

Background: Site-specific hydrogen-deuterium exchange of factor VIIa (FVIIa) was measured to elucidate the mechanism by which FVIIa is up-regulated by tissue factor (TF).
Results: Individual residues in FVIIa whose interaction pattern changes when TF induces the active form are pinpointed.
Conclusion: A picture of the activation signal from TF contact site to active site emerges.
Significance: Guidance for future work on FVIIa allostery is provided.
Keywords: Allosteric Regulation, Blood, Coagulation Factor, Conformational Change, Hydrogen Exchange Mass Spectrometry, Serine Protease, Electron Transfer Dissociation
Abstract
Factor VIIa (FVIIa) is a trypsin-like protease that plays an important role in initiating blood coagulation. Very limited structural information is available for the free, inactive form of FVIIa that circulates in the blood prior to vascular injury and the molecular details of its activity enhancement remain elusive. Here we have applied hydrogen/deuterium exchange mass spectrometry coupled to electron transfer dissociation to pinpoint individual residues in the heavy chain of FVIIa whose conformation and/or local interaction pattern changes when the enzyme transitions to the active form, as induced either by its cofactor tissue factor or a covalent active site inhibitor. Identified regulatory residues are situated at key sites across one continuous surface of the protease domain spanning the TF-binding helix across the activation pocket to the calcium binding site and are embedded in elements of secondary structure and at the base of flexible loops. Thus these residues are optimally positioned to mediate crosstalk between functional sites in FVIIa, particularly the cofactor binding site and the active site. Our results unambiguously show that the conformational allosteric activation signal extends to the EGF1 domain in the light chain of FVIIa, underscoring a remarkable intra- and interdomain allosteric regulation of this trypsin-like protease.
Introduction
Factor VIIa (FVIIa)2 is a trypsin-like serine protease found in the blood. It is formed by endo-proteolytic cleavage of zymogen factor VII (FVII), which is a single chain polypeptide. Upon damage to a blood vessel, both FVIIa and FVII are exposed to the cofactor tissue factor (TF), normally located inside or on the exterior of the blood vessel wall, and FVIIa becomes activated which triggers blood coagulation (1). FVIIa consists of four domains; a γ-carboxyglutamic (Gla) domain, two epidermal growth factor (EGF)–like domains, and a protease domain (2). Despite the suffix “a,” FVIIa is predominantly in a zymogen-like state. FVIIa is highly cofactor-dependent and only exhibits biologically relevant enzymatic activity upon binding to TF. Compared with FVIIa, zymogen FVII was previously observed to have a crippled allosteric response to TF binding (3).
Similar to the activation mechanism of the trypsinogen/trypsin system, that has been extensively characterized, zymogen FVII is converted to FVIIa by endoproteolytic cleavage of a peptide bond which creates the N terminus of the protease domain. This cleavage leaves the two chains connected through an inter-domain disulfide bridge from residue Cys-135 to Cys-262. Unlike trypsin, however, free FVIIa is latent and zymogen-like and TF binding is required for initiation of the blood coagulation cascade. Like in the trypsinogen/trypsin system, the newly generated N terminus inserts into a region close to the three activation loops, termed the activation pocket, during activation (4). This insertion results in the formation of a critical salt bridge between the N-terminal Ile-16 (153 in FVIIa) and Asp-194 (343 in FVIIa) at the bottom of the active site S1 pocket and several hydrogen bonds to residues in the activation domain. The salt bridge leads to the formation of a rigid and correctly designed specificity pocket. Free FVIIa, in contrast, retains zymogen-like properties following limited proteolysis and does not spontaneously rearrange into the active form. The x-ray crystallographic structure of FVIIa bound to TF has provided invaluable structural details of the complex and the active form of FVIIa (5). Several structures of FVIIa in the absence of TF have also been solved, but so far, it has only been possible to crystallize FVIIa in the presence of active site inhibitors that maintain FVIIa in the active form (6, 7). When the inhibitor benzamidine is soaked out of the active site of crystallized FVIIa, the resulting structure reveals a slightly rotated TF-binding helix and a disordered 170-loop (6), indicating larger conformational flexibility in the absence of the inhibitor. The N terminus, however, remained buried in the activation pocket presumably because of constraints exerted by crystal packing. Hence no structural information is available for the free, zymogen-like form of FVIIa that patrols the blood prior to vascular injury. It has been shown that TF changes FVIIa and FVII structurally upon binding (3, 8), but more detailed information on the precise trigger points and solution-phase dynamics for TF-induced FVIIa activation is needed.
The role of subtle changes in protein mobility and structural flexibility in the regulation of enzyme function is becoming increasingly evident (9, 10). Sensitive, high-resolution techniques are needed to detect the elusive dynamic interplay of distant sites critical for protein function. The hydrogen/deuterium exchange (HDX) of main-chain amides is highly sensitive to dynamic changes in conformation between protein states, and report on the overall flexibility of the protein backbone and local hydrogen bonding (i.e. conformational dynamics). Mass spectrometry (MS) has evolved to be a powerful technique to measure protein HDX and thus monitor protein dynamics in solution, due to tolerance to complex protein systems, buffer composition, and low sample concentration. More recently, the integration of electron transfer dissociation (ETD) into the HDX-MS workflow has enabled the mapping of conformational changes in proteins at a spatial resolution down to individual residues (11, 12, 31–35). During HDX-ETD measurements, peptides generated from solution-phase proteolysis are further fragmented in the gas-phase by ETD, and through mass analysis of fragment ions deuterium contents can often be assigned to individual sites.
Numerous studies in recent years have yielded substantial insight into the complex conformational changes governing FVIIa regulation (reviewed in Refs. 13, 14). However, mechanistic details concerning allostery in the protease domain still remain unclear due to insufficient experimental structural information. Furthermore, the presence of inter-domain allostery between the protease domain and the light chain of FVIIa is disputed due to a lack of experimental evidence. In light of earlier studies (3, 15, 16), the current work seeks to address some of these questions by applying HDX-MS to map the conformational rearrangements occurring in both chains of FVIIa during TF- and inhibitor-mediated activation. Moreover, we perform a targeted high-resolution HDX-ETD analysis of regions of FVIIa observed to undergo reduced HDX upon TF association to reveal changes in conformational dynamics pertinent to the mechanism of activation. Our results provide much needed experimental evidence that will be instrumental in the development of a more elaborate and comprehensive view of FVIIa activation.
