

Background: Phage hyaluronate lyases (HLs) of streptococci are intracellular and specifically cleave hyaluronan.
Results: HLs completely degrade glycosaminoglycans, have catalytically independent TSβH domains, and are functional in physiological conditions.
Conclusion: Phage HL has a broad glycosaminoglycan degrading activity that is regulated by its own N-terminal region.
Significance: Degradation of ECM components by HLs may facilitate the diffusion of toxins etc.
Keywords: Bacteriophage, Enzyme Catalysis, Glycosaminoglycan, Hyaluronate, Streptococcus, Domain, Hyaluronic Acid
Abstract
Streptococcus equi is the causative agent of the highly contagious disease “strangles” in equines and zoonotic meningitis in human. Spreading of infection in host tissues is thought to be facilitated by the bacterial gene encoded extracellular hyaluronate lyase (HL), which degrades hyaluronan (HA), chondroitin 6-sulfate, and dermatan sulfate of the extracellular matrix). The clinical strain S. equi 4047 however, lacks a functional extracellular HL. The prophages of S. equi and other streptococci encode intracellular HLs which are reported to partially degrade HA and do not cleave any other glycosaminoglycans. The phage HLs are thus thought to play a role limited to the penetration of streptococcal HA capsules, facilitating bacterial lysogenization and not in the bacterial pathogenesis. Here we systematically looked into the structure-function relationship of S. equi 4047 phage HL. Although HA is the preferred substrate, this HL has weak activity toward chondroitin 6-sulfate and dermatan sulfate and can completely degrade all of them. Even though the catalytic triple-stranded β-helix domain of phage HL is functionally independent, its catalytic efficiency and specificity is influenced by the N-terminal domain. The phage HL also interacts with human transmembrane glycoprotein CD44. The above results suggest that the streptococci can use phage HLs to degrade glycosaminoglycans of the extracellular matrix for spreading virulence factors and toxins while utilizing the disaccharides as a nutrient source for proliferation at the site of infection.
Introduction
Many pathogenic bacteria produce extracellular products that have a tissue damaging effect. Streptococcus equi (group C Streptococcus) causes a number of suppurative mucosal diseases, including strangles (1). The nasopharyngeal infection with S. equi spreads rapidly to the lymph nodes of the head and neck and is usually associated with depression, loss of appetite, pyrexia, etc. Abscesses can form in other body organs and turn fatal when ruptured (1, 2). The organism can cause zoonotic meningitis in humans associated with infected horses (3). The current antibiotic therapy to treat these mucosal diseases is ineffective and necessitates effective treatment by other strategies (4). The development of an effective vaccine has been slow, and the novel vaccine targets include cell surface and extracellular virulence factors (4, 5). S. equi has many virulence factors and toxins in common with the closely related StreptococcuS. pyogenes (group A Streptococcus) such as M protein, hyaluronan capsule, hyaluronate lyase (HL),4 C5a peptidase, DNases, pyrogenic exotoxins, fibronectin binding proteins, etc. (1, 6–9). S. pyogenes causes a wide range of mucosal and invasive human infections including scarlet fever, pharyngitis, and necrotizing fasciitis. Several of the virulence factors, toxins, etc., produced by these two species are now known to be bacteriophage-encoded and include the HL, super antigen pyrogenic exotoxins (Spe) A and C, DNase, etc. (9, 10).
HLs show considerable complexity and heterogeneity and are encoded by the bacterial genes or the prophage genes (9). The streptococcal extracellular HL (encoded by bacterial gene) degrade hyaluronan (HA), chondroitin 6-sulfate (CS-C), and dermatan sulfate (DS), the major components of the host extracellular matrix (ECM) (10, 11). As HA degradation directly helps in the invasion of bacteria and their toxins, HL are also called “spreading factor” (9). The prophage encoded HLs of S. equi 4047 (SEQ2045) and S. pyogenes (HylP, HylP1, and HylP2) lack the signal peptide, and hence, HL activity has not been observed in the extracellular milieu of either S. equi or S. pyogenes infected with temperate bacteriophage (5, 9, 12, 13). The phage HLs have been reported to be specific to HA and do not cleave CS-C and DS (5, 12, 13). Furthermore, unlike the extracellular bacterial HLs of pathogenic streptococci (Streptococcus agalactiae, Streptococcus pneumoniae, S. pyogenes etc.), which completely degrade HA to disaccharides (ΔHA2) by processive mode of cleavage, the phage HLs are reported to produce tetra-decasaccharides (ΔHA4-ΔHA10) only by random cleavage (5, 12–14). The disaccharides are useful nutrients that support bacterial proliferation in the host tissue (9). The potential role of phage HLs has thus been postulated to cleave streptococci HA capsule to reduce the viscosity of HA in the immediate vicinity of phage particles for bacterial lysogenization. The role of phage HLs in pathogenesis or generating nutrient source was ruled out (5, 9, 12–14). Conversely, a few studies have reported an antibody response to phage-encoded HLs during S. equi and S. pyogenes infection, increased tissue permeability after exposure to HL, and conversion of S. pyogenes from non-infective to toxigenic after infection with bacteriophage (9, 15, 16). Most importantly, in the clinical strain S. equi 4047, the only functional HL is of phage origin, and the bacteria-encoded HL is nonfunctional due to the truncation of the catalytic C-terminal region (5, 17). In light of the above observations, it appeared necessary to re-evaluate the role of phage HL as a virulence factor.
The SEQ2045 HL is ∼67% identical to S. pyogenes HylP1 or HylP2, and the difference is mostly due to the presence of (Gly-X-Y)10 motif at the N-terminal region of SEQ2045 (5, 13, 14, 18). The HylP1 and HylP2 are ∼97% identical. The structures of HylP1 and HylP2 are known, and these homo-trimeric enzymes have an unusual triple-stranded β-helical (TSβH) domain. The N-terminal domain of the enzyme forms a mixed globular α/β capping region followed by a coiled region and a segmented α helical coiled-coil region extending up to the core region. The central TSβH domain provides an extended groove in which long stretches of HA could bind for catalysis. The remainder of the structure of the C terminus is composed of a random coil and an α-helical nose (13, 19). Fig. 1 shows the modeled structure of trimeric SEQ2045 using the structure coordinates of HylP1. The streptococcal extracellular HLs are monomeric with globular domains (14).
FIGURE 1.

Phage HL has broad substrate specificity. A, diagrammatic representation of the structures of HylP1 and SEQ2045. The model of SEQ2045 was generated by the Swiss model using HylP1 (PDB ID 2C3F) as template (33). The trimeric assembly was generated by PISA, and the resultant trimeric model was visualized through PyMOL (34). B, SDS-PAGE analysis of the purified protein samples (10 μg). Lanes 1–9 represent molecular mass marker, SEQ20451–372, SEQ204586–372, SEQ2045132–337, SEQ20451–372 (untagged), HylP21–337, GST-SEQ20451–372, GST-SEQ20451–85, and HylP258–337, respectively. The inset shows the enzymatic activity of His-tagged SEQ20451–372 and untagged SEQ20451–372 in the presence of 0.2 mg/ml HA. C, relative enzymatic activity of SEQ20451–372 with CS-C at different enzyme and substrate concentrations. The control represents the activity without the enzyme. D, the relative rate of product appearance as a function of substrate concentration (CS-C) with 1 μg (○), 3 μg (●), and 5 μg (▴) of SEQ20451–372. The enzyme activity values are the mean ± S.D. of minimum three independent experiments.
