

Abstract
In reply to internal or external danger stimuli, the body orchestrates an inflammatory response. The endogenous triggers of this process are the damage-associated molecular patterns (DAMPs). DAMPs represent a heterogeneous group of molecules that draw their origin either from inside the various compartments of the cell or from the extracellular space. Following interaction with pattern recognition receptors in cross-talk with various non-immune receptors, DAMPs determine the downstream signaling outcome of septic and aseptic inflammatory responses. In this review, the diverse nature, structural characteristics, and signaling pathways elicited by DAMPs will be critically evaluated.
Keywords: ATP, Autophagy, Biglycan, Decorin, Heat Shock Protein (HSP), Heparan Sulfate, Hyaluronan, Inflammasome, Necrosis (Necrotic Death), Small Leucine-rich Proteoglycan (SLRP), Toll-like Receptor (TLR)
Introduction
Inflammation is the body's defense to microbial invasion or a critical response to tissue injury under aseptic or sterile conditions. Foreign agents (e.g. bacteria or viruses) release pathogen-associated molecular patterns (PAMPs)2 that are recognized by pattern recognition receptors (PRRs) of the immune system and thus trigger an inflammatory response (1). In contrast, sterile inflammation, an important characteristic of myocardial infarction, atherosclerosis, several autoimmune diseases, and cancer, is induced by the release of endogenous molecules called DAMPs following tissue stress or injury (2–5). PAMPs and DAMPs are highly conserved motifs derived from the pathogens themselves (PAMPs) or from self-molecules that are normally hidden from the PRRs by compartmentalization (intracellular DAMPs) or sequestration in the ECM (extracellular DAMPs). DAMPs, similar to PAMPs, are recognized by PRRs. Five classes of PRRs have currently been described: cell surface or endosomal Toll-like receptors (TLRs), cytoplasmic NOD-like receptors (NLRs) and inflammasome, intracellular RIG-I (retinoic acid-inducible gene-I)-like receptors, transmembrane C-type lectin receptors (e.g. dectin-1), and AIM2 (absent in melanoma 2)-like receptors (6). It is not well understood why, despite the structural heterogeneity of DAMPs, they share the capacity to bind and activate the same PRRs, frequently TLR2 and TLR4, to trigger an immune response (7) (Tables 1 and 2). The complexity of DAMP-TLR-mediated immune signaling is further increased by accessory molecules (e.g. cluster of differentiation CD14, CD36, and myeloid differentiation 2 (MD2)) and by the CD14 preference for DAMPs (8, 9). Additionally, after TLR dimerization, the adaptor molecules myeloid differentiation factor 88 (MyD88), TIR domain-containing adaptor-inducing interferon (TRIF), TRIF-related adaptor molecule (TRAM), MyD88-adaptor like (Mal), and sterile α and armadillo motif-containing protein (SARM) are recruited, resulting in activation of NF-κB pathway (via MyD88) and interferon (via TRIF) (10).
TABLE 1.
The nature and sources of extracellular DAMPs
EDA, extra domain A.
| Cell compartment/ organelle | DAMP | Mechanism of release | Chemical nature | Receptor | Ref. |
|---|---|---|---|---|---|
| Extracellular matrix | Biglycan | MMP cleavage, de novo synthesis | Proteoglycan | TLR2, TLR4, NLRP3 | (16, 18) |
| Decorin | MMP cleavage, de novo synthesis | Proteoglycan | TLR2, TLR4 | (14) | |
| Versican | Secretion | Proteoglycan | TLR2, TLR6, CD14 | (22) | |
| LMW hyaluronan | Hyaluronidase cleavage | Glycosaminoglycan | TLR2, TLR4, NLRP3 | (7, 23, 24, 81) | |
| Heparan sulfate | Heparanase cleavage | Glycosaminoglycan | TLR4 | (25, 29, 30) | |
| Fibronectin-EDA+ | MMP cleavage | Glycoprotein | TLR4 | (31) | |
| Fibrinogen | Extravasation | Glycoprotein | TLR4 | (33) | |
| Tenascin C | De novo synthesis | Glycoprotein | TLR4 | (34) |
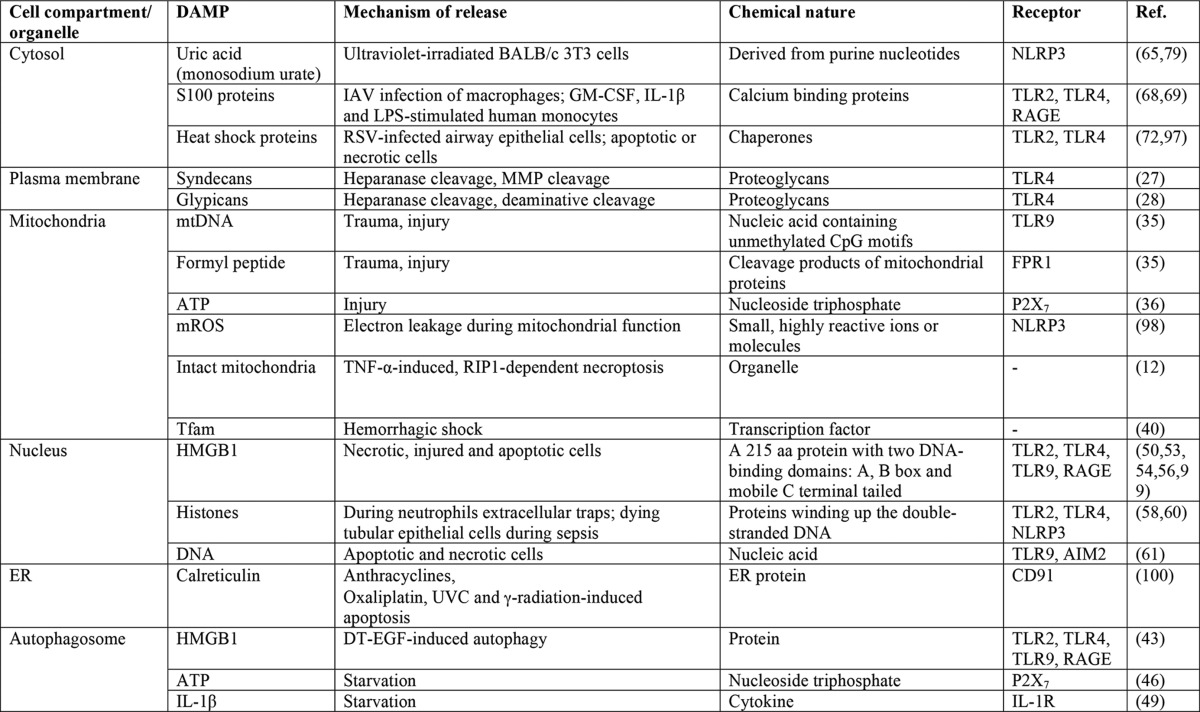
TABLE 2.
