

Background: LDL and modified LDL may be cleared via the adherence receptor CR1.
Results: LDL bound C1q, whereas acLDL bound IgM, C1q and properdin. Both became opsonized by C3b.
Conclusion: Binding of opsonized LDL and acLDL to CR1 is mediated via different complement pathways.
Significance: CR1 is a novel receptor for cell-bound lipoproteins.
Keywords: Complement, Erythrocyte, Leukocyte, Lipoprotein, Low-density Lipoprotein (LDL), C3b, CR1, Complement Activation, Receptor
Abstract
Lipoproteins can induce complement activation resulting in opsonization and binding of these complexes to complement receptors. We investigated the binding of opsonized native LDL and acetylated LDL (acLDL) to the complement receptor 1 (CR1). Binding of complement factors C3b, IgM, C1q, mannose-binding lectin (MBL), and properdin to LDL and acLDL were investigated by ELISA. Subsequent binding of opsonized LDL and acLDL to CR1 on CR1-transfected Chinese Hamster Ovarian cells (CHO-CR1) was tested by flow cytometry. Both native LDL and acLDL induced complement activation with subsequent C3b opsonization upon incubation with normal human serum. Opsonized LDL and acLDL bound to CR1. Binding to CHO-CR1 was reduced by EDTA, whereas MgEGTA only reduced the binding of opsonized LDL, but not of acLDL suggesting involvement of the alternative pathway in the binding of acLDL to CR1. In vitro incubations showed that LDL bound C1q, whereas acLDL bound to C1q, IgM, and properdin. MBL did neither bind to LDL nor to acLDL. The relevance of these findings was demonstrated by the fact that ex vivo up-regulation of CR1 on leukocytes was accompanied by a concomitant increased binding of apolipoprotein B-containing lipoproteins to leukocytes without changes in LDL-receptor expression. In conclusion, CR1 is able to bind opsonized native LDL and acLDL. Binding of LDL to CR1 is mediated via the classical pathway, whereas binding of acLDL is mediated via both the classical and alternative pathways. Binding of lipoproteins to CR1 may be of clinical relevance due to the ubiquitous cellular distribution of CR1.
Introduction
The complement system is part of the innate immune system, which provides protection against micro-organisms. Complement activation occurs via three different pathways: the classical pathway that depends on IgG and/or IgM antibodies and C-reactive protein (CRP),2 the alternative pathway initiated by factor B, factor D and properdin and the lectin pathway initiated by the recognition of neutral sugars like mannose and fructose via mannose-binding lectin (MBL) and ficolins (1–3). All three pathways lead to the conversion of complement component (C) 3, the most abundant complement component in serum, into C3a and C3b via surface-bound C3-convertases. The final step of the complement cascade is the formation of the membrane attack complex, resulting in removal of pathogens and immune complexes from the circulation (2, 4, 5).
The complement system and its activation are not only important in microbial defense but also in the development of atherosclerosis, both by anti- and pro-atherogenic properties (6, 7). In addition, the complement system has been linked to lipoprotein metabolism, which plays a role in the development of atherosclerosis, e.g. by binding of complement factors to lipoproteins. C3 and C4 have been found on high density lipoproteins (HDL), especially in subjects with coronary artery disease (8). MBL may also be involved in postprandial lipid metabolism with effects on chylomicron and very low density lipoprotein (VLDL) handling (9). Properdin has recently been linked to postprandial lipid metabolism as well (10). Modified forms of low density lipoproteins (LDL), like acetylated LDL (acLDL), oxidized LDL (oxLDL) or enzymatically degraded LDL have been shown to induce complement activation (6, 11), which was enhanced by the addition of CRP (6). Finally, modified forms of LDL bind C3b and C1q (12).
Although binding of lipoproteins to specific lipoprotein receptors and heparan sulfate proteoglycans is well established (13, 14), binding to complement receptors may also occur as suggested previously by our group (15). In theory, C3b-opsonized lipoproteins may bind to the complement receptor 1 (CR1, also known as CD35 or C3b receptor), which is present on many types of cells such as erythrocytes, endothelial cells and inflammatory cells (16, 17). CR1 on erythrocytes is important for “immune adherence,” which functions as a clearing mechanism for immune complexes (18, 19) and microorganisms (20). CR1 also facilitates phagocytosis of immune complexes by monocytes, macrophages, and neutrophils (17). Therefore, CR1-mediated cellular binding of opsonized lipoproteins may also contribute to the removal of lipoproteins from plasma (15).
The aim of this study was to investigate whether CR1 can bind native LDL and acLDL and which complement activation pathways may be involved. The clinical relevance of this question is based on the recent finding that circulating leukocytes and erythrocytes carry apolipoprotein B-containing lipoproteins on their surface (21–23). Leukocytes express lipoprotein binding receptors, but erythrocytes do not. CR1-mediated lipoprotein binding by erythrocytes and leukocytes may contribute to the clearance of lipoproteins similarly to the earlier mentioned process of immune adherence. For this purpose, experiments were conducted with a cell model with CR1-transfected Chinese ovarian hamster cells and human leukocytes.