Here we have studied the mechanistic details of phage HLs and show that they have broad substrate specificity, can completely degrade not only HA but also the other GAGs, and function at physiological conditions. The phage HLs also interact with CD44, a polymorphic transmembrane glycoprotein of humans.
EXPERIMENTAL PROCEDURES
Cloning and Site-directed Mutagenesis
The His-tagged seq20451–372/seq2045, seq204586–372, and seq2045132–337 and untagged seq2045 were amplified from synthesized pIDTSmart-seq2045 (accession number YP_002747260.1). The forward primers used for the seq20451–372/seq2045-untagged, seq204586–372, and seq2045132–337 were 5′-GCA GCT AGC ATG TCA AAA GAA GTT GCA TCA GCT AGG ATA-3′, 5′-GCA GCT AGC ATG ACG ACA GAT TAT AAC CAA CTC CAA AAT AAA CCA-3′, and 5′-GCC GCT AGC ATG ATA GAA CTA GAC AAA AAA TTG AGT TTA ACA-3′, respectively. Similarly the reverse primers used for seq20451–372/seq204586–372, seq2045132–337, and seq2045-untagged were 5′-GCC CTC GAG TTT TTT TAG TAT TAG TTT TTT TAA CTC AGA AAT-3′, 5′-ATT CTC GAG GGG GTC CTT GAG TTT CAG GTT GCC ATC AAT-3′, and 5′-GCC CTC GAG CTA TTT TTT TAG TAT TAG TTT TTT TAA CTC AGA AAT-3′, respectively. For all the samples, PCR reactions were carried out in a total volume of 50 μl with Platinum Pfx DNA polymerase (Invitrogen). The amplification condition was 94 °C for 3 min, 94 °C for 30 s, 53 °C for 1 min, and 68 °C for 1 min (30 cycles) and 68 °C for 10 min. These amplified gene fragments were digested with NheI and XhoI and then ligated into the pET-21d(+) vector (Novagen) cut with the same enzymes. Competent Escherichia coli DH5-α cells were transformed with the plasmid constructs and screened for positive clones. The double mutant seq20451–372(P65D/P74D) and triple mutant seq20451–372(P65D/P74D/P80D) were generated from pIDTSmart-seq2045 using the GeneTailorTM site-directed mutagenesis system (Invitrogen) with mutagenic primer pairs 5′-GGA CCT CAA GGT GAT TCA GGA GAA AGA GGC CTA ACT GGC GAT ATT GGT CCT-3′/5′-ATC GCC AGT TAG GCC TCT TTC TCC TGA ATC ACC TTG AGG TCC CAT AGG GCC GCG CAA GTC-3′ and 5′-CCT CAA GGT GAT TCA GGA GAA AGA GGC CTA ACT GGC GAT ATT GGT CCT CGG GGC GAT GCT GGC AAG CCT-3′/5′-ATC GCC CCG AGG ACC AAT ATC GCC AGT TAG GCC TCT TTC TCC TGA ATC ACC TTG AGG TCC CAT AGG GCC GCG CAA-3′, respectively. For mutants, the amplification condition was 94 °C for 3 min, 94 °C for 30 s, 53 °C for 1 min, 68 °C for 6.5 min (30 cycles) and 68 °C for 10 min. For GST-seq20451–372, the cloned fragment from pET-21d(+) was ligated into pET-41a(+) between NcoI and XhoI sites. GST-seq20451–85 was also cloned in the same vector using primers 5′-GCA CCA TGG ATG TCA AAA GAA GTT GCA TCA GCT AGG ATA-3′ and 5′-ATT CTC GAG TGT CGT TCC AGG CTT GCC AGC AGG TCC CCG-3′ respectively. For FLAG-tagged CD44 (GenBankTM accession number BC004372.1), the nucleotide 56–530 was amplified using human blood cDNA using primers 5′-GCA GCT AGC ATG GCG CAG ATC GAT TTG AAT ATA ACC TGC CGC-3′ and 5′-GCA CTC GAG TCA CTT GTC ATC ATC ATC CTT GTA ATC CAC GTC ATC ATC AGT AGG GTT GCT GGG GTA GAT-3′ and cloned into the pET-21d(+) vector. The PCR condition was 94 °C for 3 min, 94 °C for 30 s, 44.5 °C for 40 s, 68 °C for 1 min (5 cycle) followed by 94 °C for 30 s, 52.5 °C for 40 s; 68 °C for 1 min (30 cycles) and 68 °C for 10 min. The resultant plasmid was termed CD44158FT. The His-tagged CD44 (CD44158His) was cloned as described before (20). The His-tagged isocitrate lyase (21) was subcloned into pET41a downstream of GST (GST-ICL). For the expression of SEQ2045 in the HCT 116 cells, the seq2045 gene was cloned in pEGFP-C1 vector (Clontech) downstream of GFP using forward primer 5′-CCG TCG AAT TCT TCA AAA GAA GTT GCA TCA GCA AGG ATA CAG-3′ and reverse primer 5′-CTA GGA TCC CTA TTT TTT TAG TAT TAG TTT TTT TAA CTC AGA AAT-3′ between the EcoRI and BamHI restriction sites. The DNA sequencing of pIDTSmart-seq2045 and other amplified genes confirmed the homogeneity of the sequences.
Preparation of Recombinant Proteins
Plasmid constructs expressing SEQ20451–372/SEQ2045 (full-length SEQ2045 with the C-terminal His tag), SEQ204586–372 (without N-terminal α/β capping region and Gly-X-Y motif), SEQ2045132–337 (TSβH domain), SEQ20451–372(P65D/P74D), SEQ20451–372(P65D/P74D/P80D), SEQ20451–372 (untagged), GST-SEQ20451–372, GST-SEQ20451–85, SagHL, HylP21–337, and HylP258–337 were introduced into the BL21(DE3) cells and initially grown in Luria-Bertani media with the desired antibiotics at 30 °C until A600 ∼ 0.6 and later induced at 20 °C with 0.5 mm isopropyl-1-thio-β-d-galactopyranoside. Induced cultures were further grown for 8 h with shaking at the same temperature. The cell lysates for His-tagged proteins were loaded onto a nickel-nitrilotriacetic acid column pre-equilibrated with buffer containing 20 mm Tris-Cl, pH 7.5, and 0.4 m NaCl and washed with the same buffer followed by 20 and 40 mm imidazole buffer. The proteins were eluted with a linear gradient of 40–400 mm imidazole buffer and dialyzed against buffer containing 20 mm Tris-Cl, pH 7.2, 0.1 m NaCl. The cell lysate of untagged SEQ20451–372 was suspended in the lysis buffer containing 50 mm HEPES, 10 mm EDTA, pH 7.0, cleared, and loaded onto a CM-Sepharose column equilibrated with same buffer. The column was initially washed with 100 ml of lysis buffer and subsequently with the same buffer containing 200 mm NaCl. The protein was eluted using a linear gradient of 200–500 mm NaCl, and fractions obtained were tested for the required protein by enzymatic assay and SDS-PAGE. The protein was dialyzed, concentrated, and finally purified by passing through the size exclusion column pre-equilibrated with buffer containing 20 mm Tris-Cl, pH 7.2, and 0.1 m NaCl. The purity of recombinant proteins was checked by SDS-PAGE. FLAG-tagged CD44 plasmids were further introduced into RosettaTM (DE3) pLysS and grown in Luria-Bertani media containing 34 μg/ml chloramphenicol plus 100 μg/ml ampicillin at 37 °C until A600 ∼ 0.6. The cells were induced with 0.1 mm isopropyl-1-thio-β-d-galactopyranoside at 20 °C and further grown for 4 h at the same temperature. The cells were harvested and washed, and the resultant pellet was stored at minus 80 °C until further use. The CD44158His was purified as described previously (20).