The nature and sources of intracellular DAMPs
Abbreviations: aa, amino acid; DT-EGF, epidermal growth factor receptor-targeted diphtheria toxin; ER, endoplasmic reticulum; FPR1, formyl peptide receptor; IAV, influenza A virus; IL-1R, interleukin 1 receptor; mROS, mitochondrial reactive oxygen species; RIP1, receptor-interacting protein; RSV, respiratory syncytial virus.
Several classes of DAMPs can be identified based on their origin or chemical nature (Tables 1 and 2). The molecular makeup of DAMPs is quite heterogeneous, ranging from the small uric acid or ATP molecules to large proteins >100 kDa in size and even organelles (11, 12). In turn, this high structural diversity allows DAMPs to achieve cross-talk between PRRs and a wide variety of “non-immune” receptors, finally impacting on the complexity of DAMP signaling (5, 7). In this review, I provide a critical appraisal of the diverse modalities of DAMP generation and discuss the major receptors and signaling pathways operating in DAMP-mediated inflammation.
Origin of DAMPs
DAMP liberation occurs from the extracellular or intracellular space following tissue stress and injury or cell death.
DAMPs Derived from the Extracellular Matrix (ECM)
There is growing evidence that a series of molecules that are under normal conditions sequestered components of the ECM can be proteolytically released from the ECM following tissue injury to act in their soluble form as DAMPs (Table 1 and Fig. 1) (7, 13). Some of these soluble ECM fragments engage multiple PRRs, initiating a rapid inflammatory reaction. To ensure continuation of the inflammatory response, the cytokine-activated macrophages followed by tissue-resident cells start the de novo synthesis of intact and soluble ECM-derived DAMPs (Table 1) (14–16).
FIGURE 1.

Cell- and ECM-derived DAMPs activate TLRs and inflammasome. Following cell death or injury, various intracellular DAMPs are released extracellularly from mitochondria, the autophagosome, the nucleus, and the cytosol. In turn, HMGB1, histones, HSPs, and S100 proteins target the cell surface TLRs. mtDNA activates the intracellular TLRs following endocytosis. In addition, histones and uric acid activate the inflammasome following phagocytosis, whereas ATP activates the inflammasome in a P2X7-dependent manner. Alternatively, tissue injury or stress determines the release of various ECM-derived DAMPs as a consequence of proteinase activity. Thus, SLRPs (biglycan, decorin), HA, and HS trigger cell surface TLRs activation. In addition, the SLRP biglycan activates the inflammasome via the P2X7 receptor. In contrast, HA is internalized in a CD44-dependent manner, and after fragmentation into HA-oligosaccharides, activates the inflammasome.
Proteoglycans (PGs) are the best characterized ECM-derived DAMPs (7). Based on their complex structure, containing protein core and various glycosaminoglycan side chains, proteoglycans are capable of interacting with distinct receptors and orchestrating their signaling cross-talk (Table 1 and Fig. 1) (5, 7). Upon release from the ECM by several proteolytic enzymes, such as bone morphogenetic protein (BMP)-1, matrix metalloproteinase (MMP)-2, MMP-3, MMP-13, and granzyme B (17), two chondroitin/dermatan sulfate small leucine-rich proteoglycans (SLRPs), biglycan (16) and decorin (14), act as endogenous ligands of TLR2/4 and trigger sterile inflammation (14–16). Furthermore, biglycan-dependent interaction of TLR2/4 with purinergic receptor P2X, ligand-gated ion channel, 7 (P2X7) results in the autonomous synthesis and maturation of IL-1β via activation of the leucine-rich repeat (LRR)- and PYD domain-containing protein (NLRP)3 inflammasome and caspase-1 (18). In contrast, soluble decorin stimulates signaling of TLR2/4 and inhibits TGF-β1 activity, thereby creating a pro-inflammatory environment that appears to be beneficial in the treatment of established tumors (14). Thus, both SLRPs as ligands of the same TLRs trigger different downstream signaling outcomes due to their interaction with other receptors, growth factors, and ECM components. The data regarding other SLRPs in maintaining danger signals are sparse. Interestingly, instead of being a canonical DAMP, lumican, a keratan sulfate member of the SLRP family, acts as a helper of PAMP via binding to and presenting LPS to the TLR adaptor CD14 (19). For further details regarding the mechanism of SLRP release and their use as biomarkers in inflammation, please refer to recent more specific reviews (5, 7, 13, 20, 21).
Versican, a chondroitin sulfate PG and the largest member of the hyaluronan- and lectin-binding proteoglycans, has been suggested to act as a ligand to the TLR2/TLR6 heterodimer and to its adaptor CD14, thereby promoting metastatic spread of cancer (22). Besides “true” PGs, hyaluronan (HA), a non-sulfated glycosaminoglycan that is never covalently linked to a protein core, is capable of evoking a danger signal in response to injury following partial enzymatic cleavage by hyaluronidases (23). The resulting low molecular weight (LMW)-HA fragments accumulate and stimulate TLR2/4 and inflammatory gene expression, thereby promoting angiogenesis and cancer metastasis (7, 23, 24). Furthermore, active heparan sulfate (HS) fragments generated by heparanase-1 from extracellular barriers (25) and HS proteoglycan sources, such as the basement membrane perlecan (26), the cell surface syndecans (27), and glypicans (28), serve as TLR4-interacting DAMPs (29, 30) (Fig. 1). Several glycoproteins, such as MMP-cleaved extra domain A of fibronectin (31, 32), extravascularly deposited fibrinogen (33), and tenascin C, can act as DAMPs via TLR4 (34) (Table 1).
Despite initial skepticism regarding potential contaminations, it is now broadly accepted that the ECM serves as a reservoir of sequestered DAMPs that upon release facilitate a rapid response to tissue injury. Thus, there is much stored information in the ECM that is not only structural in nature but also functional when released by activated enzymes.
Intracellular DAMPs
Besides ECM-derived DAMPs, there is a vast and heterogeneous group of intracellular danger molecules. Cells dying from classical, accidental, and secondary necrosis or apoptosis release endogenous molecules from different compartments or organelles (Table 2 and Fig. 1) that can act as extracellular soluble DAMPs (11).
Mitochondrial DAMPs
During various types of cell death, both programmed and unprogrammed, mitochondria are the major organelles that release various types of DAMPs (Table 2 and Fig. 1). Moreover, mitochondria are the only organelles that act as DAMPs by themselves following release during necroptosis (12). Intact mitochondria purified from necrotic cells are engulfed by human monocyte-derived macrophages and induce the production of pro-inflammatory cytokines (12). When injured, mitochondria can release into the circulation various intra-mitochondrial components such as mtDNA (35), formylated peptides (35), and ATP (36), all of which are recognized by PRRs of the immune systems and act as DAMPs (37, 38). mtDNA can also be detected in transfusion blood and may contribute to transfusion-related acute lung injury (39). In the human brain, mitochondrial transcription factor A (Tfam) can be released into the extracellular space and can be recognized by microglia, thereby inducing a pro-inflammatory response (40).
