipid-linked sugar, β-d-arabinofuranosyl-1-monophosphoryldecaprenol, as a donor substrate. Three catalytic or substrate-binding motifs required for the function of EmbC have been suggested: the GT-C DDX motif characteristic of glycosyl transferases, the proline-rich motif found in polysaccharide polymerases (19, 27, 28), and a lectin-like carbohydrate binding module in the C-terminal domain (29). Much of what is known about the function of EmbC comes from studies of M. smegmatis, where deletion of embC results in the abrogation of LAM synthesis (19). Mutagenesis of the first aspartate residue in the GT-C DDX motif (D279G) in EmbCMsm results in the complete loss of LAM synthesis, likely because of inactivation of the arabinosyltransferase activity, whereas mutagenesis of the second aspartate (D280G) results in a truncated LAM (27). Mutation of the prolines within the proline-rich polysaccharide polymerase motif has no apparent effect. although mutation of a conserved tryptophan results in the synthesis of truncated LAM, mainly because of loss of incorporation of linear arabinose Ara4 chains (27). The length of the arabinan chain incorporated into LAM is dependent on the C-terminal domain of EmbC (28). Studies of EmbC function in M. tuberculosis have been considerably less extensive, although mutation of D294G results in truncated LAM and ethambutol sensitivity (14). The crystal structure of the globular C-terminal domain reveals a similarity to lectin-binding domains, and two key residues (Trp-868 and Trp-985) have been identified (29). We sought to investigate the function of EmbC in M. tuberculosis to determine its role in LAM biosynthesis and, by extension, the role of LAM in normal bacterial physiology.
EXPERIMENTAL PROCEDURES
Bacterial Culture
M. tuberculosis H37Rv was grown in Middlebrook 7H9 medium plus 0.05% w/v Tween 80 and 10% oleic acid, albumen, dextrose, and catalase supplement (BD Biosciences) or on Middlebrook 7H10 agar plus 10% v/v oleic acid, albumen, dextrose, and catalase supplement. Liquid cultures were grown in 100-ml roller cultures or 10-ml standing cultures. Selection was carried out with hygromycin at 50 μg/ml, gentamicin at 10 μg/ml, and streptomycin at 20 μg/ml as required. M. smegmatis was grown in Luria broth plus 0.05% w/v Tween 80 or on Luria broth-agar. Minimum inhibitory concentrations were determined in liquid or solid medium (30, 31).
LAM Analysis
LAM was extracted from cells grown on solid or liquid medium and analyzed by gel electrophoresis and hybridization to CS-35 or CS-40 antibodies as described previously (32).
Quantitative RT-PCR
RNA was prepared from log phase aerobic cultures. cDNA was prepared from 2 μg of total RNA using the Roche Transcriptor cDNA kit. Primer/probe sets were as follows: embC primers, 5′-CCG GCA AGA CGG TGT TGT-3′, 5′-CGG TGA CCA ACG GGA CAT-3′, and probe 5′-CGC CTA AGG CCG TCG-3′; sigA primers, 5′-CCG ATG ACG ACG AGG AGA TC-3′, 5′-GGC CTC CGA CTC GTC TTC A-3′, and probe 5′-CCT CCG GTG ATT TC-3′. Quantitative RT-PCR reactions were prepared containing Roche LightCycler 480 TaqMan Master mix, 2.5 μl of cDNA reaction, 0.9 μm of each primer, and 0.25 μm probe. A no DNA control was included for the reach run. Cycle conditions were initial denaturation at 94 °C for 10 min, 45 cycles of denaturation (94 °C for 10 s), amplification (56 °C for 1 min), and extension (72 °C for 1 s). A standard curve was generated using genomic DNA and used to calculate copy number. embC expression was normalized to sigA. For PAg85a, three biological replicates were assayed. For all other promoters, two biological replicates were prepared.
Expression of FLAG-tagged embC in M. tuberculosis
embC was PCR-amplified from genomic DNA using the primer pair 5′-GGG GAC AAG TTT GTA CAA AAA AGC AGG TGA TGG CTA CCG AAG CCG CCC CAC-3′ and 5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTA GCC GCG GCG CAA CGG CGC-3′ and cloned into pDTNF and pDTCF (33). These vectors contain an anhydrotetracycline-inducible promoter and introduce an N- or C-terminal FLAG tag into the protein. Plasmids were transformed into M. tuberculosis (34). Three independent transformants were grown in the presence or absence of 150 ng/ml anhydrotetracycline for 5 days. Cell lysates were generated, and 15 μg of total protein was run on a 12% polyacrylamide gel. Western blotting was carried out using PVDF membranes probed with α-FLAG antibodies (GenScript). The 49-kD N-terminal FLAG-BAPTM fusion protein was used a positive control.
Construction of Plasmids with Truncated Alleles of embC
Inverse PCR was used to construct truncation alleles of embC. Primers were used to amplify the plasmid pRG603, which has embC under the control of the Ag85a promoter in an integrating vector (15). The forward primer 5′-CCT CAC CGC CCT TAA GCG CGT CGC CTA CCA TCG-3′ was complementary to the plasmid sequence located downstream of embC and incorporated an AflII restriction site (boldface). Reverse primers were designed to generate embC truncations at Ser-341, Val-637, and Leu-717, terminating with a stop codon (underlined), followed by an AflII restriction site (boldface). Primers were as follows: Ser-341, 5′-GCG CTG TGG GCT CAT GTC AGC ACG GCC AGT TAG CTT AAG CGC CTA CC-3′; Val-637, 5′-G GCC CTG TCG TTC GCC AGT GTC TAG CTT AAG TGG TAC G-3′; and Leu-717, 5′-G GCA ATT GCC ACG TGG TTG CTG GTG CTT TAG CTT AAG GTA TCG C-3′. PCR products were treated with DpnI, digested with AflII, and religated. Plasmids were confirmed by enzyme digestion and sequencing.
Construction of Plasmids with Hybrid Alleles of embC/B
Hybrid alleles were constructed using “splicing by overlap extension” PCR (35). Site-directed mutagenesis using primer pair 5′-CCT CAC CGC CTT AAG CGC GTC GCC-3′ and 5′-GGC GAC GCG CTT AAG GCG GTG AGG-3′ was used to modify the pRG603 vector to include an AflII site (boldface) immediately downstream of embC to allow for directional cloning (plasmid pEAK).
