

Background: The mechanisms regulating the survival and death of neurons are poorly understood.
Results: JAZ promotes neuronal survival by stimulating the expression of p21 (WAF1/CIP1) transcriptionally, thereby inhibiting aberrant cell cycle re-entry.
Conclusion: JAZ is a novel regulator of neuronal survival.
Significance: Understanding how JAZ protects neurons could lead to the development of novel therapies for neurodegenerative diseases.
Keywords: Cell Cycle, Huntington Disease, Neurodegeneration, Neuroprotection, Sirtuin 1 (SIRT1)
Abstract
SIRT1, a class III histone deacetylase, protects neurons in various models of neurodegenerative diseases. We previously described that neuroprotection by SIRT1 is independent of its catalytic activity. To elucidate how SIRT1 protects neurons, we performed a mass spectrometric screen to find SIRT1-interacting proteins. One of the proteins identified was JAZ (Znf346), a member of a new class of Cys-2–His-2 zinc finger proteins. To investigate the significance of JAZ in the regulation of neuronal survival, we overexpressed it in neurons. We found that JAZ protects cerebellar granule neurons against potassium deprivation-induced death and cortical neurons from death resulting from oxidative stress. JAZ also protects neurons against toxicity induced by mutant huntingtin and mutant ataxin-1 expression. Although expression of endogenous JAZ does not change in neurons primed to die, knockdown of its expression promotes death of otherwise healthy neurons. In contrast to its protective effect in neurons, overexpression of JAZ in different cell lines promotes death. We find that JAZ suppresses cell cycle progression, thereby explaining its contrasting effect in postmitotic neurons versus proliferating cell lines. Although not affecting the expression of several cyclins, overexpression of JAZ stimulates expression of p21 (WAF1/CIP1), a cell cycle inhibitor known to have neuroprotective effects. Results of chromatin immunoprecipitation and transcriptional assays indicate that the stimulatory effect of JAZ on p21 expression is mediated at the transcriptional level. Furthermore, knockdown of p21 expression inhibits the neuroprotective effect of JAZ. Together, our results suggest that JAZ protects neurons by inhibiting cell cycle re-entry through the transcriptional stimulation of p21 expression.
Introduction
SIRT1, the mammalian homolog of yeast Sir2, is a class III-NAD+-dependent histone deacetylase that protects neurons from death in several tissue culture and in vivo models of neurodegenerative disease (1–3). Although neuroprotection by SIRT1 is generally believed to be dependent on its deacetylase activity, we previously reported that SIRT1 could protect neurons by a catalytic activity-independent mechanism (4). We showed that protection by SIRT1 in cultured neurons was not reduced by three separate pharmacological SIRT1 inhibitors and that mutant forms of SIRT1 deficient in catalytic activity were as neuroprotective as wild-type SIRT1 (4). More recently, we have examined the effect of a panel of SIRT1 deletion mutants and found that mutants lacking substantial portions of the catalytic domain of SIRT1 or lacking the essential for Sirt1 activity (ESA) region, recently identified as obligatory for SIRT1 deacetylase activity (5), retain full neuroprotective activity.2 To gain insight into the catalytic-independent mechanism by which SIRT1 protects neurons, we conducted a screen aimed at identifying SIRT1-interacting proteins in which SIRT1 was overexpressed in HT22 neuroblastoma cells and co-immunoprecipitated proteins identified by mass spectrometric analysis. One of the proteins identified in this screen was JAZ, also referred to as Znf346.
JAZ (Just Another Zinc finger protein) is the founding member of a new class of C2H2-type zinc finger proteins that was first identified as a gene up-regulated by interleukin-3 deprivation (6). JAZ is expressed widely and relatively highly in most organs, including the brain (6). It is localized primarily in the nucleus, but at least one study has described nucleo-cytoplasmic shuttling (7). There are only a handful of publications on JAZ, and thus its biological functions are not well understood. Although lacking a classical dsRNA-binding domain, JAZ binds dsRNA with high affinity in vitro through its C2H2 zinc fingers (6, 8). More recent work has suggested that JAZ mediates cell cycle arrest at the G1 phase (9, 10). Both p53-dependent and -independent mechanisms have been suggested for this inhibitory effect on cell cycle progression (9, 10).
We report that, like SIRT1, JAZ has strong neuroprotective activity, protecting in models where death is not due to proteinopathic stress as well as in a model of Huntington disease and spinocerebellar ataxia type-1 where accumulation of misfolded protein aggregates triggers neuronal death. We find that JAZ protects neurons by inhibiting cell cycle machinery through the up-regulation of the cyclin-dependent kinase inhibitor protein p21 (WAF1/CIP1). Indeed, knockdown of p21 blocks the neuroprotective effect of JAZ.
EXPERIMENTAL PROCEDURES
Materials
Unless stated otherwise, all of the reagents and cell culture media were obtained from Invitrogen. All chemical reagents were purchased from Sigma. Tissue culture poly-l-lysine was purchased from Trevigen (Gaithersburg, MD). Antibodies used in this paper were as follows: FLAG (catalog no. F1804, Sigma); HA (Y-11 catalog no. sc-805 and F-7 catalog no. sc-7392, Santa Cruz Biotechnology, Santa Cruz, CA); α-tubulin (TU-02 catalog no. sc-8035, Santa Cruz Biotechnology); GFP (B-2 catalog no. sc-9996a and FL catalog no. sc-8334, Santa Cruz Biotechnology); JAZ (Znf346 catalog no. SAB3500568, Sigma); p21 (catalog no. 556430, BD PharmingenTM); and SIRT1 (catalog no. 9475S, Cell Signaling). Secondary antibodies for Western blotting experiments were obtained from Pierce, and enhanced polyvinylidene difluoride membrane was from Bio-Rad. Secondary antibodies for immunocytochemistry were purchased from Jackson ImmunoResearch.
Plasmids
FLAG-tagged JAZ (JAZ-FLAG) was cloned from whole rat brain cDNA using the following primers: JAZ-FLAG forward, 5′-ATGGAGTGTCCTGTGCCC-3′, and JAZ-FLAG reverse, 5′-CGTCCTTGTAGTCGTCTTCCACTGTGCTTGAGT-3′. JAZ-HA was generated using JAZ-FLAG as a template with the following primers: JAZ-HA forward, 5′-CTAGGGATCCATGGAGTGTCCTGTGCCCGA-3′, and JAZ-HA reverse, 5′-ACATCGTATGGGTAGTCTTCCACTGTGCTTGAGT-3′. Full-length FLAG-tagged p21 (p21-FLAG) was purchased from Addgene. Full-length p21 promoter luciferase constructs (WWP-luc) were a gift from Dr. Ming Zhang (Northwestern University Feinberg School of Medicine). GFP-tagged Htt plasmids (Htt-Q15 and Htt-Q138) were a kind gift from Dr. Troy Littleton (Massachusetts Institute of Technology). FLAG-tagged Ataxin plasmids (Atx1–30Q and Atx1–82Q) were a gift from Chih-Cheg Tsai (Robert Wood Johnson Medical Center).
Adenovirus
JAZ-FLAG adenovirus (Ad-JAZ) was generated using a kit (ViraPowerTM adenoviral expression system, Invitrogen). The primers used for Ad-JAZ are as follows: Ad JAZ forward, 5′-GGGG ACA AGT TTG TAC AAA AAA GCA GGC TTC ACC ATG GAG TGT CCT GTG CCC AT-3′, and Ad JAZ reverse, 5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTC CTA CTTGTCATCGTCGTCCTTGTAGTC-3′. The PCR products were fully sequenced before utilization in virus production. Virus was amplified in the HEK293A cell line and purified using CsCl density gradient centrifugation. The final step of the purification process was dialysis using 1× PBS at 4 °C for 24–36 h with constant stirring. Cerebellar granule neurons (CGNs)3 were infected with either a GFP or JAZ adenovirus for 36 h after which mRNA or lysates were collected for RT-PCR or Western blot analysis, respectively.
Neuronal Culture, Treatments, and Viability Assay
CGNs were cultured from 7- to 8-day-old Wistar rats and were plated as described previously (11). Transient transfection was performed 5 days after plating for 8 or 24 h (see figure legends) followed by switching to HK (high potassium, 25 mm) or LK (low potassium, 5 mm) medium (12, 13). Viability of the transfected cells was determined 24 h later by DAPI staining based on the morphology of the cell nuclei. Survival was normalized to GFP in HK condition.