Despite the emerging role of mitochondrial molecules as DAMPs, it becomes clear that this pathway is operational in various inflammatory diseases. Further studies addressing cellular responsibility to mitochondrial DAMPs, their modulators of immunological activity, and their signaling pathways are required to better define their potential therapeutic target.
Autophagy-related DAMP Release
Various DAMPs can be released by stressed cells undergoing autophagy or injury without overt cell death. Evidence has been brought forth regarding the autophagy-mediated release of high-mobility group protein B1 (HMGB1), ATP, IL-1β, and DNA (41, 42) (Table 2 and Fig. 1).
The induction of autophagy in glioblastoma cells correlates with the release of HMGB1 without evoking either lysis of the plasma membrane or classical necrosis. In contrast, dying epithelial cells do not evoke autophagy but induce caspases and retain HMGB1 (43). Moreover, HMGB1 localizes to a subset of autophagosomes prior to its release from diphtheria toxin-EGF-treated cells (43). Additionally, epigallocatechin gallate, the ester of epigallocatechin and gallic acid, and a type of catechin, inhibits the release of HMGB1 from macrophages by forming a complex, which is degraded in an autophagy-dependent manner (44).
ATP is released from dying autophagic cells through the pannexin-1 channel and activates the NLRP3 inflammasome following cell engulfment by macrophages (45). Moreover, autophagic inducers determine the transfer of ATP-loaded vesicle-associated membrane protein (VAMP) 7-positive endocytic/autophagic compartments (amphisomes) to the cell periphery. There, under starvation, they fuse with the plasma membrane, thus liberating ATP extracellularly (46).
IL-1β is a DAMP that can be actively secreted from the cells in response to either PAMPs or other DAMPs, but it can also be passively released by necrotic cells (41). Many studies have shown that autophagy is a limiting factor for IL-1β release when it is induced by inflammasome activators. This may involve ubiquitination of inflammasomes, recruitment of p62, and active inflammasome elimination through autophagosome-dependent degradation (47). Furthermore, the absence of autophagic proteins microtubule-associated protein-1 light chain 3B (LC3B) and Beclin1 in macrophages determines higher activation of caspase-1 and maturation of IL-1β and IL-18 in correlation with the liberation of mtDNA in the cytosol in response to LPS/ATP stimulation (48). On the other hand, autophagy induced by starvation promotes the NLRP3 inflammasome-dependent IL-1β secretion following nigericin stimulation. This effect is based on the activation of the autophagy-mediated unconventional secretory pathway, which requires the extracellular release of IL-1β (49).
Nuclear DAMPs
The translocation of various nuclear molecules, such as HMGB1, histones, or DNA, is another mechanism described for DAMP formation during inflammation or cell death (50). HMGB1 is a nuclear non-histone DNA-binding protein that can also translocate to the cytoplasm and is released extracellularly through vesicular transport (50). Notably, HMGB1 can be produced not only by activated monocytes and macrophages, but also by necrotic or injured fibroblasts in a passive way (51–54), and to a lesser extent during late apoptosis (55). HMGB1 is also found in microparticles derived from apoptotic HeLa and Jurkat cells (56). Moreover, HMGB1 presents a different redox state during apoptosis and necrosis by being reduced or oxidized, respectively (50, 57).
Histones are nuclear proteins that form hetero-octamers to wind up double-stranded DNA to form chromatin as well as chromosomes (58). Histone release during sepsis increases endothelial cell dysfunction, organ failure, and death (59). In bacterial infections, histones are generally released either by dying neutrophils, especially during the formation process of the so-called neutrophil extracellular traps (60), or by dying tubular epithelial cells (58). In vivo DNA is also found released in the plasma of animals with hepatic models of apoptosis and necrosis (61, 62). Thus again and in analogy to extracellular and other intracellular DAMPs, a complex and expanding class of participants regulates the generation of DAMPs derived from the nucleoplasm.
Cytosolic DAMPs
Uric acid is one of the main endogenous immunological danger signals. It is constitutively present in all cells, but its levels increase following cell injury (63). It is the end product of purine nucleotide breakdown (64). Dying cells release uric acid extracellularly, which is then able to evoke immune responses such as the induction of dendritic cell maturation and increase in priming of CD8 T-cell responses to cross-presented antigens (65). In addition, uric acid is present in the circulation, and its increased levels correlate with progression of gout (66).
The Ca2+-binding proteins S100A8 and S100A9 have been extensively implicated in the induction of inflammation and fibrosis (4). They act as DAMPs after release from phagocytes in response to cell stress (67). Their secretion does not depend on the classical endoplasmic reticulum/Golgi route. Instead, it has been demonstrated that S100A8 and S100A9 are released by human monocytes after activation of protein kinase C tubulin-dependent pathway (68). A recent study has demonstrated that, following influenza A virus infection, S100A9 is released into the extracellular environment from undamaged macrophages and acts as a DAMP by augmenting pro-inflammatory responses, cell death, and virus pathogenesis (69).
Heat shock proteins (HSPs) are a class of proteins that normally play the role of chaperones and assist the biosynthetic machinery in the proper correct folding of proteins (70). However, HSPs can also act as DAMPs by interacting with TLRs following release from the intracellular space (71). HSPs are generally released from dying cells following apoptosis, necrosis, and cellular stress (72, 73) (Fig. 1). Thus, both intracellular and released products can act as DAMPs under the appropriate pro-inflammatory stimuli.
DAMP Receptors and Signaling Pathways
Toll-like Receptors
TLRs are type I integral membrane proteins that are capable of recognizing a variety of stimuli, including PAMPs or DAMPs, and trigger the immune response (74). TLRs possess one transmembrane helix, a highly glycosylated N-terminal ectodomain (ECD) responsible for ligand recognition, and a C-terminal Toll IL-1 receptor (TIR) domain responsible for signaling (74). Ten TLRs have so far been described in humans, and 13 have been described in mice (75, 76). Although most TLRs are present at the cell surface, some (TLR3, TLR7, TLR8, TLR9, and TLR13) can localize to the endosomal membrane (75, 76) (Fig. 1). The latter suggests that TLRs might have some additional functions in addition to the classical regulation of infection.
The most remarkable feature of TLRs is the presence of LRRs in their ECDs (74). These motifs, typically 22–29 residues long, are responsible for the distinct horseshoe shape adopted by TLR ECDs and mediate ligand recognition in TLRs. Note that these LRRs are designed to mediate protein-protein interactions and are also shared by decorin and biglycan, two ligands for TLR2/4.
A number of crystal structures have shown how the direct interaction of TLR ECDs and PAMPs occurs. Depending on the specific ligand, this process involves the formation of TLR hetero- or homo-dimers and sometimes utilizes accessory and adaptor molecules (see the Introduction) (2). Finally, the general end product of TLR activation is the NF-κB or interferon pathway leading to cytokine production (77).