The core and C-terminal region of embB (codons 348–1098) was amplified from H37Rv genomic DNA using primers embB-core-F 5′-CTG TGG ATG CGC CTG CCA GAC CTG GCC GCC-3′ and embB-Cterm-R 5′-CTT AAG CTA TGG ACC AAT TCG GAT CTT GCC CGG-3′ and cloned into pSC-B (Stratagene) to make pSC-B_embB. For construction of each hybrid allele, two sets of primers were designed: amplification primers that were complementary to embB or embC and ligating primers that bridged the splice sites.
The embCCB hybrid was constructed in three steps. The embB C-terminal fragment (codons 727–1098) was amplified from pSC-B_embB using the primers 5′-GCC ACG TGG TTG CTG GTG CTT GTG CGA CAG TAC CCG ACC TAC-3′ and 5′-CTT AAG CTA TGG ACC AAT TCG GAT CTT GCC CGG-3′. The embC N-terminal plus core fragment (codons 175–717) was amplified from pRG603 using primers 5′-CCC AAT GCT GAG CAC CCC GGT GCA CCG CTG-3′ and GTA GGT CGG GTA CTG TCG CAC AAG CAC CAG CAA CCA CGT GGC-3′. The two products were ligated by mixing 1:1 and running five cycles of PCR. The ligated product was amplified with primers 5′-CCC AAT GCT GAG CAC CCC GGT GCA CCG CTG-3′ and 5′-CTT AAG CTA TGG ACC AAT TCG GAT CTT GCC CGG-3′ and cloned into pSC-B. The hybrid allele was recovered from this plasmid by digestion with BlpI and AflII and directionally cloned into pEAK (replacing the embC gene). The final product pEAK-CCB was sequence-confirmed.
The embCBB hybrid was constructed in the same manner as embCCB. The embC N-terminal fragment (codons 175–341) was amplified using primers 5′-CCC AAT GCT GAG CAC CCC GGT GCA CCG CTG-3′ and 5′-GGT CTG GCA GGC GCA TCC ACA GAC TGG CCG TGC TGA CAT GAG CC-3′. The embC core plus C-terminal fragment (codons 348–1098) was amplified using primers 5′-GGC TCA TGT CAG CAC GGC CAG TCT GTG GAT GCG CCT GCC AGA CC-3′ and 5′-CTT AAG CTA TGG ACC AAT TCG GAT CTT GCC CGG-3′. The two products were ligated, amplified and cloned into pSC-B. The hybrid allele was recovered by digestion with BlpI and AflII and cloned into pEAK with final product pEAK-CBB sequence-confirmed.
The embCBC hybrid was constructed as follows. The embCB fragment (codons 175–726) was amplified from pEAK-CBB using embC-Nterm-F and embB-core/embC-Cterm-R 5′-GTC AGC GAT ACC ACC TCG AAG ATC CCG GCC ACC ATG GAC G-3′ primers. The embC C-terminal fragment (codons 718–1011) was amplified from pRG603 using primers 5′-CGT CCA TGG TGG CCG GGA TCT TCG AGG TGG TAT CGC TGA C-3′ and 5′-GAT CCG CCA CTT GGG TGT CTC GTC GAC GCC-3′. The products were ligated and cloned into pSC-B. The hybrid allele was released by digestion with BlpI and SgrDI and ligated into pEAK. The final product pEAK-CBC was sequence-confirmed.
Construction of Plasmids with Mutated Alleles of embC
Point mutations were made by site-directed mutagenesis. Amplification reactions were carried out in a 50-μl total volume containing 1× PfuUltra reaction buffer, 0.5 mm dNTPs, 160 ng of each primer, 10% dimethyl sulfoxide, 80 ng of template, and 2.5 units PfuUltra. The thermocycling program used was 94 °C for 5 min, followed by 18 cycles of 94 °C for 30 s, 65 °C for 1 min, and 68 °C for 14 min, with a final extension cycle of 68 °C for 15 min. Template DNA was degraded using 10 units of DpnI at 37 °C for 1 h. A 4-μl volume of each reaction product was used to transform competent DH5α-S.E. Escherichia coli. Recombinant plasmids were isolated and sequence-verified. The following primers were used: D293A, 5′-GGG GCC AAC ACC TCC GCC GAC GGC TAC ATC CTG ACC-3′ and 5′-GGT CAG GAT GTA GCC GTC GGC GGA GGT GTT GGC CCC-3′; D294G, 5′-GCC AAC ACC TCC GAC GGC GGC TAC ATC CTG ACC ATG G-3′ and 5′-C CAT GGT CAG GAT GTA GCC GCC GTC GGA GGT GTT GGC-3′; N638A, 5′-CG TTC GCC AGT GTC GCC GGC TGG TGG TAC G-3′ and 5′-C GTA CCA CCA GCC GGC GAC ACT GGC GAA CG-3′; and N652A, 5′-GGT GTG CCA TGG TCG GCC TCG TTT CCG AAG TGG-3′ and 5′-CCA CTT CGG AAA CGA GGC CGA CCA TGG CAC ACC-3′.
Construction of embC Expression Vectors
embC was amplified using primers 5′-GGA AGC TTG TGA TGG CTA CCG AAG CC-3′ and 5′-CAT CGA TTC GGT GGC CAC TTC TAG CC-3′ incorporating HindIII and ClaI restriction sites, respectively (boldface). The embC gene was placed under the control of a range of mycobacterial promoters (PsenX, PtrpECBA, PtrpD, PtrpE2, PRv0251c, PRv2466c, and PRv2930) in the pSM128 vector (streptomycin resistance, integrating vector).
Gene Switching to Test for Allele Functionality
Gene switching was used to test for functional complementation of mutant alleles (36). Plasmids were transformed into a recombinant strain of M. tuberculosis (del-int) in which the only functional copy of embC was present in an integrating vector carrying gentamicin resistance (the normal chromosomal copy was deleted by homologous recombination) (15). The del-int strain was electroporated with 1 μg of plasmid carrying mutant alleles (containing hygromycin or streptomycin resistance genes) and transformants selected on the appropriate antibiotic to select for the incoming plasmid. Recombinants were tested for sensitivity to gentamicin to confirm loss of the wild-type embC plasmid. Each experiment included a negative control (empty integrating vector) and a positive control (vector plus wild-type embC) to confirm that switching occurred.