EXPERIMENTAL PROCEDURES
Expression and purification of FVIIa (17) and soluble TF-(1–219) (18) were performed as described previously. Deglycosylation of FVIIa (3) and incorporation of d-Phe-Phe-Arg chloromethylketone into FVIIa have also been described (19). FVIIa was deglycosylated before incorporation of inhibitor.
Deuterium Labeling Reactions
50 μm FVIIa stock solutions (deglycosylated FVIIa, deglycosylated FVIIa bound covalently by an active site inhibitor, D-Phe-Phe-Arg (FFR) chloromethyl ketone, or wild-type FVIIa) were incubated with TF (136 μm) or HDX-MS buffer (20 mm Bis-Tris, pH 6.0, 10 mm CaCl2, 150 mm NaCl) in a volumetric ratio of 1:4 for 15 min. Amide hydrogen/deuterium exchange was initiated by a 10-fold dilution of the incubation mixture in deuterated HDX-MS buffer (20 mm Bis-Tris, pH 6.0, 10 mm CaCl2, 150 mm NaCl, 98% D2O). Exchange was quenched by removing aliquots of the reaction mixture and mixing with an equal volume of ice-cold quench buffer (i.e. 280 mm phosphate and 50 mm Tris(2-carboxyethylphosphine (TCEP)) resulting in a final pH of 2.5. Quenched samples were stored at −80 °C.
Preparation of samples for ETD followed a similar protocol. FVIIa stock solution (50 μm or 68 μm) was incubated with TF (136 μm) or HDX-MS buffer in a volumetric ratio of 3:2 for 15 min. HDX was initiated by a 10-fold dilution of the incubation mixture in deuterated HDX-MS buffer. The exchange reaction was quenched by removing aliquots of the reaction mixture and mixing with an equal volume of ice-cold quench buffer. Quenched samples were stored at −80 °C.
MS Analysis
Deuterium-labeled and quenched samples of recombinant FVIIa and TF were loaded onto a refrigerated nanoACQUITY UPLC system (Waters Inc.) interfaced with a Synapt G2 mass spectrometer (Waters Inc.) for pepsin digestion, chromatography, and mass spectrometry. The mass spectrometer was equipped with an ESI source operated at predefined settings for minimal H/D scrambling as described previously (11, 12, 31–35). ETD was performed in the trap T-wave by reacting radical 1,4-dicyanobenzene anions with quadrupole-selected deuterium-labeled peptide ions. HDX-ETD experiments were performed using a prolonged chromatographic separation time (14–18 min) to minimize peptide coelution and maximize ETD acquisition times. A mild supplemental activation of ETD product ions performed in a data-dependent manner in the transfer T-wave was found to improve ETD efficiency for 2+ peptide ions (36).
RESULTS/DISCUSSION
HDX-MS analyses experiments were performed on the following FVIIa states: FVIIa, TF-bound FVIIa, and FVIIa bound to a covalent active site inhibitor, FFR chloromethyl ketone. Unless noted otherwise, all HDX-MS experiments made use of deglycosylated FVIIa, referred to henceforth simply as FVIIa. Wild-type FVIIa was used only in comparative experiments described later. Proteins were diluted into D2O and the resulting mass increase due to incorporation of deuterium at the main-chain amide groups was monitored as a function of time. Deuterium incorporation was localized to various regions of FVIIa by mass analysis of peptides produced by pepsin proteolysis as described earlier (3). In this manner, a complex mixture of 48 overlapping peptides of FVIIa (Fig. 1) were identified and used to monitor hydrogen exchange of 362 out of 406 amide hydrogens (75% FVIIa light chain, 98% FVIIa heavy chain).
FIGURE 1.

Map of peptides used to monitor the HDX of FVIIa. A, sequence coverage of identified peptic peptides of FVIIa with regions displaying reduced deuterium uptake upon TF binding indicated in red. B, sequence coverage of identified peptic peptides of FVIIa with regions displaying reduced deuterium uptake upon binding of a covalent active site inhibitor (FFR) indicated in red.
Comparison of the HDX of unbound and TF-bound FVIIa revealed a subset of 25 representative FVIIa peptides that displayed reduced deuterium uptake upon TF binding and thus contained residues whose conformational dynamics were affected upon TF-induced activation of FVIIa (Figs. 1A and 2 and supplemental Fig. S1). These results were in general agreement with our earlier analysis of the HDX of FVIIa upon TF binding (3). In this study, we targeted peptides displaying reduced HDX upon TF binding for ETD in follow-up experiments to account in more detail for the sites affected upon activation. Furthermore, we sought to optimize the sequence coverage of the FVIIa light chain, thus being able to provide for the first time a comprehensive analysis of any changes that might occur in this part of the molecule upon activation.
FIGURE 2.

Structural representation of the crystal structure of the FVIIa-TF complex. Regions of FVIIa exhibiting reduced deuterium incorporation due to TF binding are in red color. The active site residues of FVIIa are shown in green sticks, and TF is shown in blue. Representation is based on PDB entry 1DAN.
Finally, we also measured the HDX of FVIIa when occupied by the covalent active site inhibitor FFR. The inhibitor is known to lock FVIIa in an active conformation in the absence of TF and thus provides a unique opportunity to discriminate between the direct effects of TF binding and indirect allosteric effects of the cofactor. We observed that a similar, but not identical, subset of peptides affected by TF also displayed reduced HDX upon FFR binding (Fig. 1B). Interestingly, the reduction in HDX upon incorporation of the inhibitor was generally more pronounced compared with those seen upon TF binding (supplemental Figs. S1 and S2).
ETD Reveals Site-specific Changes in the HDX of FVIIa upon TF Binding
HDX-ETD experiments were performed using a data-dependent inclusion list to optimize precursor ion selection and ETD acquisition parameters for FVIIa peptides of interest. Using this HDX-ETD workflow, we generated ETD data for 7 peptides of FVIIa observed to be affected by TF. The peptides cover regions of interest including the 170-loop (peptide 316–325), activation loop 3 (peptide 371–377), the Ca2+-binding loop (peptide 209–227), strand B2 (peptide 298–305), strand D1 (peptide 201–208), and the interchain linkage region (peptide 134–141). Moreover, ETD measurements on peptide 288–296 (strand A2) were also acquired successfully, but the difference in deuterium uptake between unbound and bound state was not significant within the error margin of our measurements (data not shown.) The derived site-specific HDX information allow us to locate, for the first time, a comprehensive network of residues in FVIIa that relay a concerted allosteric signal from the TF binding site to distinct parts of the protease domain of FVIIa.