MATERIALS AND METHODS
CR1 Cell Model
Transfected Chinese Hamster Ovary cells expressing the complement receptor 1 (CHO-CR1) and negative controls (CHO) with CD46 cloned in reverse orientation were kindly provided by J.P. Atkinson and M.K. Liszewski (Washington University School of Medicine, St Louis) and by S. Rooijakkers (University Medical Center Utrecht, Utrecht, the Netherlands) (24, 25). The cells were cultured at 37 °C in DMEM/F12 + GLUTAMAX (HAM) (1:1) medium (GIBCO, Invitrogen, Carlsbad, CA) with 1% penicillin-streptomycin, 0.5 μg/ml G-418 (Sigma-Aldrich) and 10% heat-inactivated FCS. Trypsin was used to harvest the cells for experiments. A total of 200.000 cells per sample were used for each experiment. Expression and functionality of CR1 on CHO-CR1 was confirmed.
Binding of LDL and acLDL to Chinese Hamster Ovary Cells
BODIPY® fluorochrome-labeled LDL and acLDL were purchased (Invitrogen Molecular Probes, Eugene, OR), and all experiments were conducted within 2 weeks after arrival of the labeled lipoproteins. All experiments regarding the binding of LDL and acLDL to CHO-CR1 and CHO were conducted with these labeled lipoproteins. CHO-CR1 and CHO were incubated with 10 μg/ml LDL or acLDL or with medium as control for 30 min on ice in the dark followed by flow cytometric analysis (FACSCalibur, Becton Dickinson). To investigate whether LDL binding to CHO could be reduced by blocking the LDL receptor, CHO cells were pre-incubated with an anti-human LDL receptor blocking antibody (catalogue number AF2148, R&D Systems) for 30 min prior to LDL incubation. In addition, fluorescent LDL (10 μg/ml) was incubated with either normal human serum (NHS) or non-labeled LDL in different concentrations to test whether competition for binding to CHO cells occurred between fluorescent LDL, LDL present in NHS or with native LDL. Fluorescent LDL was incubated with NHS or with non-labeled LDL in culture medium for 60 min on 37 °C in the dark prior to incubation with CHO-CR1 and CHO.
Binding of Complement-opsonized LDL and acLDL to CR1
In experiments where it was necessary to impede complement activation heat inactivated serum (HIS) (at 56 °C for 30 min) was used. Labeled LDL and acLDL (10 μg/ml) were incubated with either 10% NHS or 10% HIS in culture medium for 60 min on 37 °C in the dark. Next CHO and CHO-CR1 were incubated with either LDL in NHS, acLDL in NHS, LDL in HIS, acLDL in HIS, or in culture medium as control for 30 min in the dark on ice. Binding of LDL or acLDL to CHO and CHO-CR1 was detected by flow cytometry (Becton Dickinson).
Inactivation of Different Complement Pathways
To inactivate the different complement pathways, the following experiments were carried out. Anti-C1q (20 μg/ml) was added to 10% NHS to inactivate the classical pathway. The lectin pathway was blocked by addition of d-mannose (200 mm) to 10% NHS. MgEGTA (20 mm) was used to block both the classical and lectin pathway. EDTA (20 mm) was used to block all three complement pathways. NHS with either anti-C1q, d-mannose, MgEGTA, or EDTA was left on room temperature for 10 min before incubation with labeled LDL (10 μg/ml) or acLDL (10 μg/ml). After the samples were incubated for 60 min on 37 °C in the dark, CHO-CR1 were incubated with the different samples for 30 min in the dark on ice, and LDL and acLDL binding were assessed by flow cytometry.
Binding of Complement Factors to LDL and acLDL by ELISA
Human lipoproteins were isolated according to the technique described by Redgrave et al. (26). In short, PBS with 0.3 mm EDTA with densities of 1.0063 g/liter, 1.019 g/liter, and 1.063 g/liter were prepared by using KBr. Serum was collected from non-fasting healthy volunteers, which was adjusted to 1.21 g/liter by the addition of KBr. A gradient was prepared and centrifuged with 40,000 rpm with slow acceleration and deceleration for 16 h at a temperature of 4 °C. The LDL fraction was aspirated with a glass pipette and subsequently dialyzed overnight against PBS pH 7.4 using a dialysis cassette (Thermo Scientific, Pierce Protein Research Products). The protein concentration was determined using the BCA protein assay kit (Thermo Scientific, Pierce Protein Research Products). In addition, a portion of LDL was acetylated according to the technique described by Van Berkel et al. (27). AcLDL was prepared by mixing LDL with saturated sodium acetate and acetic anhydride. After acetylation, the acLDL was dialyzed overnight against PBS buffer with pH 7.4.