RESULTS
To investigate the critical function and role EmbC plays in the physiology of M. tuberculosis, we looked for phenotypic changes resulting from modulating the activity of the enzyme via site-directed mutagenesis, truncations, allele hybrids, and alterations to expression level.
Identification of Key Regions in EmbC
To identify key regions in the EmbC protein, we compared its sequence with the closely related arabinosyltransferases EmbA and EmbB from the GT-C superfamily from mycobacteria and corynebacteria. The multiple sequence alignment was overlaid with data from topology prediction to identify transmembrane helices and loop regions as well as regions of conservation. From the primary sequence, all members of the Emb family are predicted to have 13 transmembrane helices and two large extracellular domains, one located near the N terminus and one at the C terminus (Fig. 1A).
FIGURE 1.

Topology prediction for M. tuberculosis EmbC. A, predicted topology of full-length protein. B, predicted topology after cleavage of the N-terminal signal sequence at position Ala-46/Thr-47.
Regions of high sequence similarity mapped to two identifiable motifs: a DDX motif (common among glycosyltransferases) located in the first extracellular loop between transmembrane helices 3 and 4 and a proline-rich polysaccharide polymerase motif in the extracellular loop between transmembrane helices 11 and 12 (Fig. 2). The proline-rich polymerase region includes two highly conserved asparagine residues (Asn-638 and Asn-652) (Fig. 2). Asn-638 is completely conserved across the three proteins (EmbA, EmbB, and EmbC) in mycobacteria and corynebacteria, whereas Asn-652 is conserved only across the mycobacterial EmbC proteins. These two motifs were separated by eight transmembrane helices connected by short loops of 5–50 amino acids with no recognizable motifs.
FIGURE 2.

Multiple sequence alignment and topology prediction of Emb proteins from mycobacteria and corynebacteria. Emb proteins from M. tuberculosis, M. smegmatis, Mycobacterium bovis, Mycobacterium marinum, Mycobacterium leprae, Mycobacterium avium, Cornyebacterium diptheriae, Cornyebacterium jeikeium, Cornyebacterium glutamicim, Cornyebacterium urealyticum, and Cornyebacterium kroppenstedtii were aligned using ClustalW (40). The alignment was imported into JalView (41) and colored according to the predicted topology from HMMTOP (39), with transmembrane regions colored light gray. Consensus scores were calculated on the basis of the alignment. Conserved regions of interest are displayed below: the DDX region with the DDX motif highlighted and the proline-rich region with M. tuberculosis EmbC Asn-638 and Asn-652 highlighted.
EmbC Contains an N-terminal Signal Peptide Sequence
Sequence analysis with the programs SignalP and SPScan suggested that EmbC has a signal peptide located in the first transmembrane helix with a potential signal peptidase cleavage site between residues Ala-46 and Thr-47. We sought to determine whether this predicted signal sequence was functional in M. tuberculosis. We expressed EmbC carrying a FLAG tag fused either to the N terminus or C terminus in M. tuberculosis. If EmbC contained a functional signal peptide, we would anticipate that the N-terminal FLAG tag would be lost upon cleavage of the signal sequence by the signal peptidase LepB. In contrast, the C-terminal FLAG tag should remain intact.
Recombinant proteins were detected by Western blotting to the FLAG tag. The C-terminal tagged protein was detected in M. tuberculosis cell lysates, whereas the N-terminal tagged protein was not (Fig. 3). These data suggest that the signal peptide is indeed functional and that it is cleaved during export of the protein. An alternative explanation is that the N-terminal tagged protein was not expressed or was unstable. This is unlikely because the two proteins (C-terminal or N-terminal tagged versions) would be expressed from the same promoter (with same transcriptional efficiency), and the presence of the tag is not expected to alter translational efficiency or protein stability. The presence of a signal peptide has important implications for the structure of the functional protein because the mature protein would have an extracytosolic N terminus rather than a cytosolic N terminus and there would only be 12 transmembrane helices, leading us to predict a revised topology (Fig. 1B).
FIGURE 3.
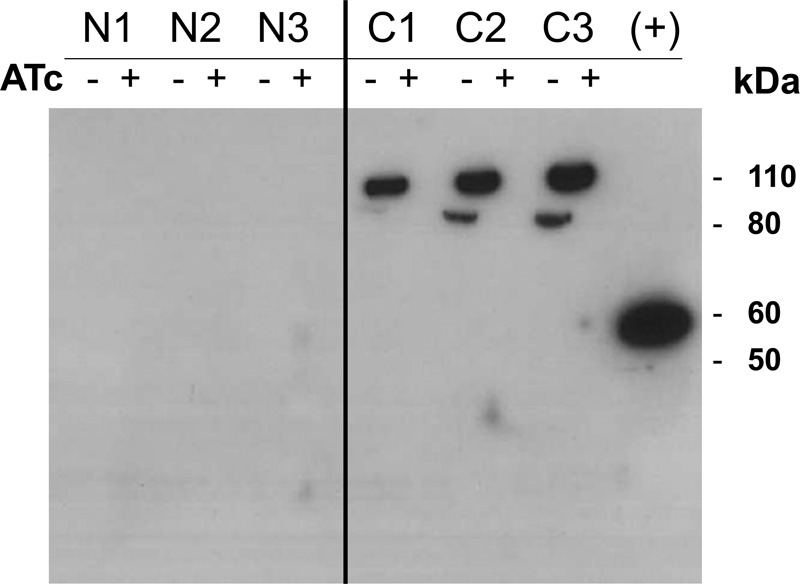
EmbC has an N-terminal signal sequence. EmbC fused with either an N-terminal or C-terminal FLAG tag was expressed in M. tuberculosis recombinants. Expression of tagged EmbC was induced by the addition of anhydrotetracycline (ATc) in three independent transformants. Lysates were probed using α-FLAG tag antibodies. N1–3, N-terminal tagged recombinants; C1–3, C-terminal tagged recombinants; +, 49-kDA N-terminal FLAG-BAPTM positive control for the α-FLAG tag antibody.
Reduced Expression of embC Confers Sensitivity to Ethambutol in M. tuberculosis
We wanted to investigate the effect of varying the embC expression level on the viability and physiology of M. tuberculosis. Previous work demonstrated that high-level expression of embC (using the hsp60 promoter) was not tolerated in M. tuberculosis, although it was possible in M. smegmatis, where it resulted in both increased LAM size and resistance to ethambutol (14). Because EmbC is essential, gene knockouts are not viable, but underexpression of the protein could provide valuable clues as to its function. We also postulated that there would be a minimum level of expression beyond which cells would not be viable and that underexpression might result in alterations to LAM.