Activity Assay
The activity of the enzyme was determined by measuring its ability to break down HA or CS-C or DS to unsaturated disaccharides (5, 22). The polymeric HA of 3–5.8 MDa, 851 kDa–1.19 MDa, 66–90 kDa, 21–40 kDa, and 10 kDa were purchased from Sigma and Lifecore Biomedicals. The approximate monosaccharide contents (mers) were calculated considering the molecular mass of a monosaccharide is ∼200 Da (14). The theoretical monomer contents in the purchased polysaccharides are approximately HA15000–29000, HA4250–6000, HA330–450, HA100–200, and HA50.
For kinetic measurements, 30 nm concentrations of enzyme and varying concentrations of HA15000-29000/HA (0.025–0.6 mg/ml) were taken. The progress of the reaction was linear over the time of measurement within the precision of the instrument. The initial velocity of the reaction was determined from the increase in absorbance over the first 1 min of the reaction. The data for initial velocity (Vi) from each experiment in which the concentration of substrate (S) was varied were fitted into the Michaelis-Menten equation with a nonlinear regression program using Graph Pad Prism 3 and expressed as the mean ± S.D. from at least three independent experiments. For all experiments goodness of fit statistics showed that R2 and correlation values were >0.9. For enzymatic activity measurements, the required chemicals, buffers, and other components were treated to remove metal ion contamination as described previously (22).
The enzymatic activity of SEQ20451–372 (30 nm) with 0.2 mg/ml of HA was taken with varying concentrations of CD44158His (5–40 nm) by using two approaches. In the first assay, SEQ20451–372 was preincubated with varying concentrations of CD44158His in HEPES, pH 7.2, 0.1 m NaCl for 1 h at 25 °C. Similarly, in second assay HA was preincubated with varying concentrations of CD44158His. In both the assays, the reaction was initiated by adding either HA or HL exactly after 1 min at 25 °C to the reaction mixture and monitoring the absorbance at 232 nm (22).
Fluorescence Anisotropy Experiment
The SEQ20451–372 and SEQ204586–372 (30 nm) were labeled with fluorescein isothiocyanate (FITC) and titrated with polymeric HA and CS-C (0.025–0.8 mg/ml) at 4 °C with and without EDTA (2 mm). The reading was taken on PerkinElmer Life Sciences LS 50B luminescence spectrometer in a 10-mm path length quartz cell with excitation at 495 nm and emission at 525 nm.
Characterization of GAG Fragments
Size exclusion chromatography experiments were carried out with Superdex Peptide 10/300 GL column on AKTA FPLC (GE Healthcare). The column was calibrated with various molecular mass standard markers and equilibrated with 0.1 n acetate buffer, pH 3.0, before use. ∼600 μl of the sample was loaded onto the column at 25 °C with a flow rate of 0.5 ml/min and detection at 232 nm. Unsaturated HA/CS-C oligosaccharides (ΔHA10, ΔHA8, HA6, ΔHA4, and ΔHA2/ΔCS-C2–10) were obtained from the cleavage of HA/CS-C (0.5 mg/ml) by 3.5 μg of enzyme for 16 h at 25 °C. The molecular masses of purified fragments were determined by electrospray ionization-mass spectrometry (MS). The purified HA and CS-C oligosaccharides were lyophilized, and MS detection was performed to direct infusion of compound using a Harvard infusion pump (Bedford, MA) on API 4000 (Q-trap) triple quadruple instrument (AB-SCIEX, Toronto, Canada). A turbo electrospray interface was used in both positive and negative ionization modes.
The extensively digested sample of HA and CS-C was also analyzed by the liquid chromatography system (Shimadzu, Kyoto, Japan) composed of quaternary pump LC-20AD, thermostated auto sampler SIL-HCT, degasser DGU-20A3, and column oven CTO-20A. The chromatographic separation was achieved with automated injection of 20-μl samples on reversed phase phenomenex column, Luna 5u C8 (250 × 4.6 mm, 5 μm; Hyderabad, India) at 40 °C with the run time of 10 min. The mobile phase eluent was acetonitrile, methanol, 5 mm ammonium acetate solution with 0.1% acetic acid 30:30:40 (v/v/v) with a flow rate of 0.5 ml/min. UV detection was at 232 nm.
Co-immunoprecipitation Assays
The cleared and pretreated E. coli cell lysate (with testicular hyaluronidase) of FLAG-tagged CD44158FT was incubated with the 100 μg of His-tagged SEQ20451–372 and SEQ2045132–337 for 2 h at 25 °C with gentle shaking. Subsequently it was passed through a nickel-nitrilotriacetic acid affinity column (Qiagen) equilibrated with lysis buffer (50 mm Hepes, pH 7.2, 0.1 m NaCl) and washed with the same buffer containing 20 and 40 mm imidazole. The proteins were eluted using lysis buffer containing 300 mm imidazole. The interacting partners were resolved by SDS-PAGE and detected by standard Western blotting techniques using corresponding primary and secondary antibodies and developed in LAS4000 ImageQuant.
For other assays the mammalian cells were grown overnight in a 150-mm tissue culture dish and transfected with expression plasmids using Lipofectamine 2000 reagent. After 48 h, cells were washed twice in cold phosphate-buffered saline (PBS) and lysed in buffer (25 mm HEPES, 0.4 m NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 1% Nonidet P-40) containing protease inhibitor for 1 h on ice. Cell lysates were clarified by centrifugation at 16,000 × g for 15 min at 4 °C, and protein content of the supernatant was determined by BCA assay kit (Thermo Scientific). Six hundred microgram of total protein was diluted in 500 μl of low salt lysis buffer (50 mm HEPES, 15–150 mm NaCl, 1.5 mm MgCl2, 1 mm EDTA, 10% glycerol, 1% Triton X-100) and immunoprecipitated with 3 μg of anti-GFP antibody by overnight incubation at 4 °C. Immunocomplexes were then precipitated with protein A/G-agarose beads and eluted with Laemmli buffer by boiling for 10 min. Proteins were resolved by SDS-PAGE and transferred onto a PVDF membrane. After 1 h of blocking with 5% nonfat dry milk in Tris-buffered saline (TBS)-Tween at room temperature, the membrane was probed overnight with the desired antibodies at 4 °C. Membranes were then washed three times with TBS-Tween and incubated with peroxidase-conjugated secondary antibody for 1 h at room temperature, and specific protein bands were detected with an enhanced chemiluminescence (ECL) reagent.