The 170-loop and 94-shunt
These two loops are placed on rim of the active site cleft of the protease domain, with the 94-shunt forming part of the S2 site and the 170-loop bordering the S3/S4 sites. Fig. 3A displays the HDX plot for peptide 316–325, showing a reduction in deuterium uptake of the 170-loop upon TF binding (∼1D in both segment 316–325 and segment 306–316). The magnitude of this decrease is slightly less pronounced compared with the HDX-MS studies conducted earlier due to differences in the experimental setup (3). It was also proposed previously that helix α1 (307–312) was distorted and shortened by 3 residues in free FVIIa (7), and the current finding of stabilization in the 306–316 segment could indicate the existence of such a distortion. Fig. 3B shows the HDX-ETD data obtained for peptide 316–325 from the 170-loop in the presence and absence of TF after 15-s exchange time based on four c-ions. The results allow us to pinpoint that the hydrogen bond interactions involving amide hydrogen of residues Ile-323 and Glu-325 are destabilized in free FVIIa (Fig. 3C). Stabilization obviously anchors the 170-loop to the main body of the protease domain and echoes well with the proposal in a recent review suggesting that the stabilization of residues Ile-323 and Glu-325 could be part of step 2 of “Path I” (13). HDX-MS data for a total of 6 peptides covering the 94-shunt were recorded. Like previously observed, only a modest reduction in deuterium uptake was observed upon binding to TF (3) and current ETD analyses failed to sublocalize this further (data not shown).
FIGURE 3.

HDX of the 170-loop. A, HDX plot of peptide 316–325 of the 170-loop. B, localized difference in deuterium uptake for individual residues (red bars) or segments (orange bar) of peptide 316–325 from unbound versus TF-bound FVIIa after 15 s exchange time. C, structural representation of the 170-loop with individual sites shown by ETD to undergo reduced HDX upon TF binding highlighted. Based on HDX-ETD data in B, H-bonds involving amide hydrogens from residues Ile-323 and Glu-325 are likely to be destabilized or absent in unbound FVIIa.
β-Strands A2 and B2 of the Activation Pocket
The site of N-terminal insertion in FVIIa (and other trypsin-like proteases), referred to as the activation pocket, is a cavity formed by parts of activation loops 1 and 2 and β-strands A2 and B2. In addition to both activation loops, dramatic reduction of HDX in peptides of β-strands A2 and a part of B2 was observed upon TF binding to FVIIa in our current study, (Figs. 1A and 4A) which correlates well with previous HDX-MS findings in the mentioned region (3). Fig. 4B displays the HDX-ETD measurements performed on peptide 298–305 of the B2 strand, which allow us to pinpoint stabilized residues to be Leu-300, Val-302, Arg-304, and Leu-305. The observed allostery in this region may serve as the intermediate step 1 in an allosteric “Path II” heading toward the activation pocket described in a recent review (13). As illustrated in Fig. 4C, the pinpointed allosteric sites in the β-strands A2 and B2 obviously shape the secondary structure of this region while anchoring the 170-loop and activation loop 1. Specifically, main chain amides of Leu-300 and Val-302 of strand B2 form integral hydrogen bonds to the neighboring strand A in the active form of FVIIa and our HDX-ETD data show that these hydrogen bonds are significantly destabilized or even absent in the free form of FVIIa. This presumably rearranges the alignment of strands A2 and B2 with respect to each other in the free zymogen-like FVIIa.
FIGURE 4.
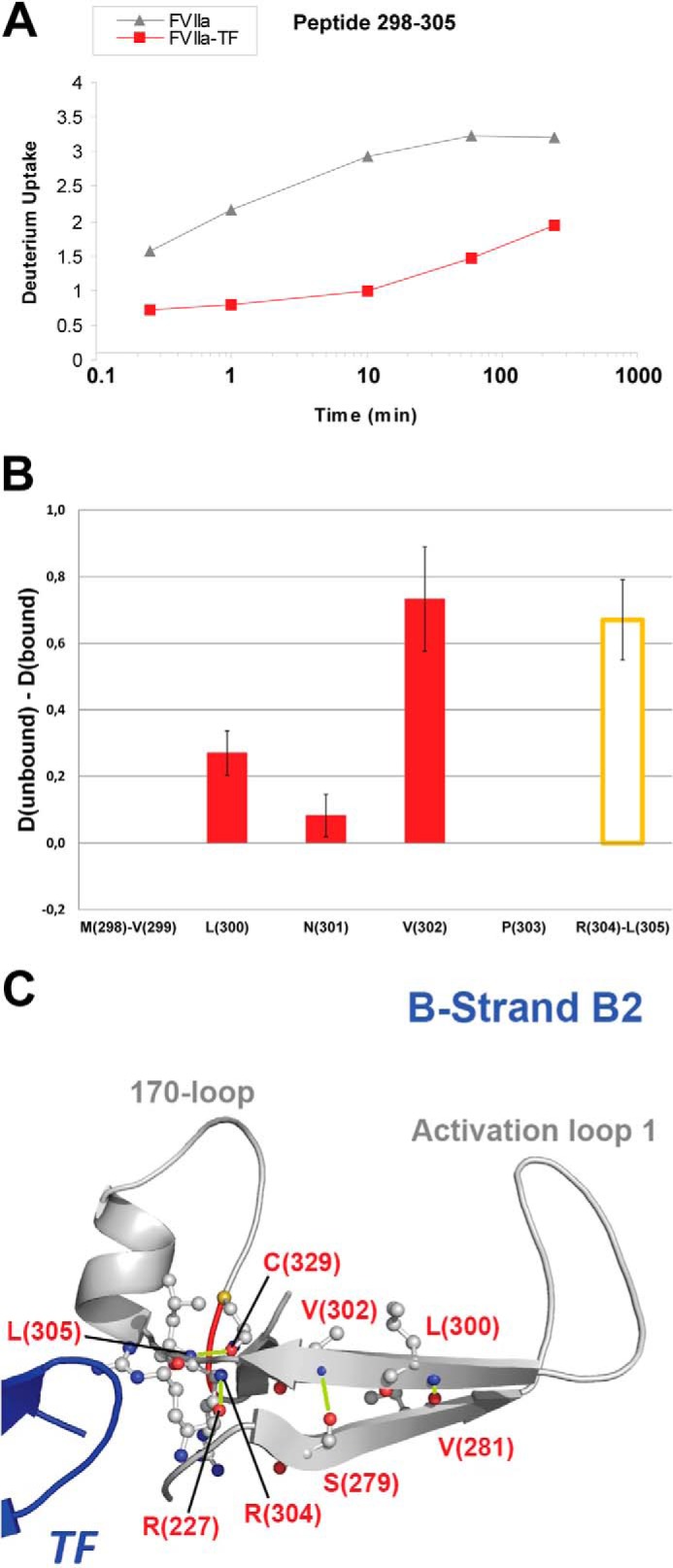
HDX of β-strand B2. A, HDX plot of peptide 298–305 from strand B2. B, localized difference in deuterium uptake for individual residues (red bars) or segments (orange bar) of peptide 298–305 from unbound versus TF-bound FVIIa after 4 h exchange time. C, structural representation of the B2 strand with individual sites revealed by ETD to undergo reduced HDX upon TF binding highlighted (PDB ID: 1DAN). Based on HDX-ETD data in B, H-bonds involving amide hydrogens from residues Leu-300, Val-302, Arg-304, and Leu-305 that are destabilized or absent in unbound FVIIa are shown in lime.