We constructed a number of strains in which embC was expressed from a heterologous promoter. We used a strain of M. tuberculosis in which the native chromosomal copy of embC was deleted (in-frame, unmarked deletion) and a functional copy was provided on an integrating (single copy) vector. In this strain, expression of EmbC was driven by the Ag85a promoter (15). Using gene switching (36), we replaced the integrated vector with alternative versions carrying embC expressed from a variety of promoters.
Strains were grown under aerobic conditions, and expression of embC was measured at the mRNA level. Interestingly, we found that expression from the Ag85a promoter was higher than from the native promoter (Fig. 4). This was surprising because PAg85a is a weak promoter and suggests that the normal level of expression of embC, at least at the mRNA level, is low. Therefore, the recombinant del-int is overexpressing EmbC ∼10- to 20-fold. Expression levels from other promoters were also measured (Fig. 4). As expected, we saw a range of expression ranging from 4-fold up-regulation to 2-fold down-regulation, with the lowest level of expression driven by the Rv2466c promoter.
FIGURE 4.
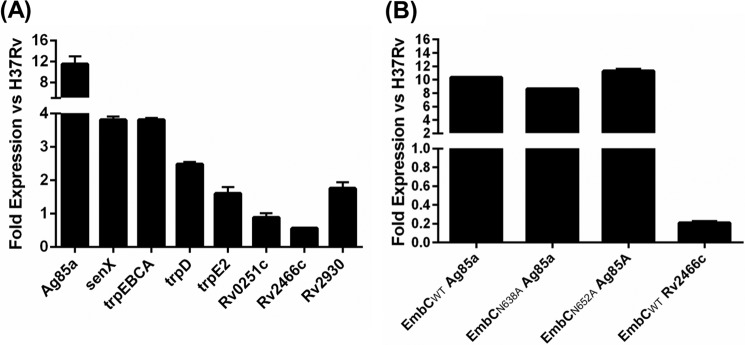
Expression level of EmbC in recombinant strains. RNA was purified from M. tuberculosis strains carrying recombinant alleles of embC in which expression was from the indicated promoter. RNA was extracted, and embC mRNA was measured using RT-qPCR and normalized against sigA. Results are given for each promoter and are expressed as fold change compared with the wild-type strain. Each sample was assayed at least twice. A, promoter variants. B, mutant alleles expressed from the Ag85a promoter. A and B are from independent experiments.
Because the strain carrying PRv2466c-embC was underexpressing embC mRNA, we selected it for phenotypic studies. We also included the original del-int strain (PAg85a-embC) because it overexpresses embC mRNA. Because EmbC is one of the targets of ethambutol, we looked at the sensitivity of each strain to ethambutol. Overexpression of embC led to increased ethambutol resistance, with a 2-fold shift in minimum inhibitory concentration (4.9 ± 0.2 μm, n = 10) compared with the wild-type (2.6 ± 0.6 μm, n = 19). Similarly, the underexpressor strain was slightly more sensitive to ethambutol (1.7 ± 0.2 μm, n = 7). Although the absolute differences were small, they were reproducible and statistically significant (p < 0.001 using Student's t test). The data show a correlation between embC expression level and ethambutol sensitivity, with lower levels of embC expression leading to ethambutol sensitivity and higher levels leading to resistance.
The C Terminus of EmbC Is Required for Its Function
EmbC is essential in M. tuberculosis, but it is not known whether the entire protein is required for its functionality. To determine which region(s) of EmbC is/are essential, we constructed truncated alleles and tested these for functional complementation in M. tuberculosis. The truncations were Leu-717, which removed 377 amino acids from the C terminus and the complete C-terminal domain; Val-637, which removed the polysaccharide polymerase loop and the C-terminal domain; and Ser-341, which removed everything downstream of the DDX glycosyltransferase loop. Truncated alleles were cloned into a single copy (integrating) complementing vector under the control of the Ag85a promoter and were tested for their ability to complement an embC deletion in M. tuberculosis using gene switching (36). We were unable to obtain viable strains with any of the truncated alleles, indicating that the C terminus is essential for function.
Because we were unable to look at the effect of truncations on LAM biosynthesis directly in M. tuberculosis, we used M. smegmatis as a model. The three truncated alleles were tested for their ability to complement embC deletion in M. smegmatis. Plasmids were transformed into an M. smegmatis embC knockout strains, and LAM was analyzed (15). None of the truncated alleles were able to restore LAM production in this strain, although the wild-type M. tuberculosis allele was functional (Fig. 5) (15). It is unlikely that the loss of function in the truncated alleles is due to lack of transcription or translation because the promoter used in all cases was the same, and there is no reason to suppose changes in translational efficiency. Alternatively, it could be due to altered protein folding and/or increased susceptibility to proteolytic degradation, which is possible. We made several attempts to measure protein by Western blot analysis but were unable to make antibodies with sufficient specificity for EmbC, and, as such, we are unable to directly measure protein levels. However, regardless of the mechanism, we can conclude that the C terminus is required for proper protein function. Therefore, the C-terminal domain of EmbC is essential for bacterial viability in M. tuberculosis and is necessary to complement LAM production in M. smegmatis.
FIGURE 5.

Analysis of LAM from M. smegmatis recombinants. The M. smegmatis embCΔ deletion strain was complemented with the indicated embCMtb alleles. LAM was analyzed in wild-type and recombinant strains grown on agar. A, periodic acid-Schiff staining. B, Western blotting using antibody CS-35. Lane 1, wild-type M. smegmatis; lanes 2–9, M. smegmatis embCΔ complemented with wild-type embCMtb (lane 2), no allele (lane 3), embCBBMtb hybrid (lane 4), embCMtb N638A (lane 5), embCMtb N652A (lane 6), embCMtb Ser-341 truncation (lane 7), embCMtb Val-637 truncation (lane 8), and embCMtb Leu-717 truncation (lane 9); and lane M, markers. C, Western blotting using antibody CS-35. Lanes 1 and 2, wild-type M. smegmatis; lane 3, wild-type embCMtb; lane 4, empty; lane 5, no allele; and lane 6, embCMtb D293A. The expected range for migration of LAM and LM is shown for the PAS-stained gel. The Western blot detects only LAM.