Cortical cultures were obtained from the cerebral cortex of Wistar rats (day 17 or 18 of gestation) using neurobasal media with B27 supplement. Five days after plating, the cultures were transfected using the calcium phosphate method as described previously (13, 14). 8 h after transfection, 1 mm homocysteic acid (HCA) was added for 15–18 h. HCA induces death as a result of oxidative stress as described previously (15–17). HCA stock solution was 100 mm at pH 7.5.
RNA Preparation and RT-PCR
RNA was extracted from cell lines or cultured neurons according to the manufacturer's instructions using TRIzol (Invitrogen). cDNA was prepared using the Superscript First Strand Synthesis system for RT-PCR kit (Invitrogen), and PCR was performed with GoTaq Green Master Mix (Promega, Madison, WI). The primers used for PCR amplification were as follows: actin forward, 5′-AGGACTCCTATGTGGGTGACGA-3′, and actin reverse, 5′-CGTTGCCAATAGTGATGACCTG-3′; JAZ forward, 5′-AGTGTCCTGTGCCCGATTT-3′, and JAZ reverse, 5′-AGTTCCTTGCGTGGGTCTT-3′; JAZ-MR forward, 5′-GATCCAGAAGAACCAGTGTCTCTTC-3′, and JAZ-MR reverse, 5′-GTGCTTGAGTCCTTTGGAGTGG-3′; GFP forward, 5′-GCACGACTTCTTCTTCAAGTCCGCCA-3′, and GFP reverse, 5′GCGGATCTTGAAGTTCACCTTGA-3′; c-Jun forward, 5′-GATGGAAACGACCTTCTACG-3′, and c-Jun reverse, 5′-GTTGAAGTTGCTGAGGTTGG-3′; CycA1 forward, 5′-TGGTGGAGGTTGGGGAAGAATA-3′, and CycA1 reverse, 5′-AGCTCACTCAGGCAAGGCACA-3′; CycB1 forward, 5′-TTGTGTGCCCAAGAAGATGCT-3′, and CycB1 reverse, 5′-TTCTTAGCCAGGTGCTGCAT-3′; CycD1 forward, 5′-CTCCTCAACGACCGGGTGCT-3′, and CycD1 reverse, 5′-CGGAGGCAGTCCGGTCACA-3′; CycE forward, 5′-CTTTTACAGCTTATTGGGATTTCA-3′, and CycE reverse, 5′-AATGGAACCATCCACTTGACACA-3′; p15-MR forward, 5′-CGGTGCGGCAGCTCCTGGAAG-3′; p15-MR reverse, 5′-GCAGCGTGCAGATACCTCGC-3′; p15-H forward, 5′-GGACTAGTGGAGAAGGTGCG-3′, and p15-H reverse, 5′-GCAGGTACCCTGCAACGTC-3′; p21-H forward, 5′-GAGCAGCTGAGCCGCGA-3′, and p21-H reverse, 5′-CTGGTCTGCCGCCGTTTTC-3′; p27 forward, 5′-CCGGGACTTGGAGAAGCACTGC-3′, and p27 reverse, 5′-GGCTTCTTGGGCGTCTGCTCCAC-3′; and p57 forward, 5′-CTACCGCGAGACGGTGCAGGTG-3′, and p57 reverse, 5′-GCGCGGGGTCTGCTCCACCG-3′.
Western Blotting
Cultured neurons or cell lines were lysed in 1× cell lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 μg/ml leupeptin, and protease inhibitor mixture tablets). The lysates were then kept at −80 °C for at least an hour before proceeding to Western blot analysis. Western blots were performed as described previously (13, 14). Primary antibodies were used in a 1:1000 dilution except JAZ antibody at 1:5000 and tubulin at 1:10,000, and secondary antibodies were used in a 1:10,000 dilution.
Cell Line and Transfection
For viability experiments involving JAZ overexpression, HT22 (mouse hippocampal cell line) and HEK293T (human embryonic kidney cell line) cells were 25–30% at the time of transfection, and viability was quantified 24 h later. For co-immunoprecipitation and other analyses with these cell lines, cells were at 40–50% confluence when transfected.
shRNA Knockdown Experiment
For knockdown analysis of JAZ, five shRNAs were obtained from Sigma (TRCN0000099256, TRCN0000099258, TRCN0000099259, TRCN0000316265, and TRCN0000348955) denoted as JAZ Sh1, JAZ Sh2, JAZ Sh3, JAZ Sh4, and JAZ Sh5, respectively. The pLKO.1-TRC control vector, which contains a non-hairpin 18-bp insert, was used as a transfection control. Five different shRNAs were obtained for p21 knockdown from Sigma (TRCN0000042583, TRCN0000042585, TRCN0000042587, TRCN0000054901, and TRCN0000054902) denoted as ShA, ShB, ShC, ShD, and ShE. The efficiency of the shRNAs was checked in both HEK293T (by suppression of ectopically expressed rat JAZ construct) and HT22 cell line by overexpressing shRNAs for 3 days.
BrdU Assay
BrdU assay was performed in cultures that had been transfected as described previously (18). Briefly, 20 μm BrdU was added to the medium 22 h after transfection for 2 h. Cells were then fixed, washed with 0.5% Triton X-100, and followed by washes with 1 and 2 n HCl. Immunocytochemistry was performed using BrdU antibody (catalog no. B8434, Sigma) at a 1:100 dilution along with another antibody to the tag of the transfected protein. Incubation with secondary antibody was performed the following day before the slides were analyzed by fluorescence microscopy.
Immunoprecipitation
Cell lysates for performing immunoprecipitation were collected as described under “Western Blotting.” An aliquot of the pre-immunoprecipitation whole cell lysate (10% input) was mixed with 6× SDS and prepared for Western blot analysis. The rest of the lysates were subjected to pulldown with the respective antibodies and protein A/G beads. The bead-bound antibodies were allowed to bind to protein at 4 °C overnight with constant stirring followed by washes with 1× cell lysis buffer. The lysate was then subjected to Western blot analysis. To perform endogenous co-immunoprecipitation, 17-day rat brain was collected in 1× RIPA buffer followed by homogenization. 1500 μg of protein was used for pulldown, and 150 μg of protein was used for input.
Image Analysis
ImageJ was used to quantify the immunocytochemistry panels. Area, minimum and maximum gray values, and integrated density (intensity) were selected in the set measurement option. Integrated density was divided by the area of the transfected cells and then normalized to its surrounding cells. For quantification of Western and RT-PCR data, KODAK 1D Image Analysis software was used. The intensity of the experimental image was normalized to its loading control.
Chromatin Immunoprecipitation (ChIP)
ChIP assay was performed as described previously (14). Briefly, transfection was performed in the HEK293T cell line for 40 h. For rat CGNs, 6- to 7-day-old cultures were treated with HK/LK medium for 3 or 6 h. Cells were then fixed with formaldehyde, lysed, and sonicated. The lysates were then subjected to immunoprecipitation with the respective antibody. Reverse cross-linking was performed using elution buffer (10 mm Tris-HCl, pH 8, 1 mm EDTA, pH 8, 1% SDS) at 65 °C. After several rounds of purification (with proteinase and phenol chloroform), the DNA was subjected to PCR analysis using primers designed for the human p21 gene promoter: p21 pro-forward, 5′-CGCACACGGTGTCTCTAAGT-3′, and p21 pro-reverse, 5′-ACGAACTTACTCCACTCCGC-3′; p27 promoter primers, p27 pro-forward, 5′-CTGGGTTAAGGCTGAGCGAA-3′, and p27 pro-reverse, 5′-GGCTGGCCTCGGAGAAATTA-3′; p53 promoter primers, p53 pro-forward, 5′-CAGGCTTCAGACCTGTCTCC-3′, and p53 pro-reverse, 5′-GTGAGGGCAGAATTGGTGGA-3′; and cyclin D1 promoter primers, cycD1 pro-forward, 5′-ACCGACTGGTCAAGGTAGGA-3′, and cycD1 pro-reverse, 5′-TATCCAAGCCGGCAGAATGG-3′. Primers designed to the rat p21 promoter are as follows: p21 pro-forward, GCCCCTGTGCTTAGGTCATT, and p21 pro-reverse, AAGGAAGGCTCCGTGTTAGC.
Luciferase Assay
For transcriptional assays, the luciferase reporter plasmid (WWP-luc) was co-transfected with either GFP or JAZ along with Renilla. The cells were lysed 36 h after transfection, and assays were performed (19, 20) using the Promega Dual-Luciferase® reporter assay system (catalog no. E1910) according to the manufacturer's instructions. Briefly, the cultures were lysed with 1× lysis buffer provided in the kit. Substrate was added to the lysate and luciferase activity was measured in a luminometer. Stop and Glow reagent was then added to quench the luciferase activity followed by measurement of Renilla activity. Fold induction indicates the ratio of luciferase activity to Renilla activity.