Almost all classes of DAMPs can bind to TLRs (Tables 1 and 2), and a selection of those classes is shown in Fig. 1. TLR9 recognizes DNA containing unmethylated CpG motifs that are usually derived from bacteria or viruses. However, it can also recognize mitochondrial DNA that contains similar CpG sequences and activate immune pathways identical to those elicited in sepsis (35). HMGB1 can associate with TLR9 and CpG-DNA and hasten redistribution of TLR9 to early endosomes in response to the CpG-DNA analog CpG-oligodeoxynucleotide (CpG-ODN) (78). Additionally, HMGB1 serves as a ligand for TLR2/4 (50), a feature it shares with histones (58) and decorin/biglycan (14, 16).
Notably, HMGB1 bioactivity is regulated via redox modifications of Cys residues, insofar as either oxidation or reduction of essential Cys residues can inhibit cytokine production (57). Monosodium urate can also activate macrophages through TLR2/4 (79). Although S100 proteins are known activators of TLR4 (69), S100B can also bind TLR2 and inhibit its activity (80). HSPs are also ligands of TLR2/4 (72). In the case of biglycan and decorin, interaction with TLRs is most likely mediated by LRRs present in the ligands (14–16). HS and LMW-HA both activate TLR4 (25, 81).
Although structures of TLR-DAMP complexes have not yet been determined, it is conceivable that at least some of the binding sites for PAMPs can also be employed by DAMPs. However, evidence also exists that DAMPs can utilize binding sites different from those utilized by PAMPs (82) and that DAMPs can utilize different accessory proteins for binding to TLRs than those used by PAMPs (2). Also interesting is the fact that some DAMPs can trigger from downstream responses different from those evoked by PAMPs binding to the same TLR (81). Collectively, these findings point to a central paradigm whereby TLR activation is more than a simple on/off event.
It is particularly interesting that signaling initiated by activation of the same receptors results in different downstream effects depending on the initial DAMP. Employing various accessory and adaptor molecules of TLRs, some DAMPs (e.g. PGs) can target diverse non-immune receptors, thereby refining the final outcome of combined signaling cascades (7). In contrast to PAMPs, selected DAMPs have the unique ability to interact with two TLRs, frequently TLR2/4, sensing Gram-positive as well as Gram-negative pathogens. The fact that the affinity binding of two DAMPs, biglycan and decorin, to TLR4 is comparable with those of LPS (14, 83) further underscores how powerful these DAMPs are. Thus, it is conceivable that the DAMP-PAMP interaction defines the biological outcome of TLR signaling.
The Receptor for Advanced Glycation End Products (RAGE)
One of the best-studied DAMP receptors is RAGE. The full-length receptor contains three parts: an extracellular region that is responsible for ligand interaction through its V domain, a transmembrane domain to anchor the protein to the cell surface, and a cytoplasmic domain that is responsible for downstream signaling (84). RAGE can also exist in truncated forms following alternate splicing or protease processing. In the absence of its V domain, it cannot bind ligands, whereas in the absence of the transmembrane domain, it becomes soluble and can bind ligands, acting as a decoy receptor by preventing them from binding and activating mature RAGE (84). RAGE was initially identified as a receptor for advanced glycation end products (AGEs) (85). However, it was later shown to serve as a receptor for a number of DAMPs, including HMGB1 (53), S100 proteins (86), and amyloid-β protein (87).
An intriguing aspect of RAGE is that its expression can be up-regulated by the presence of its ligands, leading to an amplification of the initial response (84). Interestingly, despite the structural diversity of RAGE ligands, activation of RAGE leads to a common pathway: the activation of NF-κB, cell proliferation, and TGF-β production (84). This makes RAGE a valuable target not only in Type 1 diabetes (88), but also in Type 2 diabetes, atherosclerosis, pulmonary fibrosis, and cancer (89).
The Inflammasome
A particular role in DAMP signaling is played by the NLRP3 inflammasome. The NLRP3 inflammasome contains the NLRP3 protein, the adaptor protein apoptosis-associated speck-like protein containing a CARD (ASC), and the cysteine protease caspase-1 (90). Assembly of the inflammasome leads to the cleavage of caspase-1 to its active form, which, in turn, cleaves the pro-IL-1β precursor into mature IL-1β. Interestingly, IL-1β is also classified as a DAMP, thus making NLRP3 both a receptor and a source of DAMPs.
Activation of the NLRP3 inflammasome can occur as a response to a number of distinct DAMPs. Perhaps the most investigated is the assembly of the NLRP3 inflammasome in response to ATP. Accordingly, extracellular ATP activates the purinergic receptor P2X7 present on the cell surface, causing potassium efflux from the cell. Potassium efflux, in turn, mediates the assembly of the NLRP3 inflammasome (90). Uric acid can also activate the NLRP3 inflammasome (91), a process that requires the LRR domain of NLRP3 (92). LMW-HA was also shown to activate the NLPR3 inflammasome in a process dependent on CD44 and HA internalization (93). Biglycan can also activate the NLRP3 inflammasome by clustering P2X7 with TLR2/4 (18). Finally, histones released from necrotic cells were recently shown to activate the NLRP3 inflammasome independently of P2X7 (94).
IL-1β release in macrophages, but not in monocytes, requires two signals: an induction of pro-IL-1β transcription by NF-κB activators followed by an activator of NLRP3 inflammasome formation (95). Although it is possible for a signal to activate both, as is the case for biglycan and LMW-HA (18, 93), such a requirement might reveal an interesting cooperativity between PAMPs and DAMPs that activate NF-κB and evoke the release of endogenous ATP.
Future Challenges
For far too long, immunological research has focused only on the non-self as a source for immune recognition. It is now increasingly apparent that endogenous danger signals also represent a vital component of the immune response. DAMP release can serve as a marker of disease progression and severity (13, 96). Furthermore, DAMP involvement has been shown in a large number of life-threatening sterile inflammatory conditions (for more detailed reviews, see Refs. 4, 11, and 42). During the last decade, several modes of DAMP generation and various DAMP receptors and signaling pathways have been identified. Targeting DAMPs and their receptors is a promising strategy in the treatment of inflammatory conditions (2). However, there are still several gaps in knowledge that make translation of this field to effective therapies unsatisfactory.
The list of newly described DAMPs is growing every year. This is not astonishing taking into account that the innate immune system needs to be fine-tuned by several alternate ligand-PRR interactions. Thus, it is clear that this is only the tip of the iceberg and that there are many more types of DAMPs awaiting description. In this context, due to their structural homology to TLRs and some accessory molecules, molecules containing LRRs (e.g. ECM-derived SLRPs) appear to be good candidates to investigate. Furthermore, our current knowledge, particularly regarding ECM-derived DAMPs, is still quite primitive. ECM remodeling is a normal aspect of tissue reorganization or healing and results in the deposition or degradation of the ECM components. However, it is only when this process is deregulated that ECM components are recognized as a danger signal. It is thus increasingly important to understand what keeps these potential DAMPs in check under normal conditions and how the ECM is regulated normally to prevent the triggering of inflammation.