The C-terminal Domain of EmbB Is Not Equivalent to That of EmbC
We demonstrated that the C-terminal domain of EmbC is required for its function in M. tuberculosis. It is possible that this is due to a structural requirement and that truncated alleles are unstable, do not fold correctly, or fail to insert into the membrane in the correct orientation. To address this possibility, we utilized the fact that EmbB has a very similar sequence/structure to EmbC. We constructed hybrid arabinosyltransferase alleles in which either the C terminus, the central region, or both were replaced by the corresponding region from EmbB.
We considered each protein as three regions on the basis of the topology prediction and sequence alignments. The N-terminal region was comprised of the N-terminal domain and the DDX loop (EmbC Met-1-Ser-341, EmbB Met-1-Ser-347). The core section comprised everything between the DDX loop and the C-terminal domain, including the proline-rich polysaccharide polymerase loop (EmbC Ile-342-Val-716, EmbB Leu-348-Ile-726) and the C-terminal domain (EmbC Leu-717-Gly-1094, EmbB Val-727-Pro-1098). Hybrid alleles were constructed and tested for functional complementation in M. tuberculosis.
We first tested whether the EmbB C-terminal region could be replaced by EmbB (EmbCCB allele). Despite retaining the GT-C and proline-rich motifs, EmbCCB was incapable of complementing the essential function of EmbC in M. tuberculosis. This result could be explained by the presence of EmbC-specific sequence determinants, suggesting that the essential role of the C-terminal domain is not purely structural.
We also tested a hybrid EmbCBB allele in which only the N terminus was derived from EmbC. Not surprisingly, we could not obtain viable strains expressing this allele in M. tuberculosis. In addition, EmbCBB was incapable of complementing LAM production in M. smegmatis, confirming that its key activity was lost (Fig. 5).
The core region of EmbC does not contain either of the two known functional motifs, and it is possible that this region provides structural integrity and stabilization in the membrane (because it has several transmembrane domains). We determined whether the EmbB core could replace the EmbC core by constructing an EmbCBC hybrid allele containing residues Ser-341 to Leu-717 from EmbB. This allele was unable to complement the embC deletion in M. tuberculosis, suggesting that the core also contains an EmbC-specific region that cannot be complemented EmbB.
Aspartate 293 Is Required for EmbC Activity in M. tuberculosis
We identified two key motifs in the M. tuberculosis EmbC protein. The GT-C signature motif (DDX) is known to play a role in the function of EmbC. We demonstrated previously that Asp-294 is required for correct arabinosyltransferase activity and that its mutation leads to the production of truncated LAM as well as increased sensitivity to ethambutol (14). We determined the role of the other aspartate (Asp-293) in EmbC function by constructing a D293A mutant allele. This allele was unable to complement the embC deletion in M. tuberculosis. In addition, EmbCD293A was unable to restore LAM biosynthesis in the M. smegmatis ΔembC strain, confirming that Asp-293 is critical for arabinosyltransferase activity (Fig. 5).
Asparagine N638A and N652A Are Required for EmbC Activity in M. tuberculosis
Bioinformatic analysis identified two highly conserved asparagine residues, Asn-638 and Asn-652, in the N-terminal region. In M. smegmatis, mutations in the conserved tyrosine and proline (Tyr-628, Pro-635, and Pro-641) had no observable effect on LAM synthesis, whereas mutation of a conserved tryptophan (Trp-627) resulted in a truncated LAM species, and a triple mutant, W627L/P635S/P641S, resulted in a truncated LAM lacking Ara4 (33). We hypothesized that the tryptophan and proline residues may be involved in substrate binding but that the conserved asparagines could be involved in catalysis. We determined the role of both residues by constructing mutant the alleles N638A and N652A and testing these for functionality. Both alleles were able to complement the embC deletion in M. tuberculosis because we were able to isolate strains carrying these alleles by gene switching.
We analyzed LAM production using these mutant alleles in both M. tuberculosis and M. smegmatis. In M. tuberculosis, EmbCN652A was fully functional and produced apparently normal LAM in both species, whereas EmbCN638A produced a truncated version of LAM, suggesting that this residue is key for proper activity (Fig. 6). Surprisingly, EmbCN638A was unable to restore LAM synthesis in the M. smegmatis embCΔ strain (Fig. 5).
FIGURE 6.
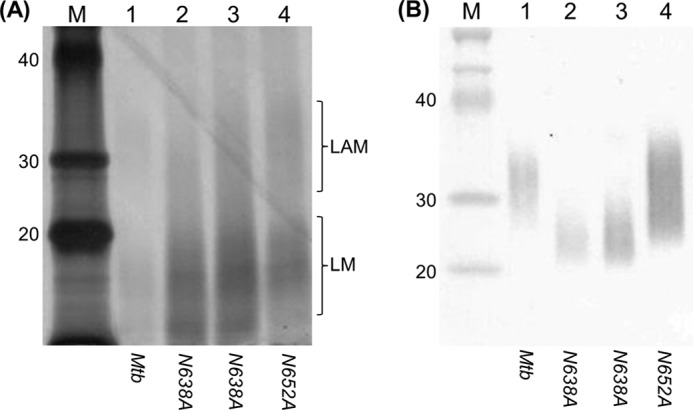
Analysis of LAM from M. tuberculosis recombinants carrying mutations in the proline motif. M. tuberculosis strains carrying the indicated embCMtb alleles were constructed by gene switching. LAM was analyzed in recombinant strains grown on agar. A, periodic acid-Schiff staining. B, Western blotting using antibody CS-35. M. smegmatis embCΔ is complemented with wild-type embCMtb (lane 1), embCMtb N638A (lane 2), embCMtb N638A (lane 3), embCMtb N652A (lane 4), and markers (lane M). The expected range for migration of LAM and LM is shown for the periodic acid-Schiff-stained gel. The Western blot detects only LAM.