Statistical Analysis
GraphPad Prism 5 software (GraphPad Software, San Diego) was used to generate all graphs used in this paper. Two-tailed Student's t test was used to perform statistical analysis unless otherwise mentioned, and the results are shown as mean ± S.E. p values of <0.05 were considered as statistically significant. Unless stated otherwise, each experiment was performed in duplicate and repeated at least three times. ≥200 cells were counted for each experiment.
RESULTS
Identification of JAZ as a SIRT1-interacting Protein
We recently performed a screen aimed at identifying SIRT1-interacting proteins by immunoprecipitating ectopically expressed SIRT1 in mouse HT22 neuroblastoma cells and identifying proteins that co-immunoprecipitated by mass spectrometry. JAZ was one of several proteins identified in this screen.4 Expression of JAZ in whole brain has previously been reported (6). We extend this observation to show that JAZ is widely expressed in the brain (Fig. 1, A and B). Although JAZ RNA is expressed at similar levels in different brain regions, JAZ protein shows differences suggesting that stability of JAZ is regulated differently in various brain regions, likely due to post-translational mechanisms. Tissue-dependent post-translational modification of JAZ is also suggested by differences in its mobility in extracts from different brain regions (Fig. 1B). We confirmed that JAZ interacts with SIRT1 by co-expressing the two proteins in the HEK293T cell and performing co-immunoprecipitation analysis (Fig. 2A). Indeed, JAZ interacted with SIRT1 more efficiently than HDAC1, a protein known to interact with SIRT1 (21). Results of co-localization studies in CGNs confirmed that JAZ and SIRT1 interact endogenously (Fig. 2B). This was further confirmed by performing co-immunoprecipitation analysis using extracts prepared from rat brain, which showed a robust level of interaction between JAZ and SIRT1 (Fig. 2C).
FIGURE 1.
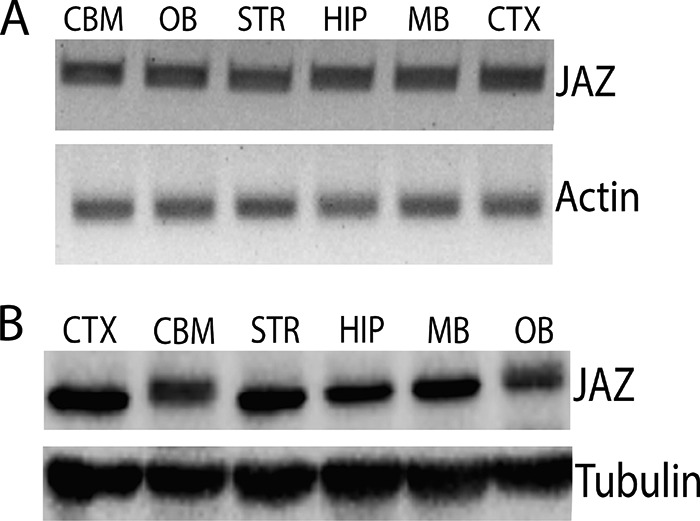
JAZ is expressed widely in the brain. A, RT-PCR analysis of JAZ mRNA in different rat brain regions (CBM, cerebellum; OB, olfactory bulb; STR, striatum; HIP, hippocampus; MB, midbrain; CTX, cortex). Actin serves as a normalization control. B, Western blot analysis of JAZ in different rat brain regions. Tubulin serves as a loading control.
FIGURE 2.
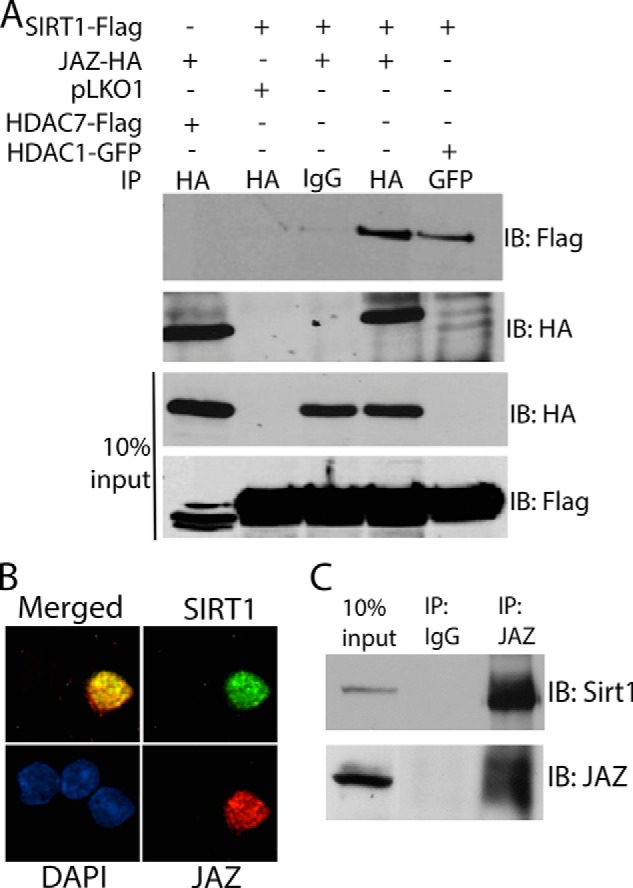
JAZ interacts with SIRT1. A, HEK293T cells were co-transfected with JAZ-HA and HDAC7-FLAG, SIRT1-FLAG and pLKO1, SIRT1-FLAG and JAZ-HA, or SIRT1-FLAG and HDAC1-GFP for 36 h. Immunoprecipitation (IP) was performed using IgG, HA, or GFP antibody. The immunoprecipitate as well as the whole cell lysate (10% input) were analyzed by Western blot analysis using FLAG and HA antibodies. SIRT1-FLAG interacted with JAZ-HA. B, rat CGNs were co-transfected with SIRT1-FLAG and JAZ-HA for 8 h and changed to HK medium for 24 h. Immunocytochemistry was performed using both FLAG and HA antibody. SIRT1 and JAZ co-localize in the nucleus. C, rat whole brain extract was used for co-immunoprecipitation assays using IgG (as control) or JAZ antibody. The immunoprecipitate and an aliquot of the pre-immunoprecipitation whole cell lysate (10% input) were subjected to Western blot analysis using SIRT1 and JAZ antibodies. IB, immunoblot.
JAZ Is a Neuroprotective Protein
SIRT1 protects neurons from death in a variety of model systems (2, 22, 23). To investigate whether JAZ also regulates neuronal survival, we used cultured CGNs. CGNs undergo apoptosis when switched from medium containing depolarizing levels of potassium (HK) to medium containing low potassium (LK) (11). We examined the effect of overexpressing JAZ on CGNs treated with HK or LK medium. Although it has no effect on neurons in HK, elevated levels of JAZ prevent LK-induced neuronal death (Fig. 3A). Additional experiments in which the epitope tag on the ectopically expressed JAZ was switched from FLAG to HA demonstrated that neuroprotection was tag-independent (Fig. 3B). To examine whether the protective effect of JAZ could be seen in paradigms other than LK-treated CGNs, we used cortical neurons treated with HCA. Treatment of cortical neurons with HCA induces death due to oxidative stress (13, 14). JAZ also protected cortical neurons from HCA-induced toxicity (Fig. 3C). JAZ has been reported to be capable of shuttling between the nucleus and cytoplasm (7). In CGNs, however, ectopically expressed JAZ and the endogenous protein localize to the nucleus (Fig. 3D). Similarly, in cortical neurons JAZ was always in the nucleus (data not shown).
FIGURE 3.

Overexpression of JAZ protects neurons from death. A, CGN cultures were transfected with GFP or JAZ-FLAG and allowed to express for 8 h followed by a switch to HK or LK medium for 24 h. Immunocytochemistry was performed using either GFP or FLAG antibody. Data represent the mean ± S.E. from eight independent experiments. ***, p < 0.0001 FLAG-tagged JAZ-transfected neurons in LK as compared with GFP-transfected neurons in LK. B, CGNs were transfected with either GFP or JAZ-HA. Treatment, immunocytochemistry, and viability assay was performed as described previously. Data represent the mean ± S.E. from four independent experiments. ***, p < 0.0001, HA-tagged JAZ-transfected neurons in LK as compared with GFP-transfected neurons in LK. C, cortical neuron cultures transfected with GFP or JAZ-FLAG were treated with 1 mm HCA for 18 h. Data represent the mean ± S.E. from four independent experiments. *, p < 0.05, JAZ-transfected neurons treated with HCA as compared with GFP-transfected neurons treated with HCA. D, JAZ localizes inside the nucleus. CGNs transfected with JAZ-FLAG were switched to HK medium for 24 h, and immunocytochemistry was performed using FLAG antibody (top). CGN cultures were fixed, and immunocytochemistry was performed using JAZ antibody (bottom).