Activation Loops 1, 2, and 3
The three activation loops of trypsin-like serine proteases are situated in near proximity on one face of the protease domain, spanning the region between the active site cleft and the site of N-terminal insertion. Among 5 peptides covering activation loop 1, only a limited decrease in deuterium incorporation was observed (Dunbound − Dbound < 0.5 Da) upon TF binding. This observation was backed up by the ETD measurements performed on peptide 288–296, which confirmed that only a minor change in deuterium uptake took place.
Fig. 5A revealed an average 0.6 D decrease in deuterium uptake in peptide 371–377 upon TF binding, with the effect pinpointed to residues Val-376 and Tyr-377 based on ETD readings (Fig. 5B). This indicates that the hydrogen bonds that link Val-376-Trp-364 and Phe-328-Tyr-377, respectively, are likely to be absent or destabilized in the free form of FVIIa (Fig. 5C). Interestingly, the stabilization of β-strands E2 and F2 through Val-376 seems to anchor activation loop 3, while the stabilization that includes the amide hydrogen of Tyr-377 anchors the 170-loop. Moreover, it was observed in MD and SMD simulations (20) that the distance between residues Trp-364 and Pro-321/Asn-322 in FVIIa was found to vary dramatically between the free and TF-bound form and this is in accordance with our current experimental HDX-ETD data. Interestingly, the decreases in deuterium incorporation in activation loops 1 and 3 induced by the inhibitor were measured to be greater than those mediated by TF. This is possibly due to the short distance of these two loops to the active site, from where the allosteric signal is initiated by the inhibitor.
FIGURE 5.
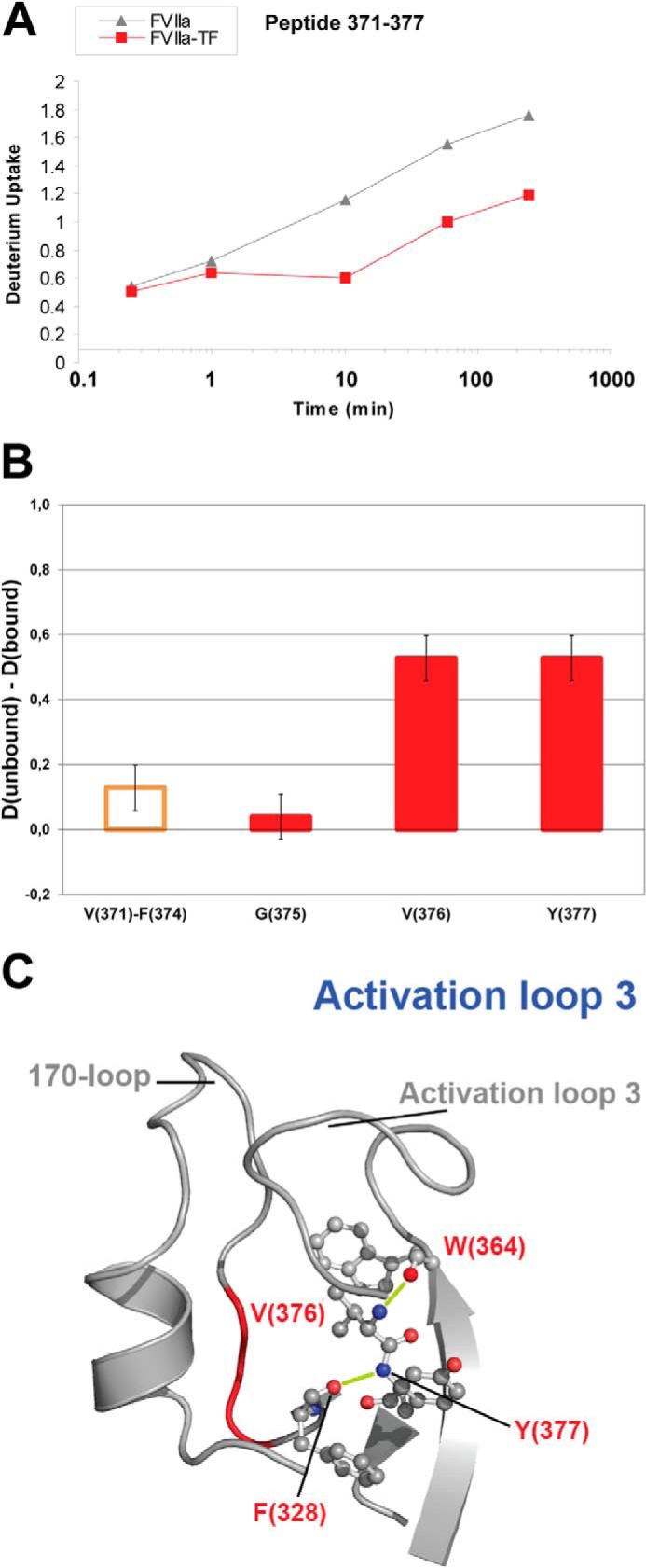
HDX of activation loop 3. A, HDX plot of peptide 371–377 from activation loop 3. B, localized difference in deuterium uptake for individual residues (red bars) or segments (orange bar) of peptide 371–377 from unbound versus TF-bound FVIIa after 4 h exchange time. C, structural representation of activation loop 3 with individual sites shown by ETD to undergo reduced HDX upon TF binding highlighted (PDB ID: 1DAN). Based on HDX-ETD data in B, H-bonds involving amide hydrogens from residues Val-376 and Tyr-377 that are destabilized or absent in unbound FVIIa are shown in lime.