To investigate this further, we analyzed the production of LAM in M. tuberculosis during growth in liquid medium. A smaller species of LAM was visible by periodic acid-Schiff stain, but this did not react with the CS-35 antibody (Fig. 7). The primary epitope for CS-35 is the terminal-branched hexa-arabinoside motif present in both LAM and arabinogalactan, although it is also known to interact with Ara4 (37, 38). We tested the CS-40 antibody, which reacts preferentially with the mannose-capped LAM, although the precise epitope is not established (7), and this did react with LAM from the EmbCN638A strain. On the basis of the lack of recognition by CS-35, it suggests that the truncated LAM synthesized by this mutant lacks either Ara4 or hexa-arabinoside subunits. This would suggest that one of the functions of EmbC is to attach these units to the linear α(1→5) arabinofuranose chain extending from the LM core of LAM. Further in-depth analysis of the key structural features of LAM produced by the N638A strain would be instructive in this regard.
FIGURE 7.
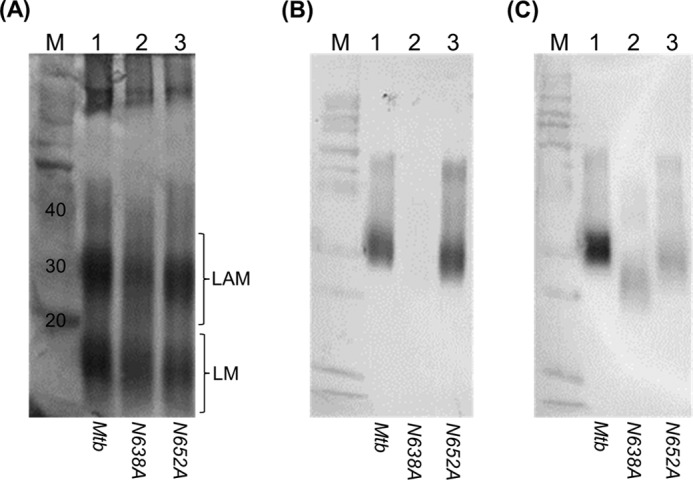
Analysis of LAM from M. tuberculosis recombinants in liquid medium. M. tuberculosis strains carrying the indicated embCMtb alleles were constructed by gene switching. LAM was analyzed in recombinant strains grown in liquid medium. A, periodic acid-Schiff staining. B, Western blotting using antibody CS-35. C, Western blotting using antibody CS-40. Lane 1, wild-type embCMtb; lane 2, embCMtb N638A; lane 3, embCMtb N652A; lane M, markers. The expected range for migration of LAM and LM is shown for the periodic acid-Schiff-stained gel. The Western blot detects only LAM.
We also determined ethambutol sensitivity in liquid culture. Strains carrying PAg85a-embCN638A had a minimum inhibitory concentration lower than the comparable parental strain (PAg85aembCwt). The values were 2.3 ± 0.5 μm (n = 14) compared with 4.9 ± 0.2 μm (p < 0.0001). In contrast, the strain with PAg85a-embCN652A was slightly more resistant, with a minimum inhibitory concentration of 7.4 ± 0.5 μm (n = 14, p < 0.0001). Therefore, mutation of Asn-638 gave rise to ethambutol sensitivity, whereas mutation of Asn-652 gave rise to resistance.
DISCUSSION
EmbC plays an essential role in the biosynthesis of LAM in M. tuberculosis. As an essential gene, deletion mutants are not viable, but we used a simple genetic approach of gene switching to probe the role of key residues and domain structure.
EmbC has a predicted signal peptide that would direct its export to the membrane via the Sec pathway. We demonstrated that the N-terminal is subject to cleavage and that, therefore, that this is likely to be functional cleavage site for the signal peptidase. This has implications for the structure of EmbC because the predicted mature protein would lose one transmembrane helix and the N-terminal domain would be free on the extracytosolic side. Releasing one of the membrane tethers from the N-terminal domain would provide additional surface area for potential interactions. Similar signal peptides are predicted in both EmbA and EmbB.
EmbC and, by extension, LAM are essential in M. tuberculosis but not in the related, non-pathogenic species M. smegmatis. The reason for this difference is not obvious, but we speculate that there may be structural differences in the cell wall and, in particular, the outer layers.
We demonstrated that the C terminus of EmbCMtb is required for activity and that the same domain from EmbBMtb is not functionally equivalent. Neither truncations nor hybrid EmbC/B alleles could support growth in M. tuberculosis or LAM biosynthesis. The lack of complementation by the C terminus of EmbB is surprising given the level of sequence identity and the conservation of most of the key residues (Trp-868, Trp-985, Asn-740, and Asp-949). A key difference may be the glutamate residue at position 899 of EmbC, which is proposed to be involved in substrate binding and is a glutamic acid in EmbBMtb. It is possible that this substitution is sufficient to disrupt an essential function of the EmbCMtb protein. Alternatively, it may be due to other differences between the two proteins domains, such as secondary or tertiary structural differences. What is evident, however, is that EmbCMtb has essential functionalities that cannot be removed nor replaced by the similar EmbBMtb domain.
These data further support the profound differences between the pathogenic and non-pathogenic species. Not only is the role of LAM different, but EmbC differs, too. For example, truncations in EmbCMsm result in synthesis of structurally altered LAM, whereas truncations in EmbCMtb result in complete abrogation of LAM synthesis (even when expressed in M. smegmatis). In addition, M. smegmatis EmbB domains are able to functionally complement for EmbCMsm. In both cases, LAM no longer contained the characteristic Ara4 units (19, 28), but it was still produced. In addition, mutation of Trp-868 or Trp-985 in the C terminus of EmbCMtb results in loss of arabinosyltransferase activity (29).
We further elaborated the required motifs in EmbC for M. tuberculosis and found that Asp-293 located in the DDX motif is required for activity. Similarly, in M. smegmatis, mutation of the same residue (Asp-279) led to a loss of LAM synthesis (27). We characterized the role of two conserved asparagines in the N terminus of the protein. Mutation of N638A disrupted a non-essential catalytic function of EmbC in M. tuberculosis that resulted in a structural change to LAM. Previous work with M. smegmatis EmbC demonstrated the role of key residues in the proline-rich motif. In particular, a triple mutant, W627L/P635S/P641S, produced LAM that lacked Ara4 (39). We hypothesize that addition of Ara4 subunits requires both the polysaccharide polymerase motif as the catalytic domain and the C-terminal domain being involved in substrate binding.