Although overexpression of JAZ could protect neurons from various death-inducing stimuli, it was unclear whether JAZ expression was necessary for the normal survival of neurons. To investigate this issue, we performed shRNA-mediated knockdown experiments. To identify shRNAs that were effective in suppressing JAZ expression, we tested the efficacy of three commercially available shRNA constructs against JAZ that are designated in this study as Sh1, Sh2, and Sh3. Experiments targeting ectopically expressed JAZ protein in HEK293T cells as well as endogenous protein in HT22 cells demonstrated that Sh1 reduced JAZ expression substantially (Fig. 4, A, B, and E). Knockdown of JAZ was confirmed in CGNs also using immunocytochemistry (Fig. 4, C and D). In comparison with Sh1, Sh2 had a partial effect of JAZ expression and Sh3 had no discernible effect. Expression of Sh1 promoted death of otherwise healthy CGNs (in HK) and increased the extent of neuronal loss in LK (Fig. 4F). While also reducing survival, the extent of neuronal loss by Sh2 was less than that observed with Sh1, whereas Sh3 had no effect. The correlation of reduced neuronal survival with knockdown efficiency of the three shRNAs indicates that elevated expression of JAZ is necessary for neuronal survival.
FIGURE 4.

Suppression of JAZ expression induces death of otherwise healthy neurons. A, whole cell lysates were collected from HEK293T cells transfected with JAZ Sh1, JAZ Sh2, and JAZ Sh3 along with rat JAZ-FLAG construct (7. 5:0.5) for 48 h. pLKO1 and water along with rat JAZ-FLAG were used as controls. Western blot analysis was performed using FLAG antibody. ERK serves as a loading control. B, quantification of JAZ protein level in the presence of JAZ shRNAs. Expression levels of JAZ shRNA-transfected cultures was normalized to that of pLKO1-transfected cells. Quantification was performed from three independent experiments. *, p < 0.05, JAZ-transfected cells as compared with control (pLKO1-transfected cells). C, CGNs were co-transfected with pLKO1 (control), JAZ Sh1, JAZ Sh2, or JAZ Sh3 along with GFP (to visualize transfected neurons) in a 6.5:1 ratio for 48 h and then switched to HK for 24 h. Immunocytochemistry was performed using JAZ antibody. D, quantification of JAZ immunoreactivity in pLKO1 and JAZ shRNA-transfected cells. The intensity of signal was normalized to pLKO1-transfected cells. Results are quantified from three independent experiments. One-way analysis of variance test with Bonferroni's multiple-comparison post-test was used to examine statistical significance. Top asterisks: ***, p < 0.001, JAZ-Sh1-transfected neurons in HK as compared with pLKO1-transfected neurons in HK. Bottom asterisks: ***, p < 0.001, JAZ Sh2-transfected neurons in HK as compared with pLKO1-transfected neurons in HK. E, whole cell lysates were collected from HT22 cells transfected with pLKO1 (control), JAZ Sh1, and JAZ Sh2 for 72 h. Western blot analysis was performed using JAZ antibody. Tubulin serves as a loading control. F, rat CGNs co-transfected with GFP (to visualize transfected cells) and pLKO1, JAZ Sh1, JAZ Sh2, or JAZ Sh3 in a ratio of 1:6.5 for 48 h and then switched to HK or LK medium for 24 h. Data represent the mean ± S.E. from seven independent experiments. Top asterisks: ***, p < 0.0001, JAZ Sh1-transfected neurons in HK as compared with control (pLKO1-transfected neurons in HK). Bottom asterisks: ***, p < 0.0001, JAZ Sh1-transfected neurons in LK as compared with pLKO1-transfected neurons in LK. *, p < 0.05, JAZ Sh2-transfected neurons as compared with pLKO1-transfected neurons in HK.
Although forced knockdown kills neurons that are normally healthy, it was unclear whether JAZ expression is normally reduced in dying neurons. We investigated this issue in LK-treated CGNs. Neither JAZ mRNA nor protein expression was reduced by LK treatment (Fig. 5, A–D). The same result was found in cortical neurons treated with HCA (Fig. 5, E–G). Taken together with reduced neuronal survival by forced knockdown of JAZ, this finding suggests that the survival-promoting activity of JAZ is regulated by post-translational mechanisms rather than at the level of its expression.
FIGURE 5.

JAZ expression does not change under apoptotic conditions. A, JAZ RNA expression under apoptotic conditions. RNA was prepared from CGNs treated with HK or LK medium for 3, 6, and 9 h. c-Jun, an apoptotic marker, is used as a control. Actin serves as a normalization control. B, quantification of the RNA level of JAZ under apoptotic conditions obtained from three independent experiments. n.s., not significant. C, JAZ protein expression under apoptotic conditions. Protein lysates were prepared from CGNs treated with HK or LK medium for 3, 6, and 9 h. Tubulin serves as a loading control. D, quantification of JAZ expression from three independent experiments, n.s. E, JAZ RNA expression in cortical neurons. RNA was prepared from cortical neurons treated with 1 mm HCA for 3 h and 6 h. c-Jun was used as apoptotic marker, and actin serves as normalization control. F, JAZ protein expression in cortical neurons. Protein lysates were prepared from cortical neurons that were either untreated (UNT) or treated with 1 mm HCA for 3, 6, and 9 h. Western blot analysis was performed using JAZ and ERK (loading control) antibodies. G, quantification of JAZ protein level in cortical neurons treated with HCA. Results are normalized to untreated conditions (UNT), and quantification was performed from three independent experiments. n.s., HCA-treated neurons as compared with control (untreated neurons).
JAZ Protects Neurons by Inhibiting Cell Cycle Progression through the Stimulation of p21 Expression
Although promoting the survival of neurons, expression of JAZ in proliferating cell lines results in their death (Fig. 6, A and B). This result is consistent with previous reports describing an inhibitory effect of JAZ on cell cycle progression (9, 10). Aberrant activation of the cell cycle machinery has been implicated in a variety of in vitro and in vivo paradigms of neurodegeneration. Indeed, over-expression of proteins that inhibit cell cycle progression or treatment with pharmacological inhibitors of the cell cycle protects neurons against death induced by a variety of apoptotic stimuli (18, 24–27). It was therefore possible that JAZ protected neurons by the same mechanism by which it killed proliferating cells, i.e. inhibition of the cell cycle. As described previously (9, 10), BrdU incorporation assays conducted in two separate cell lines showed that JAZ inhibits cell cycle progression (Fig. 6, C–E). Although a previous publication has described that this anti-proliferative effect involves interaction of JAZ with E2F1 (10), we were not able to detect such an interaction (data not shown). To gain insight into how JAZ inhibited cell cycle progression, we examined the effect of its expression on various molecules that regulate the cell cycle either positively or negatively. As shown in Fig. 7, no change in expression was observed on the expression of cyclins A, B1, D1, and E. Interestingly, JAZ stimulates the expression of the cyclin-dependent kinase inhibitory protein p21 (WAF1/CIP1) but not other negative regulators of the cell cycle, including p15, p27, and p57.
FIGURE 6.