No peptide covering solely activation loop 2 was identified in the current study. However, previous studies conducted on the activation of trypsinogen and HDX-MS studies of FVIIa activation suggested that activation loop 2 does not play a major role in the allosteric activation (3).
Interestingly, the stabilized hydrogen bond from Leu-305 to Cys-329 depicted in Fig. 4C is connected to the hydrogen bond network in Fig. 5C, revealing a conspicuous, direct allosteric path from the primary interaction with TF to Trp-364 positioned in the active site cleft and being the end point of the previously suggested allosteric “Path I” (13).
The Ca2+-binding Loop and Strand D1
The Ca2+-binding loop is spatially localized in the vicinity of the N-terminal tail and binding of Ca2+ has previously been correlated to an increased activity of several trypsin-like proteases including FVIIa (21–25). N-terminal insertion has been shown to stabilize the Ca2+-binding loop and TF binding apparently fails to stabilize this loop in zymogen FVII (3). Figs. 6A and 7A show the HDX plots for peptides 209–227 (Ca2+-binding loop) and 201–208 (D1 strand), which suggest a reduced uptake of 1 deuterium in the Ca2+-binding loop and 0.5 deuterium in the D1 strand upon TF-induced activation.
FIGURE 6.
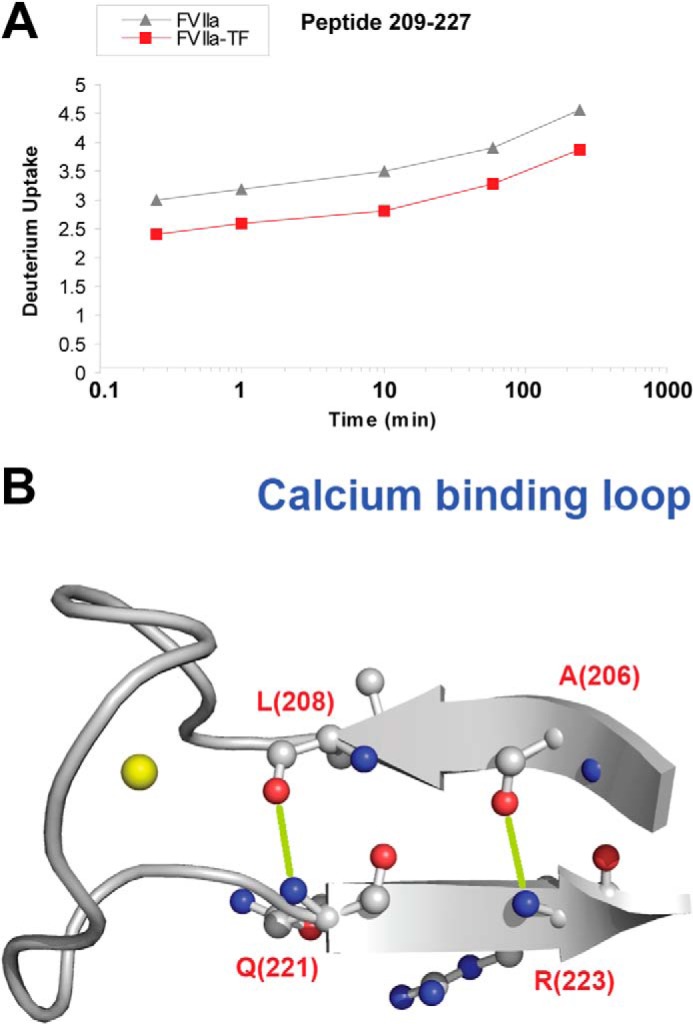
HDX of the Ca2+-binding loop. A, HDX plot of peptide 209–227 from the Ca2+-binding loop. B, structural representation of the Ca2+-binding loop. The calcium ion is depicted as a yellow sphere. Sites that undergo reduced HDX upon TF binding based on ETD measurements on peptide 209–227 are localized to residues Gln-221-Arg-224 (PDB ID: 1DAN). H-bonds involving amide hydrogens from residues Gln-221 and Arg-223 that are likely to be destabilized or absent in unbound FVIIa are shown in lime.
FIGURE 7.
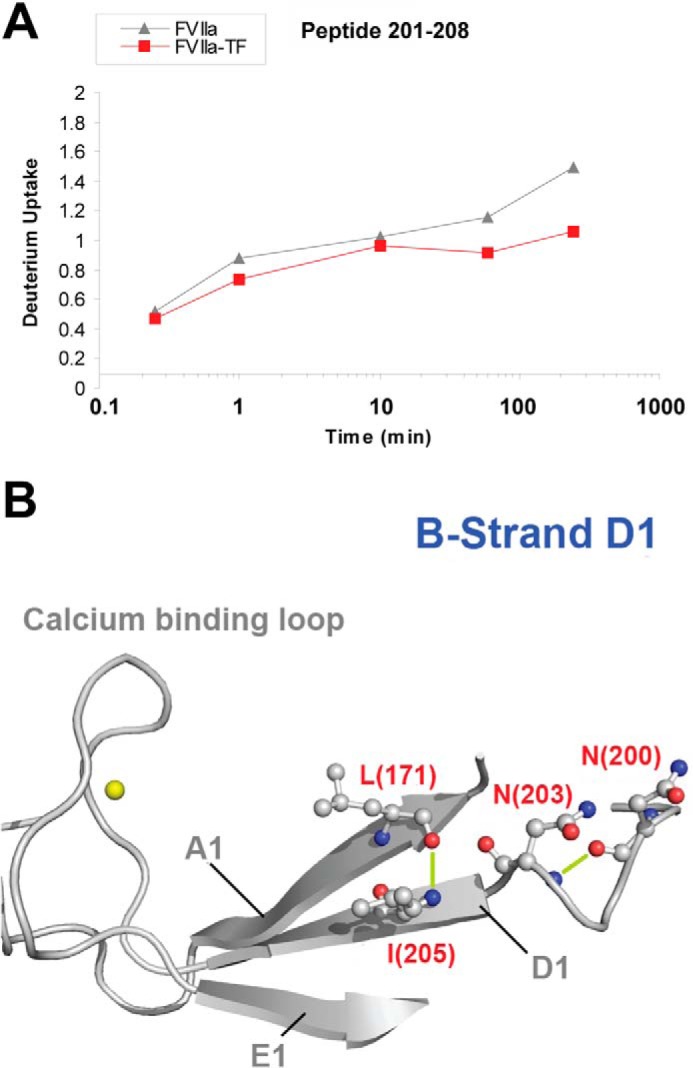
HDX of β-strand D1. A, HDX plot of peptide 201–208 from strand D1. B, structural representation of the D1 strand. The calcium ion is depicted as a yellow sphere. Individual sites that are likely to undergo reduced HDX upon TF binding based on ETD measurements on peptide 201–208 are localized to residues Asn-203-Ile-205 (PDB ID: 1DAN). H-bonds involving amide hydrogens from residues Asn-203 and Ile-205 that are likely to be destabilized or absent in unbound FVIIa are shown in lime.