Immunostaining of Cells for Confocal Microscopy
HCT 116 cells were seeded on sterile coverslips in a 6-well tissue culture plate. After overnight incubation at 37 °C, cells were transfected with EGFP-C1 or recombinant EGFP-C1 plasmid expressing SEQ20451–372 (GFP-SEQ2045) using Lipofectamine 2000 transfection reagent. At 48 h after transfection, cells were fixed with 4% paraformaldehyde in PBS and probed overnight with anti-CD44 antibody at 4 °C. After 3 washes in PBS, cells were incubated with Alexafluor 594-conjugated anti-rabbit antibody for 1 h at room temperature. Cells were then washed three times in PBS and mounted on glass slides using ProlongR Gold antifade mountant reagent with DAPI. Control samples (only EGFP-expressing cells) were used for blanking signals in the red channel by adjusting gain/offset and laser intensity before analyzing the test samples. Confocal images were acquired using Carl Zeiss LSM 510 META confocal microscope equipped with a Plan Apochromat 63× oil/1.4 NA differential interference contrast objective.
RESULTS
Unlike the case of well studied virulence factors, information regarding the role of phage HLs in spreading the streptococcal infection and in its various aspects remain controversial. On one hand the role of phage HL is proposed only in breaking the HA capsule for bacterial lysogenization, but on the other hand it appears that the phage HL may function like bacterial extracellular HLs. Here we systematically investigated why the enzyme shows absolute substrate specificity for HA, how the enzyme could act on a broad range of substrate, and other structure-function aspects.
Preparation of HLs
The cloned and overexpressed proteins SEQ20451–372, SEQ204586–372, SEQ2045132–337, SEQ20451–372 (untagged), HylP21–337, GST-SEQ20451–372, GST-SEQ20451–85, and HylP258–337 were purified by the methods described under “Experimental Procedures.” The purity and molecular masses of the purified recombinant proteins were determined by SDS-PAGE and were similar to those deduced from their primary amino acid sequences (Fig. 1B). The observed enzymatic activity of full-length SEQ2045 (SEQ20451–372) with and without the His-tag was the same (Fig. 1B, inset).
Phage HLs Show Substrate Inhibition
The HLs (bacterial as well as prophage encoded) act readily on HA, although only the bacterial extracellular HL degrades CS-C, albeit only with ∼8–10% as compared with HA (15). The reason for phage HLs to have absolute substrate specificity for HA is not known. It appears from previous studies that to determine the activities of phage HLs, a high CS-C concentration (>2 mg/ml) and low phage HL concentrations (10 ng-2 μg/ml) were used (5, 12, 13). We observed broad substrate specificity of phage HLs in degrading CS-C and DS. SEQ2045 (up to 5 μg/ml) had minimal activity with CS-C (2 mg/ml). However, the activity improved substantially when concentrations of both enzyme (3.5 μg/ml, 30 nm) and substrate (0.5 mg/ml) were decreased (Fig. 1C). A similar pattern of activity was observed with DS (data not shown). HylP21–337 also showed similar specificities for these GAGs (data not shown).
We further analyzed whether the observed substrate inhibition was due to substrate saturation or enzyme inhibition. An initial linear rate of product appearance as a function of substrate concentration was plotted, and the observed hyperbolic curve suggests the saturation of reaction rate at high substrate concentrations and no direct inhibition due to substrate binding (Fig. 1D). We also obtained the enzyme kinetics of HLs with a broader range of substrate concentrations as per the Michaelis-Menten model. For SEQ20451–372 with HA/CS-C, the observed Km and Vmax were 0.357 ± 0.03/0.182 ± 0.002 mm and 43.34 ± 1.5/6.3 ± 0.27 μmol.l−1·min−1, respectively. Similarly Km and Vmax for HylP21–337 with HA/CS-C were 0.445 ± 0.03/0.186 ± 0.002 mm and 55.6 ± 1.54/7.5 ± 0.25 μmol.l−1·min−1, respectively.
N-terminal Region Modulates the Substrate Specificity
SEQ2045 has a long and flexible N-terminal region that can potentially span the central TSβH domain. To study the structural flexibility and the role of the N-terminal domain in affecting the saturation of reaction rate, the enzymatic activities of SEQ20451–372 and SEQ204586–372 were measured with different HA-mers (Fig. 2A). SEQ20451–372 showed almost similar activity with all the substrates, whereas the SEQ204586–372 showed comparatively enhanced activity with larger HA-mers only. The activities were nearly the same for HA50. The results suggest that either the flexible N-terminal domain interacts non-specifically with large polymeric HA or the enzyme active site pocket is enlarged in the truncated enzyme and shows higher affinity for HA or other GAGs. Furthermore, the observed Kcat/Km of SEQ20451–372 (17.74) and SEQ204586–372 (28.50) for HA15000–29000 also suggests an influence of the N-terminal region in the enzyme specificity.
FIGURE 2.

Phage HLs activity is influenced by the flexible N-terminal region. A, enzymatic activity profile of SEQ20451–372 and SEQ204586–372 with different HA-mers (500 nm). The activity of SEQ20451–372 with HA15000–29000/HA was taken as 100%. B, enzymatic activity profile of SEQ20451–372, SEQ204586–372, and GST-SEQ20451–372 with HA, CS-C, and DS. The activity was measured with 3.5 ng of enzyme and 0.2 mg/ml substrate. C, enzymatic activity profile of SEQ204586–372 in the presence of GST or GST-SEQ20451–85. D, binding of SEQ20451–372 with HA and CS-C, as measured by the fluorescence anisotropy titration experiment at 4 °C in the presence or absence of 2 mm EDTA. For both the figures the data points of FITC-labeled SEQ20451–372 titrated with HA and CS-C are represented as filled squares (■) and open squares (□), respectively. The data points of FITC-labeled SEQ204586–372 titrated with HA and CS-C are represented as filled circles (●) and open circles (○), respectively. The lines represent the best fit to the average data of three individual measurements. The enzyme activity values are the mean ± S.D. of minimum three individual experiments.
Because the N-terminal region does not directly influence catalysis, we tested whether native and truncated SEQ20451–372 have variable specificities for CS-C and DS. Although the full-length SEQ20451–372 showed a preference for HA and only limited activity against other GAGs (15–18%), the truncated SEQ204586–372 showed substantial activity (>40%) with CS-C and DS (Fig. 2B). HylP2 constructs also showed a similar activity pattern (data not shown). We subsequently measured the enzymatic activity of GST-SEQ20451–372 and observed that the construct has broad substrate specificity and its activity pattern is also similar to SEQ204586–372 (Fig. 2B). The above results further suggest that the observed substrate inhibition for phage HLs (Fig. 1B) is due to the conformational flexibility rendered by the N-terminal domain.