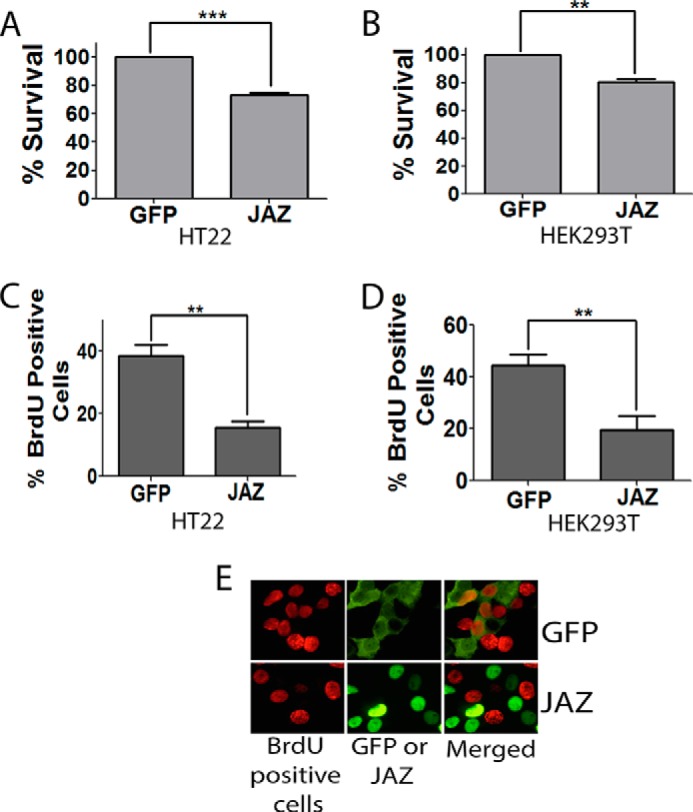
JAZ kills cell line and inhibits cell cycle progression. HT22 (A) and HEK293T (B) cell lines were transfected with GFP or JAZ-FLAG for 24 h. Immunocytochemistry was performed using GFP or FLAG antibody. Data represent the mean ± S.E. for four independent experiments. ***, p < 0.0001, or **, p < 0.01, JAZ-transfected cells as compared with controls (GFP-transfected cells). C and D, BrdU assay of JAZ in HT22 (C) and HEK293T (D) cell lines. Both cell lines were transfected with GFP and JAZ-FLAG for 22 h, and BrdU was added in 20 μm concentration for 2 h. Immunocytochemistry was performed using BrdU and FLAG or GFP antibody. The proportion of transfected cells that were BrdU-positive was quantified. **, p < 0.01, JAZ-transfected/BrdU-positive cells as compared with GFP-transfected/BrdU-positive cells. E, representative picture of BrdU and JAZ or GFP transfected cells.
FIGURE 7.
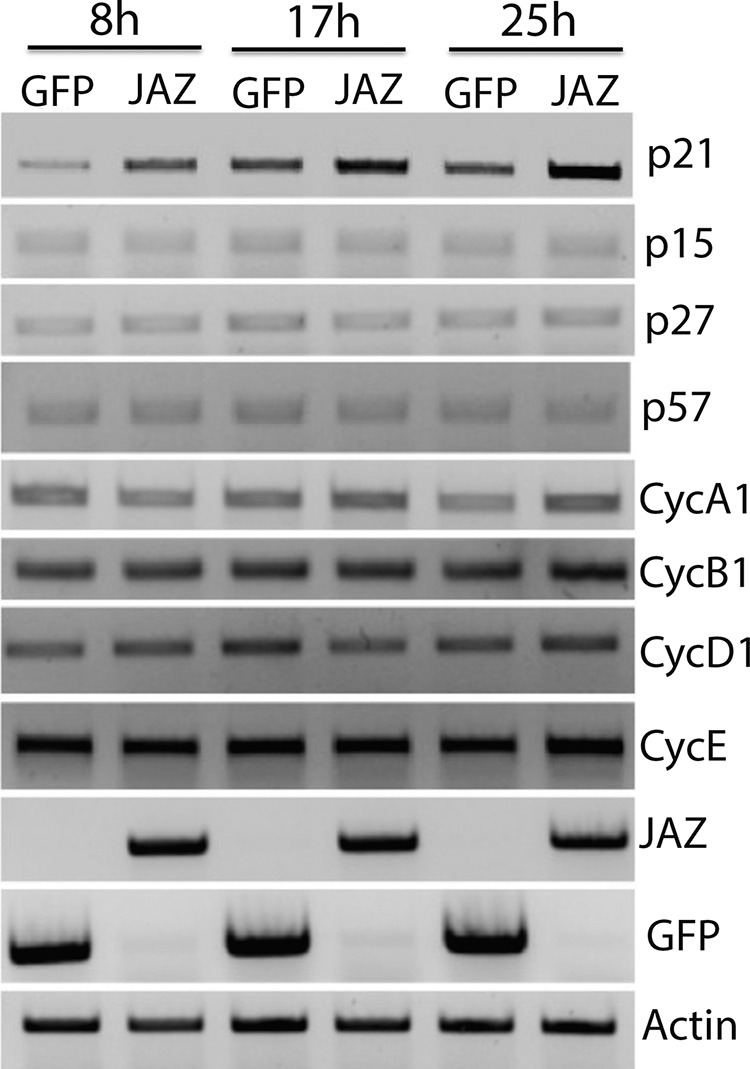
Expression of different cyclins and CDK inhibitors in the presence of JAZ. Expression pattern of different cyclins and cyclin-dependent kinase inhibitors in the presence of GFP or JAZ transfection. HEK293T cells were transfected with either GFP or JAZ-FLAG for 8, 17, and 25 h. Cells were then collected for RT-PCR analysis. Primers for cyclins and cyclin-dependent kinase inhibitors were then used along with GFP and JAZ primers. Actin serves as a normalization control.
Because p21 has previously been implicated in neuroprotection (15, 28–32), we investigated whether the neuroprotective effect of JAZ was mediated by the stimulation of p21. We first examined whether overexpression of JAZ increases p21 expression in neurons. As shown in Fig. 8, A and B, JAZ overexpression in CGNs results in a robust stimulation of p21 expression. Furthermore, knockdown of JAZ results in the reduction of p21 levels both in the HT22 cell line (Fig. 8C) and in CGNs (Fig. 8, D and E). To investigate whether the effect of JAZ on p21 expression was mediated directly, we examined whether JAZ associates with the p21 gene promoter. Results of ChIP assays indicated that JAZ does bind to the p21 gene promoter. In contrast, binding was not observed with the promoters of the cyclin D1, p27 or p53 genes (Fig. 9A). To verify that JAZ regulates p21 promoter activity, we performed transcriptional assays using a luciferase reporter. As shown in Fig. 9B, expression of JAZ stimulates the transcriptional activity of the p21.
FIGURE 8.

JAZ stimulates p21 expression. A, CGNs were infected with Ad-GFP or Ad-JAZ-FLAG for 36 h after which RNA was prepared and RT-PCR analysis was performed using primers for p21, GFP, and JAZ. Actin serves as a normalization control. B, cell lysates were prepared from CGNs infected with Ad-GFP or Ad-JAZ-FLAG for 36 h. Western blot analysis was performed using a p21 antibody. GFP and FLAG antibodies were used to evaluate infection efficiency of both GFP and JAZ adenovirus. ERK was used as a loading control. C, HT22 cell lines were transfected with either pLKO1 or JAZ Sh1 for 72 h. Cell lysates were collected, and Western blot analysis was performed using JAZ and p21 antibodies. ERK was used as a loading control. D, CGNs were co-transfected with pLKO1 (control) and JAZ Sh1 along with GFP (6.5:1) for 48 h and then switched to HK for 24 h. Immunocytochemistry was performed with JAZ (top) and p21 (bottom) antibodies. GFP fluorescence was used to visualize shRNA-transfected cells. E, quantification of JAZ and p21 immunoreactivity in cells overexpressing JAZ Sh1 or pLKO1 (control). Left asterisk: *, p < 0.05 JAZ immunoreactivity in JAZ Sh1-transfected cells as compared with pLKO1-transfected cells. Right asterisk: *, p < 0.05, p21 immunoreactivity in JAZ Sh1-transfected cells as compared with pLKO1-transfected cells.
FIGURE 9.