Based on our ETD results with peptide 209–227 (data not shown), the increased stabilization during N terminus insertion was localized to be within residues Gln-221 to Arg-224. Interestingly, according to the information provided by the crystal structure (PDB code 1DAN) (5) of the FVIIa-TF complex, the main-chain amides of Gln-221 and Arg-223 engage in integral hydrogen bonds that shapes the secondary structure of β-strands D1 and E1, while also anchoring the Ca2+-binding loop (Fig. 6B). Present HDX-ETD data indicate that these interactions are destabilized in the latent, zymogen-like form of FVIIa thus impacting the conformation of the Ca2+ loop and strands D1 and E1.
From the ETD measurements on peptide 201–208, the structural stabilization was pinpointed to lie between Asn-203 and Ile-205. Combined with the information provided by the crystal structure of FVIIa:TF (5), our present findings indicate that the hydrogen bonds connecting Ile-205-Leu-171 and Asn-203-Asn-200 are likely to be weakened or absent in free FVIIa. As shown in Fig. 7B, it is apparent that this stabilization shapes the framework of the local secondary structures around the Ca2+-binding loop. This proves that and further reveals how the region surrounding the Ca2+-binding loop and the N-terminal tail undergoes extensive structural rearrangements upon activation.
The N-terminal Tail and the Interdomain Linkage
Insertion of the N terminus into the activation pocket and the subsequent formation of a salt bridge between Ile-153 and Asp-343, neighboring the active site residue Ser-344, has been postulated as an essential requirement for activation of trypsin-like proteases (26, 27). Consequently, we observed significantly reduced HDX rates in peptide 153–169 of the N-terminal tail of FVIIa following the activation by TF, which is in good agreement with previous results from HDX-MS and carbamylation experiments that reported on the existence and the extent of burial of the N terminus (16, 26). ETD experiments failed to sublocalize this any further.
Located in the proximity of the C-terminal end of the FVIIa light chain, the interdomain linkage is situated at residue Cys-135 where a disulfide bridge with Cys-262 holds the heavy and light chains together. In previous HDX-MS measurements conducted on FVIIa upon activation, a significant decrease in deuterium uptake was observed in this region. Observations from our current study (Fig. 8, A and B) agree with this finding, with significant reduction in deuterium incorporation following TF binding in peptides 133–141 and 134–141.
FIGURE 8.

HDX of the interdomain linkage. HDX plots of (A) peptide 133–141 and (B) peptide 134–141 of the interdomain linkage region that contain the S-S bond that connects the light and heavy chain of FVIIa. C, structural representation of the interdomain linkage. Individual sites that are likely to undergo reduced HDX upon TF binding are localized by ETD to residues Ile-138-Leu-141 (PDB ID: 1DAN). The H-bond involving amide hydrogen from residue Leu-141 that is likely to be destabilized or absent in unbound FVIIa is shown in lime.
Our ETD measurements performed on peptide 134–141 (data not shown) suggest that the hydrogen bond between residues Ile-138 and Leu-141 is stabilized or formed during TF-induced FVIIa activation. As shown in Fig. 8C, this stabilization obviously anchors the secondary structure in the surroundings of the interdomain linkage.
Analyzing the Conformational Effect of FVIIa Glycosylation
To gain insight into the functional role and impact of the N-linked glycan on Asn-322 in the 170-loop of FVIIa, we have measured the HDX of FVIIa in its wild-type glycosylated form and in a state where the N-linked glycans on Asn-322, and Asn-145 in the C-terminal tail of the light chain, have been removed. The deuterium uptake pattern of glycosylated wild-type FVIIa and that of deglycosylated FVIIa were similar but showed significant minor local differences. Primarily we observed an increase in the deuterium uptake upon deglycosylation in peptides 61–68 (EGF1 domain), 370–377 (activation loop 3) and 298–305 (B2-strand). This would indicate that the glycans are stabilizing the conformational framework in the mentioned areas located in the vicinity of the N-linked glycans at Asn-145 and Asn-322. To our knowledge, this represents the first experimental evidence for a localized structural role of the N-linked glycans of FVIIa. Notably, we did not observe a significant difference in activity between glycosylated and deglycosylated FVIIa (data not shown).
Detection of Long-range Interdomain Allostery in FVIIa
To also investigate the presence of allosteric pathways propagating from the active site, we analyzed the HDX of FVIIa in an inhibitor-bound form. The inhibitor used was a tripeptide chloromethylketone with the sequence D-Phe-Phe-Arg, which binds covalently to His-193 and Ser-344 in the active site and occupies the S1, S2, and aryl-binding subsites (7). Because of its limited size, the inhibitor is unlikely to have steric effects on the HDX of FVIIa, and any reduction in deuterium uptake it might induce will thus be due to allosteric effects. Moreover, by using a covalent inhibitor like this, and ensuring total inhibition of FVIIa, we can investigate solely the inhibitor-occupied form of the enzyme without interference from the unbound state, as is the case when using a non-covalent inhibitor (or binding to TF). As shown in Fig. 1B, we observed reduction in deuterium uptake of peptides 25–61 and 61–68, indicating the propagation of long-range allosteric signals. In a previous study, we observed a similar effect in a peptide covering residues 63–80 of the first EGF-like domain upon binding of a non-covalent inhibitor to FVIIa as well as the physiological cofactor TF. The present finding of decreased HDX for an overlapping peptide in the EGF-like domain upon incorporation of a covalent active-site inhibitor into FVIIa corroborates earlier indications and lends support to the presence of a remarkable long-range allosteric link between the heavy and light chains of FVIIa. Interestingly, the peptide 61–68 is part of the Ca2+-binding site in the first EGF-like domain (5). As no effects are detected in the second EGF-like domain, the crosstalk is likely mediated through a direct interaction between the protease domain and the first EGF-like domain. This suggests that this part of the light chain has substantial flexibility in solution relative to the protease domain. Data from other studies of FVIIa in solution and in silico favor this interpretation (7, 28–30).