To see the influence of N-terminal domain (1–85 amino acids) in enzyme catalysis, the activity of SEQ204586–372 with and without 1–85 amino acids (GST-SEQ20451–85) was measured. No significant change in the enzymatic activity of SEQ204586–372 was observed in the presence of the 1–85-amino acid domain, emphasizing its nonspecific regulatory role in vitro (Fig. 2C). We also checked the enzyme specificities to help differentiate the HA binding ability from HA cleavage using fluorescence anisotropy at 4 °C with and without a chelator (Fig. 2D). The binding efficiency of GAGs to HL is similar irrespective of the presence or absence of EDTA at 4 °C. CS-C seems to bind to HL even at low concentrations. The observed Kd for SEQ20451–372 at 4 °C with and without EDTA were 0.44 ± 0.068 (HA)/0.089 ± 0.014 (CS-C) and 0.42 ± 0.07 (HA)/0.089 ± 0.014 (CS-C), whereas the corresponding values for SEQ204586–372 were 0.55 ± 0.067 (HA)/0.19 ± 0.027 (CS-C) and 0.52 ± 0.053 (HA)/0.17 ± 0.03 (CS-C), respectively.
These findings suggest that, the flexibility of the N-terminal α/β domain of SEQ20451–372 modulates the conformation of the catalytic pocket for better positioning and catalysis of substrates. When the conformational flexibility of the N-terminal domain is absent/restricted as in case of SEQ204586–372/GST-SEQ20451–372, the enzyme catalytic pocket attains a conformation more suitable for binding and processing of other GAGs. It is reasonable to think that in vivo there may be some proteins associated with the large flexible N-terminal region of the enzyme, and it may not be as flexible as observed in vitro.
Phage HL Completely Degrades the GAGs
The recombinant HLs (SEQ20451–372, SEQ204586–372, andGST-SEQ20451–372), despite having similar active site residues, show different catalytic properties. This implies differences in the active site geometry and hence the GAGs degradation products. We analyzed the digested products of HA and CS-C using Superdex Peptide 10/300 GL size exclusion column (fractionation range of 100–7000 Da) on AKTA FPLC (Fig. 3, A and B) followed by MS/MS. SEQ20451–372 degrades polymeric GAGs into ΔHA2, ΔHA4, ΔHA6, ΔHA8, and ΔHA10/ΔCS-C2–10. In previous reports the phage HL-mediated digestion of HA is limited to ΔHA4-ΔHA8 only as no ΔHA2 was observed (5, 15). SEQ204586–372 produced ΔHA2-ΔHA8/ΔCS-C2–6. Surprisingly, the GST-SEQ20451–372 exclusively produced disaccharides (ΔHA2/ΔCS-C2) as the end product of HA/CS-C digestion. The disaccharide is a suitable carbon source for bacteria at the site of proliferation and is the exclusive digestion product of bacterial extracellular HLs. The controls in the void volume (Fig. 3, A and B) show some auto degradations of HA/CS-C. TheHylP21–337 also produced ΔHA2-ΔHA10/ΔCS-C2-10 (data not shown). The extensive digestion products of HA and CS-C by HLs were further analyzed and confirmed by HPLC (Fig. 3C).
FIGURE 3.

Phage HL completely degrades GAGs. A, the size-exclusion chromatography profiles represent undigested polymeric HA and HA digested by the S. agalactiae hyaluronate lyase (SagHL), SEQ20451–372, SEQ204586–372, and GST-SEQ20451–372, respectively. The table shows the molecular mass of HA fragments (disaccharide to octasaccharide) obtained after digestion with SEQ20451–372. B, the size-exclusion chromatography profiles represent undigested polymeric CS-C and CS-C digested by SagHL, SEQ20451–372, SEQ204586–372, and GST-SEQ20451–372. The table shows the molecular masses of CS-C fragments (disaccharide to octasaccharide) obtained after digestion with SEQ20451–372. C, high performance liquid chromatography analysis of the extensive digestion products of HA (a) and CS-C (b) by SEQ2045 and SagHL.
Gly-X-Y Motif Has a Role in Substrate Regulation and Stability
Gly-X-Y repeat motifs in certain proteins make them adaptable to a range of protein assemblies (23). The Gly-X-Y motif may provide structural constraints to HLs needed for substrate selection, anchoring, targeting, etc.
Calcium ions have been reported to influence the activity of phage HLs (5, 12–13, 17). We checked the influence of CaCl2 on the activity of SEQ20451–372 (contains the Gly-X-Y motif), SEQ204582–372 (without Gly-X-Y motif), and HylP21–337 (does not contain Gly-X-Y motif) (Fig. 4A). For SEQ20451–372, an initial increase in enzyme activity up to 20 mm CaCl2 followed by a plateau and then a steady decrease in activity was observed. The reduced activity only in the case of SEQ20451–372 suggests a direct interaction of calcium ions with the Gly-X-Y motif. HylP also shows a similar interaction with Ca2+ (24). The inset of Fig. 4A summarizes the result of the effect of calcium chloride on the function of SEQ20451–372. The activity of SEQ20451–372 was independent of Ca2+ and was the same for smaller HA-mer. The result suggests that calcium ions at low concentration change the conformation of polymeric substrates suitable for optimal for maximum catalysis.
FIGURE 4.

Gly-X-Y motif influences the enzymatic activity and stability of phage HLs. A, enzymatic activity profile of SEQ20451–372 (▴), SEQ204586–372 (●), and HylP21–337 (■) increasing concentrations of CaCl2. The inset shows the enzymatic activity profile of SEQ20451–372 with HA-mers in the presence and absence of CaCl2. B, ClustalW alignment of the Gly-X-Y region of phage HLs. Prolines of the Gly-X-Y motif are marked as red. C, enzymatic activity profile of SEQ20451–372, double mutant SEQ20451–372(P65D/P74D), and triple mutant SEQ20451–372(P65D/P74D/P80D) with HA-mers. D, plot of the fractional change in the fluorescence wavelength maxima at increasing concentrations of urea for SEQ20451–372 (●), SEQ204586–372 (○), and HylP21–337 (■), respectively. The excitation and emission wavelengths to record the spectra were 280 and 300–400 nm, respectively.
SEQ2045 and HylP both consist of the (Gly-X-Y)10 repeat motif near the N terminus and contain seven (at position 59, 62, 65, 74, 77, 80, and 84) and five (at position 59, 62, 77, 80, and 84) prolines (bold numbers represent conserved prolines), respectively (Fig. 4B) (5, 12). To check the binding or interaction of HA with Gly-X-Y motif of SEQ2045, we introduced double point mutations (P65D/P74D) and triple point mutations (P65D/P74D/P80D) in the motif. The mutations are expected to affect the local electrostatic potential, although the non-conserved mutation should not affect the overall motif structure. As shown in Fig. 4C, the activity of mutants was reduced up to 30%, suggesting a direct interaction of HA with the Gly-X-Y motif. Furthermore the full-length SEQ20451–372 and the mutants showed no marked difference in the enzymatic activity with smaller HA-mers, suggesting a nonspecific regulatory role of Gly-X-Y motif in HA binding during catalysis.