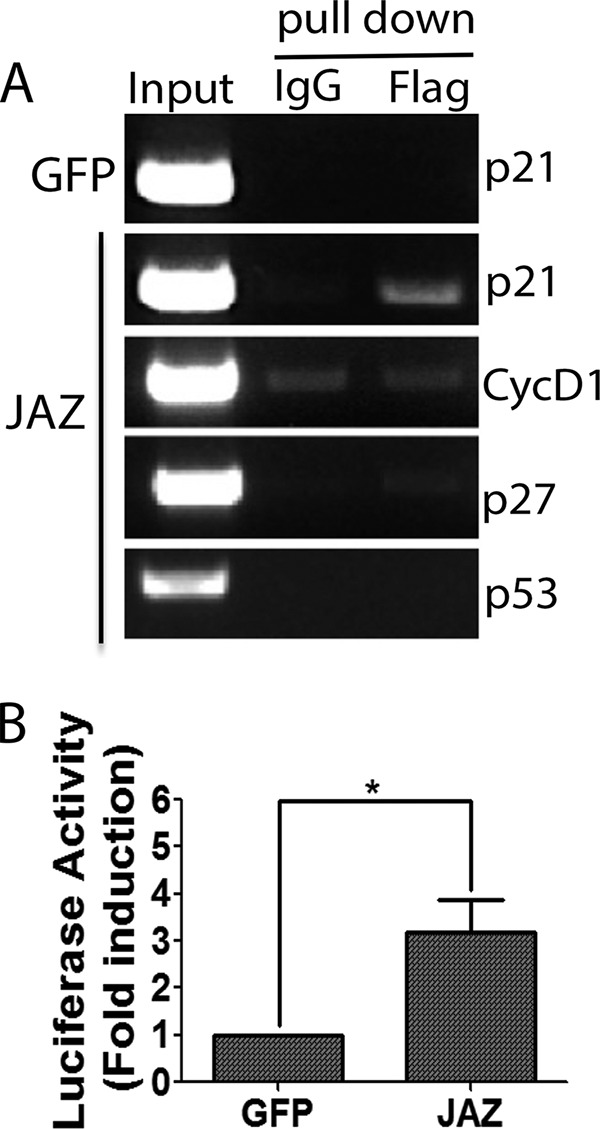
JAZ binds to the p21 promoter and stimulates its activity. A, HEK293T cells were transfected with GFP or JAZ-FLAG for 36 h. Cells were processed for ChIP analysis as described in “Experimental Procedures.” Immunoprecipitation was performed using FLAG antibody or IgG (as control). Primers designed in the promoter regions of human p21, cyclin D1 (CycD1), p27, and p53 were used to amplify the DNA pulled down with the antibody. B, HEK293T cells were transfected with GFP or JAZ-FLAG along with p21 luciferase construct (WWP-luc) for 36–40 h. Fold induction represents the ratio of luciferase to Renilla activity. Results shown are from at least four independent experiments. *, p < 0.05, JAZ luciferase activity as compared with control (GFP luciferase activity).
As described previously in other paradigms of neuronal death, the expression of p21 in LK-treated CGNS is reduced at 6 h and is even lower at 9 h after LK treatment (Fig. 10, A and B). We examined the relationship of reduced p21 expression to the binding of JAZ to the p21 gene promoter. Results of ChIP analysis showed a reduction in the level of interaction of JAZ with the p21 gene promoter following LK treatment of CGNs (Fig. 10C). The reduced interaction of JAZ with the p21 promoter strengthens the conclusions (Fig. 9B) that JAZ stimulates p21 gene transcription in healthy neurons and that its ability to do so is reduced when neurons are primed to die.
FIGURE 10.
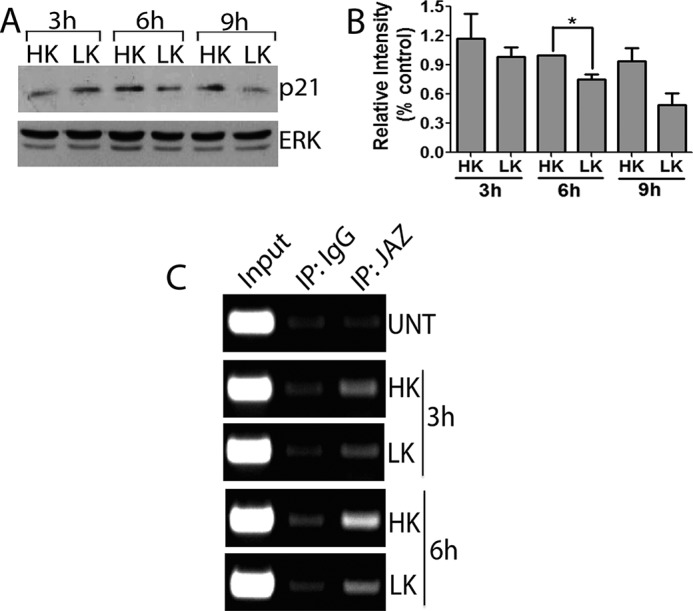
Binding of JAZ to the p21 promoter correlates with p21 expression during apoptosis. A, lysates from CGNs treated with HK or LK medium for 3, 6, and 9 h were used in Western blot analysis. The blots were probed sequentially with p21 and ERK antibodies. ERK serves as a loading control. B, quantification of p21 levels in CGNs treated with HK or LK for 3, 6, and 9 h and performed from three independent experiments. *, p < 0.05, 6 h LK-treated cells as compared with 6 h HK-treated cells. C, CGN cultures were either untreated (UNT) or treated with HK or LK medium for 3 and 6 h. ChIP assay was performed using IgG and JAZ antibodies for immunoprecipitation (IP). Primers to the promoter region of the rat p21 genes were used to amplify the input and pulldown DNA.
If JAZ works by stimulating p21 expression, then direct overexpression of p21 should protect neurons from death without overexpressing JAZ. Indeed, overexpression of p21 in CGNs protects against LK-induced death (Fig. 11A). Furthermore, stimulation of p21 is necessary for the neuroprotective effect of JAZ, knockdown of p21 expression should reduce JAZ-mediated neuroprotection. To perform this experiment, we tested three commercially available p21 shRNA constructs. We found that one of two of these shRNAs, p21-shB and p21-shC, could knock down p21 expression, with shC being more effective (Fig. 11B). Although p21-shA had no effect on the viability of neurons in HK or LK medium, p21-shB and p21-shC reduced survival in HK and potentiated death in LK (Fig. 11C). More importantly, knockdown of p21 expression abrogated the neuroprotective effect of JAZ both in HK- and LK-treated CGNs (Fig. 11C).
FIGURE 11.
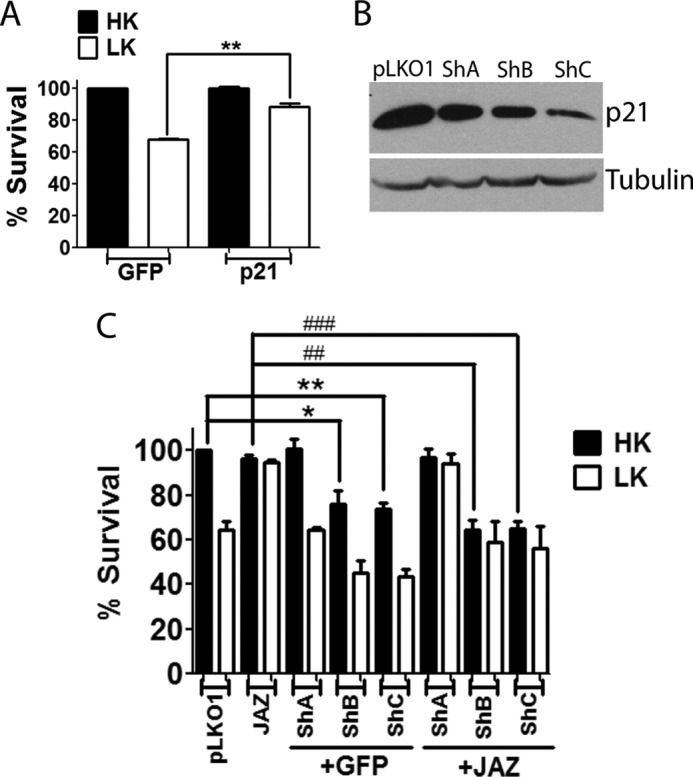
Knockdown of p21 blocks JAZ-mediated neuroprotection. A, overexpression of p21 protects neurons from LK-induced death. CGNs transfected with GFP or p21-FLAG for 8 h were switched to either HK or LK medium for 24 h. Viability of transfected neurons was evaluated by immunocytochemistry as described under “Experimental Procedures.” Data represent the mean ± S.E. for four independent experiments. **, p < 0.01, p21-transfected neurons as compared with control (GFP-transfected neurons in LK). B, HT22 cell lines were transfected with pLKO1 (control) or three different p21 shRNAs named ShA, ShB, and ShC for 72 h. Cell lysates were collected, and Western blot analysis was performed using a p21 antibody. Tubulin serves as a loading control. C, CGNs were co-transfected with p21 shRNAs and either GFP (6.5:1) or JAZ-FLAG (5:2.5) for 48 h and then switched to HK or LK medium for 24 h. Immunocytochemistry was performed using GFP or FLAG antibody. Data represent the mean ± S.E. of four independent experiments. One-way analysis of variance was performed with Bonferroni's multiple comparison test. *, p < 0.05, GFP/ShB transfected in HK as compared with GFP/pLKO1-transfected neurons in HK. **, p < 0.01, GFP/ShC-transfected neurons in HK as compared with GFP/pLKO1-transfected neurons in HK. ##, p < 0.01, JAZ/ShB-transfected neurons in HK as compared with JAZ/FLAG-transfected neurons in HK. ###, p < 0.001, JAZ/ShC-transfected neurons in HK as compared with JAZ-FLAG-transfected neurons in HK.