Remarkably, the allosteric activation signal induced by TF or an active-site inhibitor is not confined to the heavy chain (protease domain) as structural stabilization is observed even in remote regions of FVIIa such as that containing the disulfide bond linking the heavy and light chains and in the first EGF-like domain. This interdomain signaling extends approx. 50 Å through space from the active site. It constitutes an impressive example of concerted interplay between functional sites in this coagulation factor and reveals the potential for allosteric regulation of the family of trypsin-like proteases.
CONCLUSION
A challenge in understanding the activation and function of trypsin-like serine proteases is to define the roles played by key residues. By monitoring the HDX of FVIIa in selected conformational states, we are able to provide a detailed map of the regions in both chains of FVIIa that display conformational stabilization upon activation. By high-resolution HDX analysis, we have pinpointed the location of residues responsible for the observed widespread reduction in HDX in the protease domain. In short, FVIIa displayed conformational stabilization in the TF binding loop, sheet A2 and B2 and the activation loops and most of these stabilizations could be further pinpointed to distinct residues or short segments by ETD. They appear to be due to optimized hydrogen bonding at the base of loops or between β-strands, which explains the key role of the identified residues in shaping the local conformational environments. Our results show that TF stabilizes the active site region via strengthening of a sophisticated hydrogen bonding network between key sites connecting the TF binding site and the active site. In particular, the increased protection of the main chain hydrogen bond from Val-376 to Trp-364 by TF reveals a direct allosteric path from the primary interaction with TF to the active site. Finally, analysis of the HDX of FVIIa with a covalent inhibitor demonstrated a long-range allosteric signal from the active site to multiple sites in the protease domain and extending to the first EGF-like domain in the light chain. Our results provide strong experimental evidence of an allosteric linkage between conformational stabilizations of the protease domain and distinct parts of the light chain. The functional consequences of this hitherto unappreciated interdomain crosstalk warrant further studies.
Supplementary Material
This work was supported by the Marie Curie Actions Programme of the E.U Grant No. PCIG09-GA-2011-294214 (to K. D. R.) and the Danish Council for Independent Research Natural Sciences (Steno Grant No. 11-104058, to K. D. R.).

This article contains supplemental Fig. S1 and S2.
- FVII(a)
- (activated) coagulation factor VII
- EGF
- epidermal growth factor
- ETD
- electron transfer dissociation
- Gla
- γ-carboxyglutamic acid
- HDX
- hydrogen/deuterium exchange
- MS
- mass spectrometry
- TF
- tissue factor.
REFERENCES
- 1. Davie E. W., Fujikawa K., Kisiel W. (1991) The coagulation cascade: initiation, maintenance, and regulation. Biochemistry 30, 10363–10370 [DOI] [PubMed] [Google Scholar]
- 2. Hagen F. S., Gray C. L., O'Hara P., Grant F. J., Saari G. C., Woodbury R. G., Hart C. E., Insley M., Kisiel W., Kurachi K. (1986) Characterization of a cDNA coding for human factor VII. Proc. Natl. Acad. Sci. U.S.A. 83, 2412–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rand K. D., Jørgensen T. J. D., Olsen O. H., Persson E., Jensen O. N., Stennicke H. R., Andersen M. D. (2006) Allosteric activation of coagulation factor VIIa visualized by hydrogen exchange. J. Biol. Chem. 281, 23018–23024 [DOI] [PubMed] [Google Scholar]
- 4. Fehlhammer H., Bode W., Huber R. (1977) Crystal structure of bovine trypsinogen at 1–8 A resolution. II. Crystallographic refinement, refined crystal structure and comparison with bovine trypsin. J. Mol. Biol. 111, 415–438 [DOI] [PubMed] [Google Scholar]
- 5. Banner D. W., D'Arcy A., Chène C., Winkler F. K., Guha A., Konigsberg W. H., Nemerson Y., Kirchhofer D. (1996) The crystal structure of the complex of blood coagulation factor VIIa with soluble tissue factor. Nature 380, 41–46 [DOI] [PubMed] [Google Scholar]
- 6. Sichler K., Banner D. W., D'Arcy A., Hopfner K. P., Huber R., Bode W., Kresse G. B., Kopetzki E., Brandstetter H. (2002) Crystal structures of uninhibited factor VIIa link its cofactor and substrate-assisted activation to specific interactions. J. Mol. Biol. 322, 591–603 [DOI] [PubMed] [Google Scholar]
- 7. Pike A. C., Brzozowski A. M., Roberts S. M., Olsen O. H., Persson E. (1999) Structure of human factor VIIa and its implications for the triggering of blood coagulation. Proc. Natl. Acad. Sci. U.S.A. 96, 8925–8930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Persson E., Nielsen L. S., Olsen O. H. (2001) Substitution of aspartic acid for methionine-306 in factor VIIa abolishes the allosteric linkage between the active site and the binding interface with tissue factor. Biochemistry 40, 3251–3256 [DOI] [PubMed] [Google Scholar]
- 9. Hoofnagle A. N., Resing K. A., Goldsmith E. J., Ahn N. G. (2001) Changes in protein conformational mobility upon activation of extracellular regulated protein kinase-2 as detected by hydrogen exchange. Proc. Natl. Acad. Sci. U.S.A. 98, 956–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kamata K., Mitsuya M., Nishimura T., Eiki J.-i., Nagata Y. (2004) Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure 12, 429–438 [DOI] [PubMed] [Google Scholar]
- 11. Rand K. D., Zehl M., Jensen O. N., Jørgensen T. J. (2009) Protein hydrogen exchange measured at single-residue resolution by electron transfer dissociation mass spectrometry. Anal. Chem. 81, 5577–5584 [DOI] [PubMed] [Google Scholar]
- 12. Rand K. D., Pringle S. D., Morris M., Brown J. M. (2012) Site-specific analysis of gas-phase hydrogen/deuterium exchange of peptides and proteins by electron transfer dissociation. Anal. Chem. 84, 1931–1940 [DOI] [PubMed] [Google Scholar]