To develop selective DAMP-targeted therapy, it is clear that several key questions need to be addressed in the future, including the following. 1) What are the triggers of DAMP release and de novo synthesis upon tissue injury? 2) What is the mode of interaction between several DAMPs? 3) Do they have additive effects, or do they instead compete for binding to the same PRR? Preliminary unpublished data suggest a hierarchical mode of DAMP interactions.3 Therefore, approaches to identify an initial DAMP trigger and the following DAMP-mediated “chains of command” should be the focus of future scientific attention. 4) How do DAMPs interact with PRRs? It is of note that the interactions of DAMPs with their receptors, in particular TLRs, are still not fully elucidated. Although clinical and immunological studies are invaluable in showing the importance and role of DAMPs, the details of these interactions need a closer examination, involving basic biochemical and structural biology approaches to characterize the assembly of DAMP-PRR complexes. 5) Is DAMP capable of choosing various accessory molecules by interacting with the same PRR to achieve different downstream effects? 6) How does DAMP-dependent cross-talk between PRRs and non-immune receptors influence signaling outcome? 7) What are the effects of DAMPs on PRRs in non-immune cells? 8) How do DAMPs and PAMPs interact during PAMP-dependent inflammation? It is conceivable that DAMPs may potentiate the effects of PAMPs via a second PRR that is not involved in pathogen sensing. However, when DAMPs and PAMPs are interacting with the same PRR, contact of PRRs with DAMPs prior to microbial invasion could lower the responsiveness of this PRR to PAMPs. Such studies might also reveal the molecular basis for some differential effects observed between PAMP and DAMP activation of the same receptors and their interplay in orchestrated signaling via distinct PRRs. Thus, answers to these questions would crucially contribute to therapeutic targeting of the PAMP-DAMP-PRR interactions and should open an extremely interesting avenue in clinical research.
Once the role of a molecule as a DAMP and its corresponding PRR(s) have been identified, it is at this level that any potential treatment for sterile inflammation has to start. It has to be restated that, under normal conditions, most molecules that can act as DAMPs exist in the organism and are essential for its continued survival. Therefore, a key aspect will be targeting these molecules specifically in their role as DAMPs, but without hindering their normal role in the body. This could be done, for example, by specifically disrupting residues or domains that are involved in the interaction of DAMPs with PRRs while leaving the rest of the molecule intact. In this respect, treating sterile inflammation seems more challenging than eliminating foreign bacteria or viruses, especially because the latter task is hardly an easy feat.
This work was supported by the German Research Council (Grant SFB 815, Project A5, Grant SFB 1039, Project B2, Excellence Cluster ECCPS, and LOEWE Program Ub-Net).
L. Schaefer, unpublished data.
- PAMP
- pathogen-associated molecular pattern
- DAMP
- damage-associated molecular pattern
- CD
- cluster of differentiation
- ECD
- glycosylated N-terminal ectodomain
- ECM
- extracellular matrix
- HA
- hyaluronan
- HMGB1
- high-mobility group protein B1
- HS
- heparan sulfate
- HSP
- heat shock protein
- LMW
- low molecular weight
- LRR
- leucine-rich repeats
- MMP
- metalloproteinase
- MyD88
- myeloid differentiation primary response gene 88
- NET
- neutrophils extracellular trap
- NF-κB
- nuclear factor κ-light-chain-enhancer of activated B cells
- NLR
- NOD-like receptor
- NLRP
- NACHT, LRR, and PYD domain-containing protein
- P2X7
- purinergic receptor P2X, ligand-gated ion channel, 7
- PG
- proteoglycan
- PRR
- pattern recognition receptor
- RAGE
- receptor for advanced glycation end products
- SLRP
- leucine-rich proteoglycan
- TLR
- Toll-like receptor
- TRIF
- TIR domain-containing adapter-inducing interferon-β
- TIR
- C-terminal Toll IL-1 receptor
- CARD
- caspase activation and recruitment domain.
REFERENCES
- 1. Mogensen T. H. (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 22, 240–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Piccinini A. M., Midwood K. S. (2010) DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010, 672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Broggi A., Granucci F. (2014) Microbe- and danger-induced inflammation. Mol. Immunol. 63, 127–133 [DOI] [PubMed] [Google Scholar]
- 4. Rosin D. L., Okusa M. D. (2011) Dangers within: DAMP responses to damage and cell death in kidney disease. J. Am. Soc. Nephrol. 22, 416–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moreth K., Iozzo R. V., Schaefer L. (2012) Small leucine-rich proteoglycans orchestrate receptor crosstalk during inflammation. Cell Cycle 11, 2084–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen G. Y., Nuñez G. (2010) Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frey H., Schroeder N., Manon-Jensen T., Iozzo R. V., Schaefer L. (2013) Biological interplay between proteoglycans and their innate immune receptors in inflammation. FEBS J. 280, 2165–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chun K. H., Seong S. Y. (2010) CD14 but not MD2 transmit signals from DAMP. Int. Immunopharmacol. 10, 98–106 [DOI] [PubMed] [Google Scholar]
- 9. Escamilla-Tilch M., Filio-Rodríguez G., García-Rocha R., Mancilla-Herrera I., Mitchison N. A., Ruiz-Pacheco J. A., Sánchez-García F. J., Sandoval-Borrego D., Vázquez-Sánchez E. A. (2013) The interplay between pathogen-associated and danger-associated molecular patterns: an inflammatory code in cancer? Immunol. Cell Biol. 91, 601–610 [DOI] [PubMed] [Google Scholar]
- 10. O'Neill L. A., Bowie A. G. (2007) The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364 [DOI] [PubMed] [Google Scholar]