JAZ Is Protective in Tissue Culture Models of Neurodegenerative Diseases
HK mimics the survival-promoting effect of neuronal activity during the development of the nervous system. Although this paradigm models developmentally regulated neuronal death, it is not a model of any neurodegenerative disease. To examine whether the neuroprotection by JAZ also extends to disease models, we tested its ability to protect against the toxic effect of mutant huntingtin and mutant ataxin. Neuronal death resulting from mutant-huntingtin (mut-Htt) is frequently used as a cell culture model of Huntington disease. We found that when co-expressed with JAZ, the toxicity of mut-Htt is completely abolished both in HK and LK medium (Fig. 12A). To examine whether this protective effect of JAZ extended to other proteinopathic neurodegenerative disorders, we used mutant ataxin-1 (mut-Atx1). mut-Atx1-induced neuronal death is used as a model of spinocerebellar ataxia type-1. Co-expression of JAZ protects against the neurotoxic effect of mut-Atx1 (Fig. 12B).
FIGURE 12.
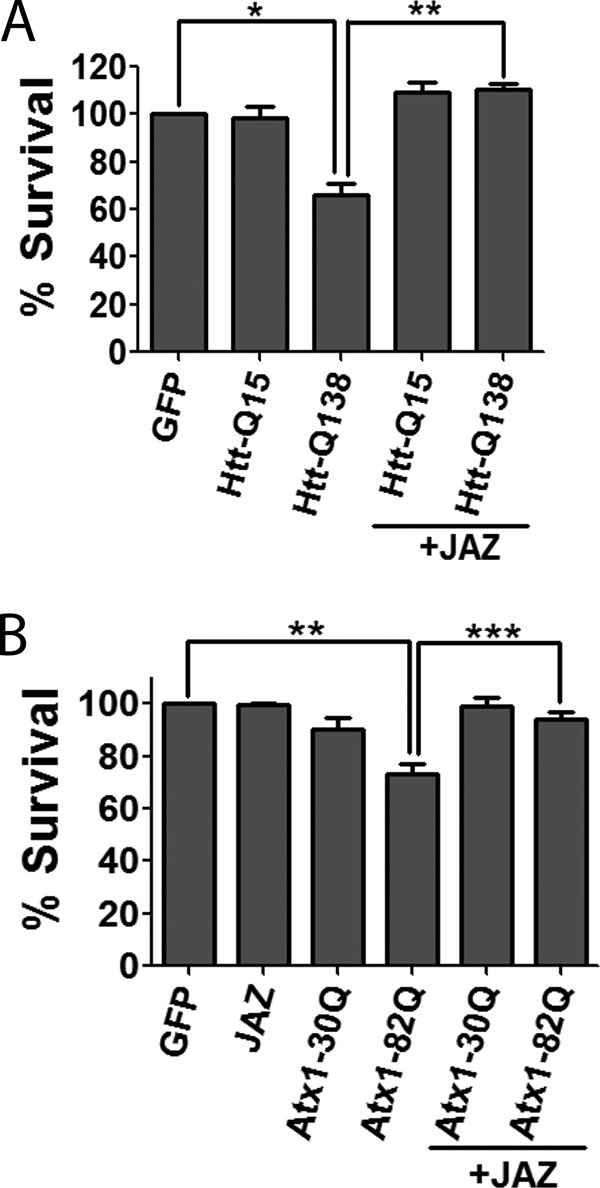
JAZ protects against mutant huntingtin and mutant ataxin-1 neurotoxicity. A, CGNs were transfected with GFP and Htt-Q15-GFP or Htt-Q138-GFP alone or co-transfected with JAZ-FLAG for 8 h followed by treatment with HK medium for 24 h. Data represent the mean ± S.E. from four independent experiments. *, p < 0.05, Htt-Q138-transfected neurons as compared with control (GFP-transfected neurons in HK). **, p < 0.01, JAZ/Htt-Q138-transfected neurons as compared with Htt-Q138-transfected neurons. B, CGNs were transfected with GFP, JAZ-HA, and Atx1–30Q-FLAG or Atx1–82Q-FLAG alone or co-transfected with JAZ-HA for 24 h followed by a switch to HK medium for 24 h. Immunocytochemistry and viability assessment was performed as described previously. Data represent mean ± S.E. from four independent experiments. **, p < 0.01, Atx1–82Q-transfected neurons as compared with control (GFP-transfected neurons under HK conditions). ***, p < 0.001, JAZ/Atx1–82Q-transfected neurons as compared with Atx1–82Q-transfected neurons.
DISCUSSION
We describe a new and important regulator of neuronal death, JAZ. Knockdown of JAZ in cultured neurons leads to their death. Although its knockdown kills neurons, JAZ expression is not reduced in neurons primed to undergo apoptosis suggesting that post-translational mechanisms are involved in its ability to maintain neuronal survival. In addition to maintaining neuronal survival, ectopic expression of JAZ protects neurons in different paradigms, including mut-Htt and mut-Atx1 toxicity. JAZ was identified in a mass spectrometric screen for interactors of SIRT1, another protein that has been shown to protect neurons in a number of different studies (22, 23). Interaction between JAZ and SIRT1 is seen in neurons and in whole brain. Although a quantitative comparison cannot be made, the level of JAZ-SIRT1 interaction in whole brain extracts was much higher than when the two proteins were overexpressed in HEK293 cells, suggesting that other factors in the brain, perhaps involving post-translation of JAZ or involvement of brain-specific proteins enhance JAZ-SIRT1 interaction. The significance of the JAZ-SIRT1 interaction to the neuroprotective activity of either JAZ or SIRT1 is not clear and remains to be elucidated in future studies.
As described previously (9, 10), we find that JAZ inhibits cell cycle progression in proliferating cells. Because abortive cell cycle reentry is widely regarded as a mode by which neurons die both in experimental paradigms and disease states, it is likely that the anti-proliferative effect of JAZ is responsible for its neuroprotective effect. Indeed, we find that JAZ expression increases the expression of p21, a potent cell cycle inhibitor. Several studies have found that elevated p21 levels have a neuroprotective effect (15, 28–32). Results of ChIP and transcriptional assays suggest that the stimulatory effect on p21 expression is transcriptionally mediated through interaction of JAZ with the p21 gene promoter. Increased p21 production in response to ectopic JAZ expression has been described by another laboratory, although in that study the stimulatory effect was believed to be indirect and to involve activation of p53 (9). Although other regulators, such as p53, have been identified as transcriptional regulators of p21, our study identifies JAZ as a novel regulator. Because dysregulation of p21 expression occurs in a variety of pathophysiological conditions, including cancer (33, 34), our finding may have implications to processes in addition to the regulation of neuronal survival and death.
JAZ belongs to a specialized class of Cys-2–His-2 zinc finger proteins that bind preferentially and strongly to double-stranded RNAs (dsRNAs) (35). It has been suggested that JAZ can shuttle between the nucleus and cytoplasm and might be involved in the nucleocytoplasmic shuttling of pre-miRNAs (6, 7). However, both in CGNs and cortical neurons, as well as in the HEK293T and HT22 cell lines, we find JAZ to be strictly nuclear under all the conditions we used. Thus, although JAZ can interact with dsRNAs, it appears unlikely that its neuroprotective effect requires dsRNA binding. Other studies have shown that JAZ can interact with proteins, such as p53 and E2F1, in a manner that is independent of dsRNA binding (9, 10). Although it is possible that the interaction of JAZ with the p21 gene promoter is also mediated through interaction with E2F1, we have not been able to detect interaction between JAZ and E2F1.4
In summary, we describe that JAZ, a poorly studied protein, maintains neuronal survival and protects neurons from death when ectopically expressed. JAZ can protect neurons against toxicity by mut-Htt and mut-Atx1 raising the possibility that finding pharmacological approaches to increase JAZ expression in the brain or gaining a better understanding of how it acts to protect neurons could lead to the identification of novel approaches to treat neurodegenerative diseases like Huntington disease and spinocerebellar ataxia type-1.
Acknowledgments
We thank Eric Medina who cloned the JAZ-FLAG construct. We also thank Chi Ma (currently at the National Institutes of Health) and Jason Pfister for early work on this project.
This work was supported, in whole or in part, by National Institutes of Health Grants NS040408 and NS066404.
J. A. Pfister and S. R. D'Mello, manuscript in preparation.
S. Mallick and S. R. D'Mello, unpublished data.
- CGN
- cerebellar granule neuron
- HCA
- homocysteic acid.