- 13. Persson E., Olsen O. H. (2011) Allosteric activation of coagulation factor VIIa. Front. Biosci. 16, 3156–3163 [DOI] [PubMed] [Google Scholar]
- 14. Olsen O. H., Persson E. (2008) Cofactor-induced and mutational activity enhancement of coagulation factor VIIa. Cell Mol. Life Sci. 65, 953–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rand K. D. (2013) Pinpointing changes in higher-order protein structure by hydrogen/deuterium exchange coupled to electron transfer dissociation mass spectrometry. Int. J. Mass Spectrom. 338, 2–10 [Google Scholar]
- 16. Rand K. D., Andersen M. D., Olsen O. H., Jørgensen T. J., Østergaard H., Jensen O. N., Stennicke H. R., Persson E. (2008) The origins of enhanced activity in factor VIIa analogs and the interplay between key allosteric sites revealed by hydrogen exchange mass spectrometry. J. Biol. Chem. 283, 13378–13387 [DOI] [PubMed] [Google Scholar]
- 17. Thim L., Bjoern S., Christensen M., Nicolaisen E. M., Lund-Hansen T., Pedersen A. H., Hedner U. (1988) Amino acid sequence and posttranslational modifications of human factor VIIa from plasma and transfected baby hamster kidney cells. Biochemistry 27, 7785–7793 [DOI] [PubMed] [Google Scholar]
- 18. Freskgård P. O., Olsen O. H., Persson E. (1996) Structural changes in factor VIIa induced by Ca2+ and tissue factor studied using circular dichroism spectroscopy. Protein Sci. 5, 1531–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sørensen B. B., Persson E., Freskgârd P. O., Kjalke M., Ezban M., Williams T., Rao L. V. (1997) Incorporation of an active site inhibitor in factor VIIa alters the affinity for tissue factor. J. Biol. Chem. 272, 11863–11868 [DOI] [PubMed] [Google Scholar]
- 20. Olsen O. H., Rand K. D., Østergaard H., Persson E. (2007) A combined structural dynamics approach identifies a putative switch in factor VIIa employed by tissue factor to initiate blood coagulation. Protein Sci. 16, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bode W., Schwager P. (1975) The single calcium-binding site of crystallin bovine β-trypsin. FEBS Lett. 56, 139–143 [DOI] [PubMed] [Google Scholar]
- 22. Rezaie A. R., Esmon C. T. (1994) Asp-70–>Lys mutant of factor X lacks high affinity Ca2+ binding site yet retains function. J. Biol. Chem. 269, 21495–21499 [PubMed] [Google Scholar]
- 23. Sabharwal A. K., Birktoft J. J., Gorka J., Wildgoose P., Petersen L. C., Bajaj S. P. (1995) High affinity Ca2+-binding site in the serine protease domain of human factor VIIa and its role in tissue factor binding and development of catalytic activity. J. Biol. Chem. 270, 15523–15530 [DOI] [PubMed] [Google Scholar]
- 24. Wildgoose P., Foster D., Schiødt J., Wiberg F. C., Birktoft J. J., Petersen L. C. (1993) Identification of a calcium binding site in the protease domain of human blood coagulation factor VII: evidence for its role in factor VII-tissue factor interaction. Biochemistry 32, 114–119 [DOI] [PubMed] [Google Scholar]
- 25. Bjelke J. R., Olsen O. H., Fodje M., Svensson L. A., Bang S., Bolt G., Kragelund B. B., Persson E. (2008) Mechanism of the Ca2+-induced enhancement of the intrinsic factor VIIa activity. J. Biol. Chem. 283, 25863–25870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Higashi S., Nishimura H., Aita K., Iwanaga S. (1994) Identification of regions of bovine factor VII essential for binding to tissue factor. J. Biol. Chem. 269, 18891–18898 [PubMed] [Google Scholar]
- 27. Higashi S., Matsumoto N., Iwanaga S. (1996) Molecular mechanism of tissue factor-mediated acceleration of factor VIIa activity. J. Biol. Chem. 271, 26569–26574 [DOI] [PubMed] [Google Scholar]
- 28. Jin J., Perera L., Stafford D., Pedersen L. (2001) Four loops of the catalytic domain of factor viia mediate the effect of the first EGF-like domain substitution on factor viia catalytic activity. J. Mol. Biol. 307, 1503–1517 [DOI] [PubMed] [Google Scholar]
- 29. Ohkubo Y. Z., Morrissey J. H., Tajkhorshid E. (2010) Dynamical view of membrane binding and complex formation of human factor VIIa and tissue factor. J. Thromb Haemost. 8, 1044–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mosbaek C. R., Nolan D., Persson E., Svergun D. I., Bukrinsky J. T., Vestergaard B. (2010) Extensive small-angle X-ray scattering studies of blood coagulation factor VIIa reveal interdomain flexibility. Biochemistry 49, 9739–9745 [DOI] [PubMed] [Google Scholar]
- 31. Rand K. D., Adams C. M., Zubarev R. A., Jorgensen T. J. D. (2008) Electron capture dissociation proceeds with a low degree of intramolecular migration of peptide amide hydrogens. J. Am. Chem. Soc. 130, 1341–1349 [DOI] [PubMed] [Google Scholar]
- 32. Abzalimov R. R., Kaplan D. A., Easterling M. L., Kaltashov I. A. (2009) Protein conformations can be probed in top-down HDX MS experiments utilizing electron transfer dissociation of protein ions without hydrogen scrambling. J. Am. Soc. Mass. Spectrom. 20, 1514–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pan J., Han J., Borchers C. H., Konermann L. (2008) Electron capture dissociation of electrosprayed protein ions for spatially resolved hydrogen exchange measurements. J. Am. Chem. Soc. 130, 11574–11575 [DOI] [PubMed] [Google Scholar]
- 34. Zehl M., Rand K. D., Jensen O. N., Jørgensen T. J. D. (2008) Electron transfer dissociation facilitates the measurement of deuterium incorporation into selectively labeled peptides with single residue resolution. J. Am. Chem. Soc. 130, 17453–17459 [DOI] [PubMed] [Google Scholar]
- 35. Rand K. D., Zehl M., Jorgensen T. J. (2014) Measuring the hydrogen/deuterium exchange of proteins at high spatial resolution by mass spectrometry: overcoming gas-phase hydrogen/deuterium scrambling. Acc. Chem. Res. 47, 3018–3027 [DOI] [PubMed] [Google Scholar]
- 36. Rand K. D., Pringle S. D., Morris M., Engen J. R., Brown J. M. (2011) ETD in a traveling wave ion guide at tuned z-spray ion source conditions allows for site-specific hydrogen/deuterium exchange measurements. J. Am. Soc. Mass Spectrom. 22, 1784–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.