The N638A mutant strain has the most dramatic change in LAM arabinosylation that has been seen in recombinant strains of M. tuberculosis. The N638A mutation appears to affect the structure of LAM, most likely by reducing Ara4 and hexa-arabinoside units attached to the linear α(1→5) chain, whereas the DDX mutants studied most likely truncated LAM by a shortening of the linear α(1→5) chain. Alterations to the LM/LAM structure have an effect on the physiology of mycobacteria, and alterations in the mannosyl-chain structure of both LM and LAM have consequences for cell wall integrity in M. tuberculosis.
Because EmbC is essential, it is difficult to determine whether the loss of viability after gene disruption is due to lack of LAM production or due to some other unknown essential functional of EmbC. We see a correlation between the ability of alleles to sustain LAM production (at least in M. smegmatis) and bacterial viability. The only exception to this was with EmbCN638A, which did not synthesize LAM in M. smegmatis but was able to make truncated LAM in M. tuberculosis. This correlation provides further support to our hypothesis that EmbC is essential in M. tuberculosis because its biosynthetic product, LAM, provides an essential function.
CONCLUSION
We extended our understanding of the role of EmbC in LAM biosynthesis. Using a combination of truncations, hybrids, and mutated alleles, we demonstrated that the C terminus of EmbC, as well as the DDX motif, are essential for its activity. We identified a key residue (N638A) involved in EmbC activity that likely plays a role in LAM branching. We provided further evidence that LAM itself is essential for cell viability in M. tuberculosis. Finally, we confirmed the link between EmbC expression and ethambutol sensitivity.
Acknowledgments
We thank Delphi Chatterjee and Anita Amin for discussions.
This work was supported, in whole or in part, by NIAID/National Institutes of Health Grant R21AI81111.
- LAM
- lipoarabinomannan
- LM
- lipomannan
- Araf
- arabinofuranose
- Ara4
- tetra-arabinoside.
REFERENCES
- 1. World Health Organization (2013) Global Tuberculosis Report 2013, Geneva, Switzerland [Google Scholar]
- 2. Brennan P. J. (2003) Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 83, 91–97 [DOI] [PubMed] [Google Scholar]
- 3. Brennan P. J., Crick D. C. (2007) The cell-wall core of Mycobacterium tuberculosis in the context of drug discovery. Curr. Top. Med. Chem. 7, 475–488 [DOI] [PubMed] [Google Scholar]
- 4. Lemassu A., Daffé M. (1994) Structural features of the extracellular polysaccharides of Mycobacterium tuberculosis. Biochem. J. 297, 351–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jankute M., Grover S., Rana A. K., Besra G. S. (2012) Arabinogalactan and lipoarabinomannan biosynthesis: structure, biogenesis and their potential as drug targets. Future Microbiol. 7, 129–147 [DOI] [PubMed] [Google Scholar]
- 6. Mishra A. K., Driessen N. N., Appelmelk B. J., Besra G. S. (2011) Lipoarabinomannan and related glycoconjugates: structure, biogenesis and role in Mycobacterium tuberculosis physiology and host-pathogen interaction. FEMS Microbiol. Rev. 35, 1126–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chatterjee D., Hunter S. W., McNeil M., Brennan P. J. (1992) Lipoarabinomannan: multiglycosylated form of the mycobacterial mannosylphosphatidylinositols. J. Biol. Chem. 267, 6228–6233 [PubMed] [Google Scholar]
- 8. Chatterjee D., Lowell K., Rivoire B., McNeil M. R., Brennan P. J. (1992) Lipoarabinomannan of Mycobacterium tuberculosis: capping with mannosyl residues in some strains. J. Biol. Chem. 267, 6234–6239 [PubMed] [Google Scholar]
- 9. Chan J., Fan X. D., Hunter S. W., Brennan P. J., Bloom B. R. (1991) Lipoarabinomannan, a possible virulence factor involved in persistence of Mycobacterium tuberculosis within macrophages. Infect. Immun. 59, 1755–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Underhill D. M., Ozinsky A., Smith K. D., Aderem A. (1999) Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc. Natl. Acad. Sci. U.S.A. 96, 14459–14463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stokes R. W., Speert D. P. (1995) Lipoarabinomannan inhibits nonopsonic binding of Mycobacterium tuberculosis to murine macrophages. J. Immunol. 155, 1361–1369 [PubMed] [Google Scholar]
- 12. Briken V., Porcelli S. A., Besra G. S., Kremer L. (2004) Mycobacterial lipoarabinomannan and related lipoglycans: from biogenesis to modulation of the immune response. Mol. Microbiol. 53, 391–403 [DOI] [PubMed] [Google Scholar]
- 13. Strohmeier G. R., Fenton M. J. (1999) Roles of lipoarabinomannan in the pathogenesis of tuberculosis. Microbes Infect. 1, 709–717 [DOI] [PubMed] [Google Scholar]
- 14. Goude R., Amin A. G., Chatterjee D., Parish T. (2009) The arabinosyltransferase EmbC is inhibited by ethambutol in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 53, 4138–4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goude R., Amin A. G., Chatterjee D., Parish T. (2008) The critical role of embC in Mycobacterium tuberculosis. J. Bacteriol. 190, 4335–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Amin A. G., Goude R., Shi L., Zhang D., Chatterjee D., Parish T. (2008) EmbA is an essential arabinosyltransferase in Mycobacterium tuberculosis. Microbiology 154, 240–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Belanger A. E., Besra G. S., Ford M. E., Mikusová K., Belisle J. T., Brennan P. J., Inamine J. M. (1996) The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc. Natl. Acad. Sci. U.S.A. 93, 11919–11924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Escuyer V. E., Lety M. A., Torrelles J. B., Khoo K. H., Tang J. B., Rithner C. D., Frehel C., McNeil M. R., Brennan P. J., Chatterjee D. (2001) The role of the embA and embB gene products in the biosynthesis of the terminal hexaarabinofuranosyl motif of Mycobacterium smegmatis arabinogalactan. J. Biol. Chem. 276, 48854–48862 [DOI] [PubMed] [Google Scholar]