REFERENCES
- 1. Donmez G., Outeiro T. F. (2013) SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Mol. Med. 5, 344–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Donmez G. (2012) The neurobiology of sirtuins and their role in neurodegeneration. Trends Pharmacol. Sci. 33, 494–501 [DOI] [PubMed] [Google Scholar]
- 3. Duan W. (2013) Targeting sirtuin-1 in Huntington's disease: rationale and current status. CNS Drugs 27, 345–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pfister J. A., Ma C., Morrison B. E., D'Mello S. R. (2008) Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS One 3, e4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kang H., Suh J. Y., Jung Y. S., Jung J. W., Kim M. K., Chung J. H. (2011) Peptide switch is essential for Sirt1 deacetylase activity. Mol. Cell 44, 203–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang M., May W. S., Ito T. (1999) JAZ requires the double-stranded RNA-binding zinc finger motifs for nuclear localization. J. Biol. Chem. 274, 27399–27406 [DOI] [PubMed] [Google Scholar]
- 7. Chen T., Brownawell A. M., Macara I. G. (2004) Nucleocytoplasmic shuttling of JAZ, a new cargo protein for exportin-5. Mol. Cell. Biol. 24, 6608–6619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Finerty P. J., Jr, Bass B. L. (1997) A Xenopus zinc finger protein that specifically binds dsRNA and RNA-DNA hybrids. J. Mol. Biol. 271, 195–208 [DOI] [PubMed] [Google Scholar]
- 9. Yang M., Wu S., Su X., May W. S. (2006) JAZ mediates G1 cell-cycle arrest and apoptosis by positively regulating p53 transcriptional activity. Blood 108, 4136–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang M., Wu S., Jia J., May W. S. (2011) JAZ mediates G1 cell cycle arrest by interacting with and inhibiting E2F1. Cell Cycle 10, 2390–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. D'Mello S. R., Galli C., Ciotti T., Calissano P. (1993) Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci. U.S.A. 90, 10989–10993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bardai F. H., D'Mello S. R. (2011) Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3β. J. Neurosci. 31, 1746–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dastidar S. G., Landrieu P. M., D'Mello S. R. (2011) FoxG1 promotes the survival of postmitotic neurons. J. Neurosci. 31, 402–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Verma P., Pfister J. A., Mallick S., D'Mello S. R. (2014) HSF1 protects neurons through a novel trimerization- and HSP-independent mechanism. J. Neurosci. 34, 1599–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Langley B., D'Annibale M. A., Suh K., Ayoub I., Tolhurst A., Bastan B., Yang L., Ko B., Fisher M., Cho S., Beal M. F., Ratan R. R. (2008) Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J. Neurosci. 28, 163–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Haskew-Layton R. E., Payappilly J. B., Smirnova N. A., Ma T. C., Chan K. K., Murphy T. H., Guo H., Langley B., Sultana R., Butterfield D. A., Santagata S., Alldred M. J., Gazaryan I. G., Bell G. W., Ginsberg S. D., Ratan R. R. (2010) Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an Nrf2-independent pathway. Proc. Natl. Acad. Sci. U.S.A. 107, 17385–17390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ratan R. R., Murphy T. H., Baraban J. M. (1994) Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J. Neurosci. 14, 4385–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Majdzadeh N., Wang L., Morrison B. E., Bassel-Duby R., Olson E. N., D'Mello S. R. (2008) HDAC4 inhibits cell-cycle progression and protects neurons from cell death. Dev. Neurobiol. 68, 1076–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Datto M. B., Yu Y., Wang X. F. (1995) Functional analysis of the transforming growth factor β responsive elements in the WAF1/Cip1/p21 promoter. J. Biol. Chem. 270, 28623–28628 [DOI] [PubMed] [Google Scholar]
- 20. Schaefer J. S., Sabherwal Y., Shi H. Y., Sriraman V., Richards J., Minella A., Turner D. P., Watson D. K., Zhang M. (2010) Transcriptional regulation of p21/CIP1 cell cycle inhibitor by PDEF controls cell proliferation and mammary tumor progression. J. Biol. Chem. 285, 11258–11269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dobbin M. M., Madabhushi R., Pan L., Chen Y., Kim D., Gao J., Ahanonu B., Pao P. C., Qiu Y., Zhao Y., Tsai L. H. (2013) SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat. Neurosci. 16, 1008–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tang B. L., Chua C. E. (2008) SIRT1 and neuronal diseases. Mol. Aspects Med. 29, 187–200 [DOI] [PubMed] [Google Scholar]
- 23. Jiang M., Wang J., Fu J., Du L., Jeong H., West T., Xiang L., Peng Q., Hou Z., Cai H., Seredenina T., Arbez N., Zhu S., Sommers K., Qian J., Zhang J., Mori S., Yang X. W., Tamashiro K. L., Aja S., Moran T. H., Luthi-Carter R., Martin B., Maudsley S., Mattson M. P., Cichewicz R. H., Ross C. A., Holtzman D. M., Krainc D., Duan W. (2012) Neuroprotective role of Sirt1 in mammalian models of Huntington's disease through activation of multiple Sirt1 targets. Nat. Med. 18, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Copani A., Uberti D., Sortino M. A., Bruno V., Nicoletti F., Memo M. (2001) Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci. 24, 25–31 [DOI] [PubMed] [Google Scholar]
- 25. Becker E. B., Bonni A. (2004) Cell cycle regulation of neuronal apoptosis in development and disease. Prog. Neurobiol. 72, 1–25 [DOI] [PubMed] [Google Scholar]
- 26. Di Giovanni S., Movsesyan V., Ahmed F., Cernak I., Schinelli S., Stoica B., Faden A. I. (2005) Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc. Natl. Acad. Sci. U.S.A. 102, 8333–8338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kabadi S. V., Stoica B. A., Hanscom M., Loane D. J., Kharebava G., Murray Ii M. G., Cabatbat R. M., Faden A. I. (2012) CR8, a selective and potent CDK inhibitor, provides neuroprotection in experimental traumatic brain injury. Neurotherapeutics 9, 405–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tomita K., Kubo T., Matsuda K., Madura T., Yano K., Fujiwara T., Tanaka H., Tohyama M., Hosokawa K. (2006) p21Cip1/WAF1 regulates radial axon growth and enhances motor functional recovery in the injured peripheral nervous system. Brain Res. 1081, 44–52 [DOI] [PubMed] [Google Scholar]
- 29. Harms C., Albrecht K., Harms U., Seidel K., Hauck L., Baldinger T., Hübner D., Kronenberg G., An J., Ruscher K., Meisel A., Dirnagl U., von Harsdorf R., Endres M., Hörtnagl H. (2007) Phosphatidylinositol 3-Akt-kinase-dependent phosphorylation of p21(Waf1/Cip1) as a novel mechanism of neuroprotection by glucocorticoids. J. Neurosci. 27, 4562–4571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Poluha W., Poluha D. K., Chang B., Crosbie N. E., Schonhoff C. M., Kilpatrick D. L., Ross A. H. (1996) The cyclin-dependent kinase inhibitor p21 (WAF1) is required for survival of differentiating neuroblastoma cells. Mol. Cell. Biol. 16, 1335–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mergenthaler P., Muselmann C., Sünwoldt J., Isaev N. K., Wieloch T., Dirnagl U., Meisel A., Ruscher K. (2013) A functional role of the cyclin-dependent kinase inhibitor 1 (p21(WAF1/CIP1)) for neuronal preconditioning. J. Cereb. Blood Flow Metab. 33, 351–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma T. C., Langley B., Ko B., Wei N., Gazaryan I. G., Zareen N., Yamashiro D. J., Willis D. E., Ratan R. R. (2013) A screen for inducers of p21(waf1/cip1) identifies HIF prolyl hydroxylase inhibitors as neuroprotective agents with antitumor properties. Neurobiol. Dis. 49, 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cox L. S. (1997) Multiple pathways control cell growth and transformation: overlapping and independent activities of p53 and p21Cip1/WAF1/Sdi1. J. Pathol. 183, 134–140 [DOI] [PubMed] [Google Scholar]
- 34. Gartel A. L., Serfas M. S., Tyner A. L. (1996) P21–negative regulator of the cell cycle. Proc. Soc. Exp. Biol. Med. 213, 138–149 [DOI] [PubMed] [Google Scholar]
- 35. Burge R. G., Martinez-Yamout M. A., Dyson H. J., Wright P. E. (2014) Structural characterization of interactions between the double-stranded RNA-binding zinc finger protein JAZ and nucleic acids. Biochemistry 53, 1495–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]