- 11. Anders H. J., Schaefer L. (2014) Beyond tissue injury: damage-associated molecular patterns, Toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J. Am. Soc. Nephrol. 25, 1387–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maeda A., Fadeel B. (2014) Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. 5, e1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hsieh L. T., Nastase M. V., Zeng-Brouwers J., Iozzo R. V., Schaefer L. (2014) Soluble biglycan as a biomarker of inflammatory renal diseases. Int. J. Biochem. Cell Biol. 54, 223–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Merline R., Moreth K., Beckmann J., Nastase M. V., Zeng-Brouwers J., Tralhão J. G., Lemarchand P., Pfeilschifter J., Schaefer R. M., Iozzo R. V., Schaefer L. (2011) Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and microRNA-21. Sci. Signal. 4, ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moreth K., Brodbeck R., Babelova A., Gretz N., Spieker T., Zeng-Brouwers J., Pfeilschifter J., Young M. F., Schaefer R. M., Schaefer L. (2010) The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J. Clin. Invest. 120, 4251–4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schaefer L., Babelova A., Kiss E., Hausser H. J., Baliova M., Krzyzankova M., Marsche G., Young M. F., Mihalik D., Götte M., Malle E., Schaefer R. M., Gröne H. J. (2005) The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J. Clin. Invest. 115, 2223–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nastase M. V., Young M. F., Schaefer L. (2012) Biglycan: a multivalent proteoglycan providing structure and signals. J. Histochem. Cytochem. 60, 963–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Babelova A., Moreth K., Tsalastra-Greul W., Zeng-Brouwers J., Eickelberg O., Young M. F., Bruckner P., Pfeilschifter J., Schaefer R. M., Gröne H. J., Schaefer L. (2009) Biglycan, a danger signal that activates the NLRP3 inflammasome via Toll-like and P2X receptors. J. Biol. Chem. 284, 24035–24048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu F., Vij N., Roberts L., Lopez-Briones S., Joyce S., Chakravarti S. (2007) A novel role of the lumican core protein in bacterial lipopolysaccharide-induced innate immune response. J. Biol. Chem. 282, 26409–26417 [DOI] [PubMed] [Google Scholar]
- 20. Schaefer L., Iozzo R. V. (2012) Small leucine-rich proteoglycans, at the crossroad of cancer growth and inflammation. Curr. Opin. Genet. Dev. 22, 56–57 [DOI] [PubMed] [Google Scholar]
- 21. Nastase M. V., Iozzo R. V., Schaefer L. (2014) Key roles for the small leucine-rich proteoglycans in renal and pulmonary pathophysiology. Biochim. Biophys. Acta 1840, 2460–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim S., Takahashi H., Lin W. W., Descargues P., Grivennikov S., Kim Y., Luo J. L., Karin M. (2009) Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457, 102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang D., Liang J., Noble P. W. (2007) Hyaluronan in tissue injury and repair. Annu. Rev. Cell Dev. Biol. 23, 435–461 [DOI] [PubMed] [Google Scholar]
- 24. Singleton P. A. (2014) Hyaluronan regulation of endothelial barrier function in cancer. Adv. Cancer Res. 123, 191–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goldberg R., Meirovitz A., Hirshoren N., Bulvik R., Binder A., Rubinstein A. M., Elkin M. (2013) Versatile role of heparanase in inflammation. Matrix Biol. 32, 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Patel V. N., Knox S. M., Likar K. M., Lathrop C. A., Hossain R., Eftekhari S., Whitelock J. M., Elkin M., Vlodavsky I., Hoffman M. P. (2007) Heparanase cleavage of perlecan heparan sulfate modulates FGF10 activity during ex vivo submandibular gland branching morphogenesis. Development 134, 4177–4186 [DOI] [PubMed] [Google Scholar]
- 27. Barbouri D., Afratis N., Gialeli C., Vynios D. H., Theocharis A. D., Karamanos N. K. (2014) Syndecans as modulators and potential pharmacological targets in cancer progression. Front. Oncol. 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mani K., Cheng F., Fransson L. A. (2006) Defective nitric oxide-dependent, deaminative cleavage of glypican-1 heparan sulfate in Niemann-Pick C1 fibroblasts. Glycobiology 16, 711–718 [DOI] [PubMed] [Google Scholar]
- 29. Brennan T. V., Lin L., Huang X., Cardona D. M., Li Z., Dredge K., Chao N. J., Yang Y. (2012) Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD after allogeneic stem cell transplantation. Blood 120, 2899–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goodall K. J., Poon I. K., Phipps S., Hulett M. D. (2014) Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS One 9, e109596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang X., Chen C. T., Bhargava M., Torzilli P. A. (2012) A comparative study of fibronectin cleavage by MMP-1, -3, -13, and -14. Cartilage 3, 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Okamura Y., Watari M., Jerud E. S., Young D. W., Ishizaka S. T., Rose J., Chow J. C., Strauss J. F., 3rd. (2001) The extra domain A of fibronectin activates Toll-like receptor 4. J. Biol. Chem. 276, 10229–10233 [DOI] [PubMed] [Google Scholar]
- 33. Smiley S. T., King J. A., Hancock W. W. (2001) Fibrinogen stimulates macrophage chemokine secretion through Toll-like receptor 4. J. Immunol. 167, 2887–2894 [DOI] [PubMed] [Google Scholar]
- 34. Midwood K., Sacre S., Piccinini A. M., Inglis J., Trebaul A., Chan E., Drexler S., Sofat N., Kashiwagi M., Orend G., Brennan F., Foxwell B. (2009) Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 15, 774–780 [DOI] [PubMed] [Google Scholar]
- 35. Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., Hauser C. J. (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krysko D. V., Agostinis P., Krysko O., Garg A. D., Bachert C., Lambrecht B. N., Vandenabeele P. (2011) Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 32, 157–164 [DOI] [PubMed] [Google Scholar]
- 37. Wenceslau C. F., McCarthy C. G., Szasz T., Spitler K., Goulopoulou S., Webb R. C., and Working Group on DAMPs in Cardiovascular Disease (2014) Mitochondrial damage-associated molecular patterns and vascular function. Eur. Heart J. 35, 1172–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fernández-Ruiz I., Arnalich F., Cubillos-Zapata C., Hernández-Jiménez E., Moreno-González R., Toledano V., Fernández-Velasco M., Vallejo-Cremades M. T., Esteban-Burgos L., de Diego R. P., Llamas-Matias M. A., García-Arumi E., Martí R., Boscá L., Andreu A. L., López-Sendón J. L., López-Collazo E. (2014) Mitochondrial DAMPs induce endotoxin tolerance in human monocytes: an observation in patients with myocardial infarction. PLoS One 9, e95073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee Y. L., King M. B., Gonzalez R. P., Brevard S. B., Frotan M. A., Gillespie M. N., Simmons J. D. (2014) Blood transfusion products contain mitochondrial DNA damage-associated molecular patterns: a potential effector of transfusion-related acute lung injury. J. Surg. Res. 191, 286–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Little J. P., Simtchouk S., Schindler S. M., Villanueva E. B., Gill N. E., Walker D. G., Wolthers K. R., Klegeris A. (2014) Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol. Cell. Neurosci. 60, 88–96 [DOI] [PubMed] [Google Scholar]