SEQ2045 and HylP2 are highly homologous and have similar structures except the latter lack the Gly-X-Y motif. SEQ2045 is more stable than HylP2 as observed from their denaturation patterns. The Cm (concentration of denaturant where 50% denaturation is observed) of SEQ20451–372, SEQ204586–372, and HylP21–337 were 3.25, 2.5, and 1.0 m, respectively, which primarily suggests the role of Gly-X-Y motif in HL stabilization (Fig. 4D).
CD44 Interacts with Phage HL
CD44 is a polymorphic transmembrane glycoprotein recognized as the main cell surface receptor for HA. CD44 influences the diverse physiological functions such as cell aggregation, cell-cell, and cell-substrate adhesion (20, 25). We checked the interaction of CD44158FT (first 158 residues of the human CD44 ectodomain containing the link module and HA binding residues) with the SEQ20451–372, SEQ204586–372, and SEQ2045132–337. Fig. 5A shows the result of the co-immunoprecipitation experiment where the interaction between His-tagged SEQ2045 constructs and FLAG-tagged CD44 was investigated. The result suggests an interaction of CD44158FT with the central TSβH domain (SEQ2045132–337) of the phage HL. Furthermore, the CD44158His enhanced the activity of SEQ20451–372 in a concentration-dependent manner. The activity of SEQ20451–372 gradually increased in the presence of increasing concentrations of CD44158His either when CD44158His was preincubated with SEQ20451–372 and the reaction was initiated with HA or when it was preincubated with the HA and the reaction was initiated with SEQ20451–372 (Fig. 5B and inset). The binding efficiency (Kd) of CD44158His to phage HLs was ∼1.167 nm. Similar results were obtained when HylP2 was titrated with CD44 (data not shown). The CD44 may be instrumental in optimum HA degradation by phage HLs in vivo.
FIGURE 5.
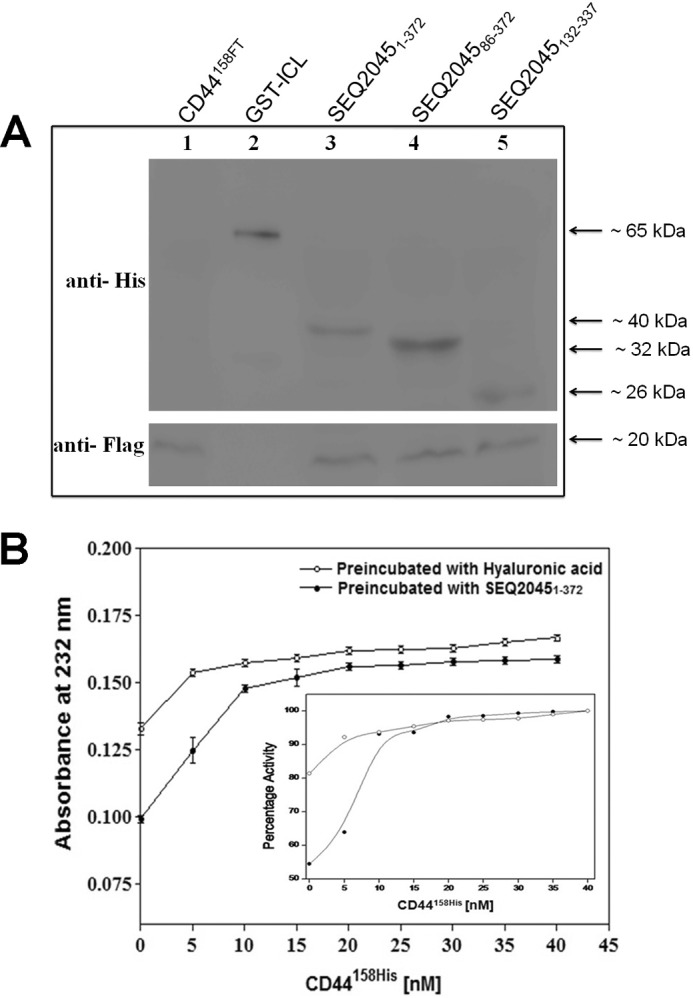
CD44 interacts and influences the activity of phage HL. A, co-immunoprecipitation from the bacterial cell lysate containing FLAG-tagged CD44 (CD44158FT) with no mock HL (lane 1), His-tagged GST-ICL (lane 2), CD44158FT + SEQ20451–372 (lane 3), CD44158FT + SEQ204586–372 (lane 4), and CD44158FT + SEQ2045132–337 (lane 5). B, enzymatic activity profile of SEQ20451–372 at increasing concentrations of CD44158His (5–40 nm). The curve (●) represents SEQ20451–372 (30 nm) preincubated with increasing concentrations of CD44158His before the reaction was initiated with 0.2 mg/ml of HA. The second curve (○) represents HA preincubated with increasing concentration of CD44158His before the reaction was initiated with SEQ20451–372. The inset shows the percent activation of SEQ20451–372 activity in presence of CD44158His.
We subsequently checked the SEQ2045-CD44 interaction in mammalian cells (HCT 116 cells) through two different approaches. In our initial experiment cells were transfected with EGFP-C1 or EGFP-C1 plasmid expressing SEQ20451–372 (GFP-SEQ2045) and analyzed microscopically after immune-labeling CD44 with Alexafluor 594.
The confocal microscopic data showed co-localization of CD44 and SEQ2045 mostly at the cell periphery (Fig. 6A). The result was further confirmed by observing the overlapping signals in co-localized cells. In the case of EGFP-C1-transfected cells, only a diffusely distributed green fluorescence was observed (Fig. 6A). To confirm the specific interactions between SEQ20451–372 and CD44, protein lysates of cells transfected with EGFP-C1 and EGFP-SEQ2045 were incubated overnight with anti-GFP antibody and probed with anti-CD44 antibody (Fig. 6Ba). The immunoprecipitation result was further confirmed by reverse co-immunoprecipitation experiments where pulldown was done with anti-CD44 antibody and probed with anti-GFP antibody (Fig. 6Bb). The Western blot analysis of immunoprecipitates together with the immunofluorescence data indicates the interaction of CD44 and SEQ2045 in the mammalian cells.
FIGURE 6.

Endogenous CD44 (90–95 kDa) interacts with SEQ2045. A, microscopic images of HCT 116 cells after 48 h of transfection with EGFP-C1expressing SEQ2045 (EGFP-SEQ2045) (upper panel) and EGFP-C1 expressing GFP only (lower panel). The cells were first incubated with anti-CD44 antibody followed by Alexafluor 594-conjugated secondary antibody. The co-localized SEQ2045 with CD44 in the cells (a and b) are also shown with corresponding overlapped signals. IP, immunoprecipitate. B, the lysates of transfected HCT 116 cells were subjected to immunoprecipitation using either anti-GFP antibody (a) or anti-CD44 antibody (b). The precipitates were subsequently probed by using anti-CD44 antibody (a) and anti-GFP antibody (b), respectively.
Phage HLs Have Broad Substrate Specificity in Physiological Buffers
The interaction of phage HLs with CD44 and their broad substrate specificity to cleave not only HA but also other GAGs suggest their possible functional role during infection. We further evaluated the enzymatic activity of SEQ20451–372 and HylP21–337 in physiological range buffers to see their ability to degrade ECM components. As shown in Fig. 7, SEQ20451–372 and HylP21–337 had substantial activities in TBS and PBS. The observed activity of HLs was not only for HA but also for CS-C and DS, suggesting its in vivo significance. We also observed substantial activity of HLs in the presence of cell lysates (data not shown).