- 19. Zhang N., Torrelles J. B., McNeil M. R., Escuyer V. E., Khoo K. H., Brennan P. J., Chatterjee D. (2003) The Emb proteins of mycobacteria direct arabinosylation of lipoarabinomannan and arabinogalactan via an N-terminal recognition region and a C-terminal synthetic region. Mol. Microbiol. 50, 69–76 [DOI] [PubMed] [Google Scholar]
- 20. Deng L., Mikusová K., Robuck K. G., Scherman M., Brennan P. J., McNeil M. R. (1995) Recognition of multiple effects of ethambutol on metabolism of mycobacterial cell envelope. Antimicrob. Agents Chemother. 39, 694–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mikusová K., Slayden R. A., Besra G. S., Brennan P. J. (1995) Biogenesis of the mycobacterial cell wall and the site of action of ethambutol. Antimicrob. Agents Chemother. 39, 2484–2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Telenti A., Philipp W. J., Sreevatsan S., Bernasconi C., Stockbauer K. E., Wieles B., Musser J. M., Jacobs W. R., Jr. (1997) The emb operon, a gene cluster of Mycobacterium tuberculosis involved in resistance to ethambutol. Nat. Med. 3, 567–570 [DOI] [PubMed] [Google Scholar]
- 23. Torrelles J. B., Khoo K. H., Sieling P. A., Modlin R. L., Zhang N., Marques A. M., Treumann A., Rithner C. D., Brennan P. J., Chatterjee D. (2004) Truncated structural variants of lipoarabinomannan in Mycobacterium leprae and an ethambutol-resistant strain of Mycobacterium tuberculosis. J. Biol. Chem. 279, 41227–41239 [DOI] [PubMed] [Google Scholar]
- 24. Verbelen C., Dupres V., Menozzi F. D., Raze D., Baulard A. R., Hols P., Dufrêne Y. F. (2006) Ethambutol-induced alterations in Mycobacterium bovis BCG imaged by atomic force microscopy. FEMS Microbiol. Lett. 264, 192–197 [DOI] [PubMed] [Google Scholar]
- 25. Sassetti C. M., Boyd D. H., Rubin E. J. (2001) Comprehensive identification of conditionally essential genes in mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 98, 12712–12717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berg S., Kaur D., Jackson M., Brennan P. J. (2007) The glycosyltransferases of Mycobacterium tuberculosis: roles in the synthesis of arabinogalactan, lipoarabinomannan, and other glycoconjugates. Glycobiology 17, 35R–56R [DOI] [PubMed] [Google Scholar]
- 27. Berg S., Starbuck J., Torrelles J. B., Vissa V. D., Crick D. C., Chatterjee D., Brennan P. J. (2005) Roles of conserved proline and glycosyltransferase motifs of embC in biosynthesis of lipoarabinomannan. J. Biol. Chem. 280, 5651–5663 [DOI] [PubMed] [Google Scholar]
- 28. Shi L., Berg S., Lee A., Spencer J. S., Zhang J., Vissa V., McNeil M. R., Khoo K. H., Chatterjee D. (2006) The carboxy terminus of EmbC from Mycobacterium smegmatis mediates chain length extension of the arabinan in lipoarabinomannan. J. Biol. Chem. 281, 19512–19526 [DOI] [PubMed] [Google Scholar]
- 29. Alderwick L. J., Lloyd G. S., Ghadbane H., May J. W., Bhatt A., Eggeling L., Fuetterer K., Besra G. S. (2011) The C-terminal domain of the arabinosyltransferase Mycobacterium tuberculosis EmbC is a lectin-like carbohydrate binding module. PLOS Pathog. 7, e1001299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ollinger J., Bailey M. A., Moraski G. C., Casey A., Florio S., Alling T., Miller M. J., Parish T. (2013) A dual read-out assay to evaluate the potency of compounds active against Mycobacterium tuberculosis. PLoS ONE 8, e60531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sirgel F. A., Wiid I. J. F., van Hleden P. D. (2008) in Mycobacteria Protocols (Parish T., Brown A. C., eds) 2nd Ed., pp. 173–186, Humana Press, New York [Google Scholar]
- 32. Shi L., Torrelles J. B., Chatterjee D. (2008) in Mycobacteria Protocols (Parish T., Brown A. C., eds) 2nd Ed., pp. 23–46, Humana Press, New York [Google Scholar]
- 33. Galagan J. E., Minch K., Peterson M., Lyubetskaya A., Azizi E., Sweet L., Gomes A., Rustad T., Dolganov G., Glotova I., Abeel T., Mahwinney C., Kennedy A. D., Allard R., Brabant W., Krueger A., Jaini S., Honda B., Yu W.-H., Hickey M. J., Zucker J., Garay C., Weiner B., Sisk P., Stolte C., Winkler J. K., Van de Peer Y., Iazzetti P., Camacho D., Dreyfuss J., Liu Y., Dorhoi A., Mollenkopf H.-J., Drogaris P., Lamontagne J., Zhou Y., Piquenot J., Park S. T., Raman S., Kaufmann S. H., Mohney R. P., Chelsky D., Moody D. B., Sherman D. R., Schoolnik G. K. (2013) The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 499, 178–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goude R., Parish T. (2008) in Mycobacteria Protocols (Parish T., Brown A. C., eds) 2nd Ed., pp. 203–216, Humana Press, New York [Google Scholar]
- 35. Horton R. M., Cai Z. L., Ho S. N., Pease L. R. (1990) Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. BioTechniques 8, 528–535 [PubMed] [Google Scholar]
- 36. Pashley C. A., Parish T. (2003) Efficient switching of mycobacteriophage L5-based integrating plasmids in Mycobacterium tuberculosis. FEMS Microbiol. Lett. 229, 211–215 [DOI] [PubMed] [Google Scholar]
- 37. Kaur D., Lowary T. L., Vissa V. D., Crick D. C., Brennan P. J. (2002) Characterization of the epitope of anti-lipoarabinomannan antibodies as the terminal hexaarabinofuranosyl motif of mycobacterial arabinans. Microbiology 148, 3049–3057 [DOI] [PubMed] [Google Scholar]
- 38. Rademacher C., Shoemaker G. K., Kim H. S., Zheng R. B., Taha H., Liu C., Nacario R. C., Schriemer D. C., Klassen J. S., Peters T., Lowary T. L. (2007) Ligand specificity of CS-35, a monoclonal antibody that recognizes mycobacterial lipoarabinomannan: a model system for oligofuranoside-protein recognition. J. Am. Chem. Soc. 129, 10489–10502 [DOI] [PubMed] [Google Scholar]
- 39. Tusnády G. E., Simon I. (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics 17, 849–850 [DOI] [PubMed] [Google Scholar]
- 40. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 41. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., Barton G. J. (2009) Jalview Version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]