- 41. Zhang Q., Kang R., Zeh H. J., 3rd, Lotze M. T., Tang D. (2013) DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy 9, 451–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hou W., Zhang Q., Yan Z., Chen R., Zeh Iii H. J., Kang R., Lotze M. T., Tang D. (2013) Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 4, e966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thorburn J., Horita H., Redzic J., Hansen K., Frankel A. E., Thorburn A. (2009) Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 16, 175–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li W., Zhu S., Li J., Assa A., Jundoria A., Xu J., Fan S., Eissa N. T., Tracey K. J., Sama A. E., Wang H. (2011) EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages. Biochem. Pharmacol. 81, 1152–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ayna G., Krysko D. V., Kaczmarek A., Petrovski G., Vandenabeele P., Fésüs L. (2012) ATP release from dying autophagic cells and their phagocytosis are crucial for inflammasome activation in macrophages. PLoS One 7, e40069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fader C. M., Aguilera M. O., Colombo M. I. (2012) ATP is released from autophagic vesicles to the extracellular space in a VAMP7-dependent manner. Autophagy 8, 1741–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shi C. S., Shenderov K., Huang N. N., Kabat J., Abu-Asab M., Fitzgerald K. A., Sher A., Kehrl J. H. (2012) Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nakahira K., Haspel J. A., Rathinam V. A., Lee S. J., Dolinay T., Lam H. C., Englert J. A., Rabinovitch M., Cernadas M., Kim H. P., Fitzgerald K. A., Ryter S. W., Choi A. M. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dupont N., Jiang S., Pilli M., Ornatowski W., Bhattacharya D., Deretic V. (2011) Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 30, 4701–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pisetsky D. S. (2014) The translocation of nuclear molecules during inflammation and cell death. Antioxid. Redox Signal. 20, 1117–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Müller S., Scaffidi P., Degryse B., Bonaldi T., Ronfani L., Agresti A., Beltrame M., Bianchi M. E. (2001) New EMBO members' review: the double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J. 20, 4337–4340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang H., Bloom O., Zhang M., Vishnubhakat J. M., Ombrellino M., Che J., Frazier A., Yang H., Ivanova S., Borovikova L., Manogue K. R., Faist E., Abraham E., Andersson J., Andersson U., Molina P. E., Abumrad N. N., Sama A., Tracey K. J. (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251 [DOI] [PubMed] [Google Scholar]
- 53. Rovere-Querini P., Capobianco A., Scaffidi P., Valentinis B., Catalanotti F., Giazzon M., Dumitriu I. E., Müller S., Iannacone M., Traversari C., Bianchi M. E., Manfredi A. A. (2004) HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 5, 825–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scaffidi P., Misteli T., Bianchi M. E. (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195 [DOI] [PubMed] [Google Scholar]
- 55. Beyer C., Stearns N. A., Giessl A., Distler J. H., Schett G., Pisetsky D. S. (2012) The extracellular release of DNA and HMGB1 from Jurkat T cells during in vitro necrotic cell death. Innate Immun. 18, 727–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Spencer D. M., Mobarrez F., Wallén H., Pisetsky D. S. (2014) The expression of HMGB1 on microparticles from Jurkat and HL-60 cells undergoing apoptosis in vitro. Scand. J. Immunol. 80, 101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang H., Lundbäck P., Ottosson L., Erlandsson-Harris H., Venereau E., Bianchi M. E., Al-Abed Y., Andersson U., Tracey K. J., Antoine D. J. (2012) Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol. Med. 18, 250–259 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58. Allam R., Scherbaum C. R., Darisipudi M. N., Mulay S. R., Hägele H., Lichtnekert J., Hagemann J. H., Rupanagudi K. V., Ryu M., Schwarzenberger C., Hohenstein B., Hugo C., Uhl B., Reichel C. A., Krombach F., Monestier M., Liapis H., Moreth K., Schaefer L., Anders H. J. (2012) Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J. Am. Soc. Nephrol. 23, 1375–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xu J., Zhang X., Pelayo R., Monestier M., Ammollo C. T., Semeraro F., Taylor F. B., Esmon N. L., Lupu F., Esmon C. T. (2009) Extracellular histones are major mediators of death in sepsis. Nat. Med. 15, 1318–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Papayannopoulos V., Zychlinsky A. (2009) NETs: a new strategy for using old weapons. Trends Immunol. 30, 513–521 [DOI] [PubMed] [Google Scholar]
- 61. Tran T. T., Groben P., Pisetsky D. S. (2008) The release of DNA into the plasma of mice following hepatic cell death by apoptosis and necrosis. Biomarkers 13, 184–200 [DOI] [PubMed] [Google Scholar]
- 62. Jiang N., Reich C. F., 3rd, Pisetsky D. S. (2003) Role of macrophages in the generation of circulating blood nucleosomes from dead and dying cells. Blood 102, 2243–2250 [DOI] [PubMed] [Google Scholar]
- 63. Kono H., Chen C. J., Ontiveros F., Rock K. L. (2010) Uric acid promotes an acute inflammatory response to sterile cell death in mice. J. Clin. Invest. 120, 1939–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Eisenbacher J. L., Schrezenmeier H., Jahrsdörfer B., Kaltenmeier C., Rojewski M. T., Yildiz T., Beyer T., Erle A., Wiegmann D. S., Grassl S., Hang R., Körper S., Wiesneth M., Lotze M. T., Lotfi R. (2014) S100A4 and uric acid promote mesenchymal stromal cell induction of IL-10+/IDO+ lymphocytes. J. Immunol. 192, 6102–6110 [DOI] [PubMed] [Google Scholar]
- 65. Shi Y., Evans J. E., Rock K. L. (2003) Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425, 516–521 [DOI] [PubMed] [Google Scholar]
- 66. Johnson R. J., Titte S., Cade J. R., Rideout B. A., Oliver W. J. (2005) Uric acid, evolution and primitive cultures. Semin. Nephrol. 25, 3–8 [DOI] [PubMed] [Google Scholar]
- 67. Foell D., Wittkowski H., Vogl T., Roth J. (2007) S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J. Leukoc. Biol. 81, 28–37 [DOI] [PubMed] [Google Scholar]
- 68. Rammes A., Roth J., Goebeler M., Klempt M., Hartmann M., Sorg C. (1997) Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J. Biol. Chem. 272, 9496–9502 [DOI] [PubMed] [Google Scholar]
- 69. Tsai S. Y., Segovia J. A., Chang T. H., Morris I. R., Berton M. T., Tessier P. A., Tardif M. R., Cesaro A., Bose S. (2014) DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog. 10, e1003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bianchi M. E. (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5 [DOI] [PubMed] [Google Scholar]
- 71. Vabulas R. M., Wagner H., Schild H. (2002) Heat shock proteins as ligands of Toll-like receptors. Curr. Top. Microbiol. Immunol. 270, 169–184 [DOI] [PubMed] [Google Scholar]