FIGURE 7.
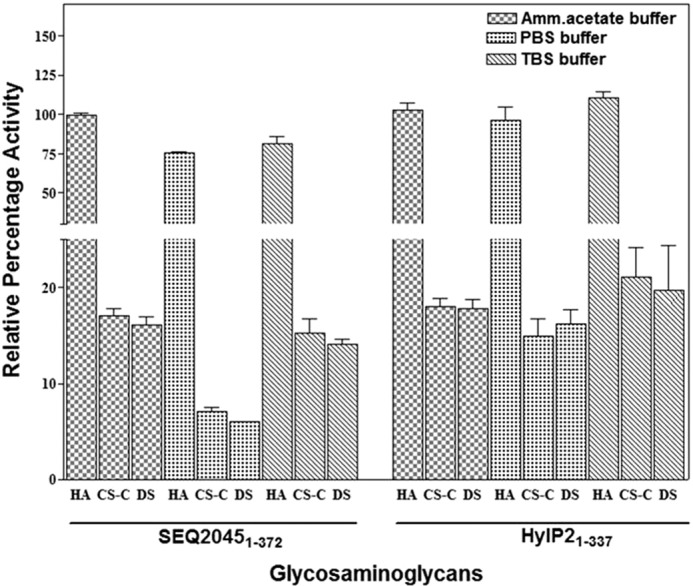
Phage HLs are active in the physiological buffers. Enzymatic activity profile of SEQ20451–372 and HylP21–337 in ammonium acetate buffer containing 10 mm CaCl2, pH 6.5, TBS, pH 7.2, and PBS, pH 7.2.
DISCUSSION
The specificity of phage HL for HA, its lack of secretion, and limited cleavage of HA (only up to tetrasaccharide and not up to disaccharide) suggested its role is limited to bacterial lysogenization. Conversely, the antibody response to phage HLs in the case of S. equi and S. pyogenes during in vivo infection, conversion of S. pyogenes from non-infective to toxigenic upon infection with bacteriophage, production of phage-encoded virulence factors and toxins, phage mobilization etc. has also been observed (5, 7, 9, 15–17, 26–28). These studies hint that the role of phage HL is perhaps not just limited to the breaking of host HA capsule for lysogenization but also in conferring virulence to the host bacterium and disease pathogenesis upon induction. Soluble phage-inducing factors of mammalian hosts influence the production of virulence factors, toxins, and lytic conversion of bacteriophage (27–30). Infection by S. equi 4047 most likely spreads with the degradation of capsule and cell lysis followed by the release of HL and other phage virulence factors and toxins to the ECM.
The major component of connective tissue ECM glycosaminoglycans is HA, and the activity of phage HLs, albeit low for other substrates like CS-C and DS, could be sufficient for spreading virulence factors and toxins required for infection. The broad substrate specificity, interaction with CD44, and activity in physiological buffer are suggestive of the role of phage HLs during infection. The phage HL may act as a primary (as in the case of S. equi 4047 infection) or supplemental (as in the case of S. pyogenes infection) virulence factor. The role of phage HLs beyond lysogenization is also supported by the fact that other phages of a similar family (C1 phage, LambdaSa1 and -2 etc.), without possessing HL in their tail fiber, are still capable of lysogenizing HA-encapsulated Group A and Group C streptococcus.
The ground substance of ECM of connective tissue provides a mechanism of defense by resisting the penetration of infectious agents and their extracellular products. The hyaluronidases depolymerize the major component of connective tissue ECM i.e. HA to the smallest possible substrate (ΔHA2-ΔHA10). Decreased viscosity results in increased permeability in ECM and potentially increased spread of the microorganism, toxins, etc. The bee venom and ovine testicular hyaluronidase produce ΔHA4, snake and spider venom hyaluronidases produce ΔHA4-ΔHA6, bacterial HLs of S. pneumoniae and S. agalactiae produce ΔHA2, and bacteriophage HL of S. pyogenes and S. equi not only produce ΔHA4-ΔHA10 (Refs. 5, 12–15, and 31 and this paper) but also ΔHA2 (this paper). This product analysis suggests that the phage HL-mediated digestion could also serve the purposes of diffusion of virulence factors and toxins, like others. In addition, the disaccharide product could also be used as a nutrient that supports bacterial growth at the site of infection. The observed exclusive ΔHA2 in the case of digestion by GST-SEQ2045 hints at a possible interaction of some host proteins with the N-terminal region of HLs and a subsequent similar function of phage and bacterial HLs.
In the mammalian system CD44 is essential for the HA degradation by endogenously (Hyal-1) and exogenously (Hyal-2) expressed hyaluronidases and contributes to the intracellular and extracellular catabolism and degradation of HA (32). The CD44 enhances the rate of HA degradation by SEQ2045 and appears to have a role in the HA degradation during streptococcal infection.
Acknowledgments
We thank Dr. Manju Y. K. and Harish Shukla for helpful discussions. We thank Prof. Bert Vogelstein for providing HCT 116 cell line, Dr. Amogh Anant Sahasrabuddhe for confocal data analysis, and Dr. Manju Y. K. and Dr. J. V. Pratap for critically reading the manuscript.
This work was supported in part by Council of Scientific and Industrial Research (India) Grant YSA001 (to M. S. A.). This is Central Drug Research Institute communication number 8839.
- HL
- hyaluronate lyase
- HA
- hyaluronan
- CS-C
- chondroitin 6-sulfate
- DS
- dermatan sulfate
- SagHL
- S. agalactiae hyaluronate lyase
- ΔHA2
- disaccharide unit of HA
- ΔCS-C2
- disaccharide unit of chondroitin 6-sulfate
- TSβH
- triple-stranded β-helix
- GAG
- glycosaminoglycan
- ECM
- extracellular matrix.