- 72. Lehnardt S., Schott E., Trimbuch T., Laubisch D., Krueger C., Wulczyn G., Nitsch R., Weber J. R. (2008) A vicious cycle involving release of heat shock protein 60 from injured cells and activation of Toll-like receptor 4 mediates neurodegeneration in the CNS. J. Neurosci. 28, 2320–2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Clayton A., Turkes A., Navabi H., Mason M. D., Tabi Z. (2005) Induction of heat shock proteins in B-cell exosomes. J. Cell Sci. 118, 3631–3638 [DOI] [PubMed] [Google Scholar]
- 74. Botos I., Segal D. M., Davies D. R. (2011) The structural biology of Toll-like receptors. Structure 19, 447–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Casanova J. L., Abel L., Quintana-Murci L. (2011) Human TLRs and IL-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu. Rev. Immunol. 29, 447–491 [DOI] [PubMed] [Google Scholar]
- 76. Oldenburg M., Krüger A., Ferstl R., Kaufmann A., Nees G., Sigmund A., Bathke B., Lauterbach H., Suter M., Dreher S., Koedel U., Akira S., Kawai T., Buer J., Wagner H., Bauer S., Hochrein H., Kirschning C. J. (2012) TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 337, 1111–1115 [DOI] [PubMed] [Google Scholar]
- 77. Kawai T., Akira S. (2007) Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 13, 460–469 [DOI] [PubMed] [Google Scholar]
- 78. Ivanov S., Dragoi A. M., Wang X., Dallacosta C., Louten J., Musco G., Sitia G., Yap G. S., Wan Y., Biron C. A., Bianchi M. E., Wang H., Chu W. M. (2007) A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 110, 1970–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Liu-Bryan R., Scott P., Sydlaske A., Rose D. M., Terkeltaub R. (2005) Innate immunity conferred by Toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. 52, 2936–2946 [DOI] [PubMed] [Google Scholar]
- 80. Sorci G., Giovannini G., Riuzzi F., Bonifazi P., Zelante T., Zagarella S., Bistoni F., Donato R., Romani L. (2011) The danger signal S100B integrates pathogen- and danger-sensing pathways to restrain inflammation. PLoS Pathog. 7, e1001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Taylor K. R., Yamasaki K., Radek K. A., Di Nardo A., Goodarzi H., Golenbock D., Beutler B., Gallo R. L. (2007) Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on Toll-like receptor 4, CD44, and MD-2. J. Biol. Chem. 282, 18265–18275 [DOI] [PubMed] [Google Scholar]
- 82. Hodgkinson C. P., Patel K., Ye S. (2008) Functional Toll-like receptor 4 mutations modulate the response to fibrinogen. Thromb. Haemost. 100, 301–307 [PubMed] [Google Scholar]
- 83. Moreth K., Frey H., Hubo M., Zeng-Brouwers J., Nastase M. V., Hsieh L. T., Haceni R., Pfeilschifter J., Iozzo R. V., Schaefer L. (2014) Biglycan-triggered TLR-2- and TLR-4-signaling exacerbates the pathophysiology of ischemic acute kidney injury. Matrix Biol. 35, 143–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lee E. J., Park J. H. (2013) Receptor for Advanced glycation endproducts (RAGE), its ligands, and soluble RAGE: potential biomarkers for diagnosis and therapeutic targets for human renal diseases. Genomics Inform. 11, 224–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schmidt A. M., Vianna M., Gerlach M., Brett J., Ryan J., Kao J., Esposito C., Hegarty H., Hurley W., Clauss M., et al. (1992) Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J. Biol. Chem. 267, 14987–14997 [PubMed] [Google Scholar]
- 86. Rani S. G., Sepuru K. M., Yu C. (2014) Interaction of S100A13 with C2 domain of receptor for advanced glycation end products (RAGE). Biochim. Biophys. Acta 1844, 1718–1728 [DOI] [PubMed] [Google Scholar]
- 87. Chaney M. O., Stine W. B., Kokjohn T. A., Kuo Y. M., Esh C., Rahman A., Luehrs D. C., Schmidt A. M., Stern D., Yan S. D., Roher A. E. (2005) RAGE and amyloid β interactions: atomic force microscopy and molecular modeling. Biochim. Biophys. Acta 1741, 199–205 [DOI] [PubMed] [Google Scholar]
- 88. Ramasamy R., Yan S. F., Schmidt A. M. (2011) Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann. N.Y. Acad. Sci. 1243, 88–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Xie J., Méndez J. D., Méndez-Valenzuela V., Aguilar-Hernández M. M. (2013) Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell. Signal. 25, 2185–2197 [DOI] [PubMed] [Google Scholar]
- 90. Agostini L., Martinon F., Burns K., McDermott M. F., Hawkins P. N., Tschopp J. (2004) NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325 [DOI] [PubMed] [Google Scholar]
- 91. Gasse P., Riteau N., Charron S., Girre S., Fick L., Pétrilli V., Tschopp J., Lagente V., Quesniaux V. F., Ryffel B., Couillin I. (2009) Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 179, 903–913 [DOI] [PubMed] [Google Scholar]
- 92. Hoffman H. M., Scott P., Mueller J. L., Misaghi A., Stevens S., Yancopoulos G. D., Murphy A., Valenzuela D. M., Liu-Bryan R. (2010) Role of the leucine-rich repeat domain of cryopyrin/NALP3 in monosodium urate crystal-induced inflammation in mice. Arthritis Rheum. 62, 2170–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yamasaki K., Muto J., Taylor K. R., Cogen A. L., Audish D., Bertin J., Grant E. P., Coyle A. J., Misaghi A., Hoffman H. M., Gallo R. L. (2009) NLRP3/cryopyrin is necessary for interleukin-1β (IL-1β) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J. Biol. Chem. 284, 12762–12771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Allam R., Darisipudi M. N., Tschopp J., Anders H. J. (2013) Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur. J. Immunol. 43, 3336–3342 [DOI] [PubMed] [Google Scholar]
- 95. Netea M. G., Simon A., van de Veerdonk F., Kullberg B. J., Van der Meer J. W., Joosten L. A. (2010) IL-1β processing in host defense: beyond the inflammasomes. PLoS Pathog. 6, e1000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Urbonaviciute V., Voll R. E. (2011) High-mobility group box 1 represents a potential marker of disease activity and novel therapeutic target in systemic lupus erythematosus. J. Intern. Med. 270, 309–318 [DOI] [PubMed] [Google Scholar]
- 97. Wheeler D. S., Chase M. A., Senft A. P., Poynter S. E., Wong H. R., Page K. (2009) Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via Toll-like receptor (TLR)-4. Respir. Res. 10, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Li X., Fang P., Mai J., Choi E. T., Wang H., Yang X. F. (2013) Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Qiu J., Nishimura M., Wang Y., Sims J. R., Qiu S., Savitz S. I., Salomone S., Moskowitz M. A. (2008) Early release of HMGB-1 from neurons after the onset of brain ischemia. J. Cereb. Blood Flow Metab. 28, 927–938 [DOI] [PubMed] [Google Scholar]
- 100. Obeid M., Tesniere A., Ghiringhelli F., Fimia G. M., Apetoh L., Perfettini J. L., Castedo M., Mignot G., Panaretakis T., Casares N., Métivier D., Larochette N., van Endert P., Ciccosanti F., Piacentini M., Zitvogel L., Kroemer G. (2007) Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61 [DOI] [PubMed] [Google Scholar]