REFERENCES
- 1. Harrington D. J., Sutcliffe I. C., Chanter N. (2002) The molecular basis of Streptococcus equi infection and disease. Microbes Infect. 4, 501–510 [DOI] [PubMed] [Google Scholar]
- 2. Sweeney C. R., Whitlock R. H., Meirs D. A., Whitehead S. C., Barningham S. O. (1987) Complications associated with Streptococcus equi infection on a horse farm. J. Am. Vet. Med. Assoc. 191, 1446–1448 [PubMed] [Google Scholar]
- 3. Waller A. S., Jolley K. A. (2007) Getting a grip on strangles: recent progress towards improved diagnostics and vaccines. Vet. J. 173, 492–501 [DOI] [PubMed] [Google Scholar]
- 4. Elsayed S., Hammerberg O., Massey V., Hussain Z. (2003) Streptococcus equi subspecies equi (Lancefield group C) meningitis in a child. Clin. Microbiol. Infect. 9, 869–872 [DOI] [PubMed] [Google Scholar]
- 5. Lindsay A. M., Zhang M., Mitchell Z., Holden M. T., Waller A. S., Sutcliffe I. C., Black G. W. (2009) The Streptococcus equi prophage-encoded protein SEQ2045 is a hyaluronan-specific hyaluronate lyase that is produced during equine infection. Microbiology 155, 443–449 [DOI] [PubMed] [Google Scholar]
- 6. Proft T., Webb P. D., Handley V., Fraser J. D. (2003) Two novel superantigens found in both group A and group C Streptococcus. Infect. Immun. 71, 1361–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Broudy T. B., Pancholi V., Fischetti V. A. (2002) The in vitro interaction of StreptococcuS. pyogenes with human pharyngeal cells induces a phage-encoded extracellular DNase. Infect. Immun. 70, 2805–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bisno A. L., Brito M. O., Collins C. M. (2003) Molecular basis of group A streptococcal virulence. Lancet Infect. Dis. 3, 191–200 [DOI] [PubMed] [Google Scholar]
- 9. Hynes W. L., Walton S. L. (2000) Hyaluronidases of gram-positive bacteria. FEMS Microbiol. Lett. 183, 201–207 [DOI] [PubMed] [Google Scholar]
- 10. Canchaya C., Desiere F., McShan W. M., Ferretti J. J., Parkhill J., Brüssow H. (2002) Genome analysis of an inducible prophage and prophage remnants integrated in the StreptococcuS. pyogenes strain SF370. Virology 302, 245–258 [DOI] [PubMed] [Google Scholar]
- 11. Menzel E. J., Farr C. (1998) Hyaluronidase and its substrate hyaluronan: biochemistry, biological activities, and therapeutic uses. Cancer Lett. 131, 3–11 [DOI] [PubMed] [Google Scholar]
- 12. Baker J. R., Dong S., Pritchard D. G. (2002) The hyaluronan lyase of StreptococcuS. pyogenes bacteriophage H4489A. Biochem. J. 365, 317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith N. L., Taylor E. J., Lindsay A. M., Charnock S. J., Turkenburg J. P., Dodson E. J., Davies G. J., Black G. W. (2005) Structure of a group A streptococcal phage-encoded virulence factor reveals a catalytically active triple-stranded β-helix. Proc. Natl. Acad. Sci. U.S.A. 102, 17652–17657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stern R., Jedrzejas M. J. (2006) Hyaluronidases: their genomics, structures, and mechanisms of action. Chem. Rev. 106, 818–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pritchard D. G., Lin B., Willingham T. R., Baker J. R. (1994) Characterization of the group B streptococcal hyaluronate lyase. Arch. Biochem. Biophys. 315, 431–437 [DOI] [PubMed] [Google Scholar]
- 16. Frobisher M., Brown J. (1927) Transmissible toxicogenicity of streptococci. Bull. Johns Hopkins Hosp. 41, 167–173 [Google Scholar]
- 17. Sting R., Schaufuss P., Blobel H. (1990) Isolation and characterization of hyaluronidases from Streptococcus dysgalactiae, S. zooepidemicus, and S. equi. Zentralbl. Bakteriol. 272, 276–282 [DOI] [PubMed] [Google Scholar]
- 18. Hynes W. L., Hancock L., Ferretti J. J. (1995) Analysis of a second bacteriophage hyaluronidase gene from StreptococcuS. pyogenes: evidence for a third hyaluronidase involved in extracellular enzymatic activity. Infect. Immun. 63, 3015–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mishra P., Prem Kumar R., Ethayathulla A. S., Singh N., Sharma S., Perbandt M., Betzel C., Kaur P., Srinivasan A., Bhakuni V., Singh T. P. (2009) Polysaccharide binding sites in hyaluronate lyase crystal structures of native phage-encoded hyaluronate lyase and its complexes with ascorbic acid and lactose. FEBS J. 276, 3392–3402 [DOI] [PubMed] [Google Scholar]
- 20. Banerji S., Day A. J., Kahmann J. D., Jackson D. G. (1998) Characterization of a functional hyaluronan-binding domain from the human CD44 molecule expressed in Escherichia coli. Protein Expr. Purif. 14, 371–381 [DOI] [PubMed] [Google Scholar]
- 21. Kumar R., Bhakuni V. (2008) Mycobacterium tuberculosis isocitrate lyase (MtbICL): role of divalent cations in modulation of functional and structural properties. Proteins 72, 892–900 [DOI] [PubMed] [Google Scholar]
- 22. Akhtar M. S., Krishnan M. Y., Bhakuni V. (2006) Insights into the mechanism of action of hyaluronate lyase: role of C-terminal domain and Ca2+ in the functional regulation of enzyme. J. Biol. Chem. 281, 28336–28344 [DOI] [PubMed] [Google Scholar]
- 23. Ramshaw J. A., Shah N. K., Brodsky B. (1998) Gly-X-Y tripeptide frequencies in collagen: a context for host-guest triple-helical peptides. J. Struct. Biol. 122, 86–91 [DOI] [PubMed] [Google Scholar]
- 24. Singh S. K., Malhotra S., Akhtar M. S. (2014) Characterization of hyaluronic acid specific hyaluronate lyase (HylP) from StreptococcuS. pyogenes. Biochimie 102, 203–210 [DOI] [PubMed] [Google Scholar]
- 25. Aruffo A., Stamenkovic I., Melnick M., Underhill C. B., Seed B. (1990) CD44 is the principal cell surface receptor for hyaluronate. Cell 61, 1303–1313 [DOI] [PubMed] [Google Scholar]
- 26. Halperin S. A., Ferrieri P., Gray E. D., Kaplan E. L., Wannamaker L. W. (1987) Antibody response to bacteriophage hyaluronidase in acute glomerulonephritis after group A streptococcal infection. J. Infect. Dis. 155, 253–261 [DOI] [PubMed] [Google Scholar]
- 27. Broudy T. B., Pancholi V., Fischetti V. A. (2001) Induction of lysogenic bacteriophage and phage-associated toxin from group a streptococci during coculture with human pharyngeal cells. Infect. Immun. 69, 1440–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Broudy T. B., Fischetti V. A. (2003) In vivo lysogenic conversion of Tox(−) StreptococcuS. pyogenes to Tox(+) with lysogenic Streptococci or free phage. Infect. Immun. 71, 3782–3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wagner P. L., Waldor M. K. (2002) Bacteriophage control of bacterial virulence. Infect. Immun. 70, 3985–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wagner P. L., Livny J., Neely M. N., Acheson D. W., Friedman D. I., Waldor M. K. (2002) Bacteriophage control of Shiga toxin 1 production and release by Escherichia coli. Mol. Microbiol. 44, 957–970 [DOI] [PubMed] [Google Scholar]
- 31. Kemparaju K., Girish K. S. (2006) Snake venom hyaluronidase: a therapeutic target. Cell Biochem. Funct. 24, 7–12 [DOI] [PubMed] [Google Scholar]
- 32. Harada H., Takahashi M. (2007) CD44-dependent intracellular and extracellular catabolism of hyaluronic acid by hyaluronidase-1 and -2. J. Biol. Chem. 282, 5597–5607 [DOI] [PubMed] [Google Scholar]
- 33. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 34. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]