Human properdin (28), human IgM (29), human C1q (30) and human MBL (29) were purified as described previously. Ninety-six well, flat bottom plates (NUNC, Thermo Fisher Scientific, Rochester, NY) were coated with either LDL (50 μg/ml), acLDL (50 μg/ml) or human serum albumin (HSA) (1 mg/ml) overnight at room temperature. The remaining sites were blocked with 1% HSA in PBS and incubated for 1 h at 37 °C. Wells were washed with PBS Tween 0.05% including 2.5 mm CaCl2 and incubated with either NHS, human IgM, C1q, MBL, or properdin diluted in Veronal buffer supplemented with 1% HSA, 2.5 mm CaCl2, 2.5 mm MgCl2, and 0.05% Tween for 1 h at 37 °C. The plate was washed again and incubated with either monoclonal antibodies against human C3 (RFK22, in-house, Leiden University Medical Center, Leiden, the Netherlands), monoclonal anti-human IgM (HB57, hybridoma obtained from the American Type Culture Collection, Manassas, VA), monoclonal antibodies against human C1q (mAb 2214, kindly provided by Prof. C. E. Hack, Sanquin Blood Supply Foundation, Amsterdam, The Netherlands) monoclonal antibodies against human MBL (3E7, Hycult Biotech, Uden, the Netherlands) or rabbit anti-human properdin (in house, Leiden University Medical Center) followed by HRP-conjugated F(ab')2 from goat IgG anti-dig (Roche Applied Science, Mannheim, Germany) or streptavidin-HRP conjugate (Zymed Laboratories Inc., South San Francisco, CA) with ABTS/H2O2 (Sigma) to measure the deposition of C3b, IgM, C1q, MBL, and properdin, respectively on LDL and acLDL. The optical density (OD) was measured at 415 nm using a microtiter plate reader (Model 680 Microtiter Reader, Bio-Rad). BSA instead of HSA was used as negative control for the determination of properdin binding. Mannan (Sigma-Aldrich) was used as a positive control for MBL deposition and IgM as a positive control for C1q binding.
The contribution of the classical and lectin pathways to C3b opsonization of LDL and acLDL were investigated. A similar kind of ELISA for the assessment of C3b deposition was done with some adjustments. Different concentrations of anti-C1q or d-mannose were added to a fixed concentration of 10% NHS and left on room temperature for 10 min, which was used to incubate the LDL- and acLDL-coated plates. Human IgM (2.6 μg/ml) and mannan (10 μg/ml) were used as controls for either inhibition of the classical or lectin pathway.
CR1 and Apolipoprotein B on Monocytes and Neutrophils after LPS Stimulation
A lipopolysaccharide (LPS)-containing solution was made from Escherichia coli (ATTC strain 25922). Grown bacteria were exposed to 4% formaldehyde for 2 h at room temperature. The formaldehyde was washed away by two cycles of spinning down and resuspending the pallet in PBS. The end suspension contained an equivalent of 2.5 × 106 bacteria per ml. The LPS suspension was checked for sterility by overnight incubation at 37 °C on blood agar.
Freshly drawn human blood samples from different healthy volunteers were collected in tubes containing 5.4 mg K2 EDTA (Becton Dickinson, Plymouth, UK). Different tubes were used to measure three different conditions: whole blood was either incubated with 0.9% NaCl or LPS for 15 min at 37 °C. The estimated bacteria to leukocyte ratio in the LPS incubation was 1:10. Whole blood without any modifications served as another control. Erythrocytes were lysed by addition of lysis solution (1.5 m ammonium chloride, 100 mm potassium hydrogen carbonate, 0.82 mm EDTA, pH 7.4) for 45 min on ice. After lysis the leukocytes were washed three times with PBS supplemented with 0.2% BSA.
To measure CR1 on human monocytes and neutrophils, tubes were prepared with phycoerythrin (PE)-conjugated anti-CR1 (catalogue no. 559872, BD Biosciences, Franklin Lakes, NJ) and phycoerythrin-Texas Red-X (ECD) conjugated anti-CD45 (catalogue no. A07784, Beckman Coulter, Miami). Mouse IgG1 kappa (catalogue no. 551436, BD Biosciences) served as the isotype control for anti-CR1. A total of 20 μl of the isolated leukocytes was added to each tube and incubated for 15 min in the dark on ice.
To measure apo B on monocytes and neutrophils, tubes with a polyclonal goat antibody directed against human apo B (catalogue no. AB742; Millipore, Billerica, MA) or with goat IgG isotype control (catalogue no. BRU5000; Linaris Biologische Produkte, Wertheim-Bettingen, Germany) were prepared and incubated with 20 μl of isolated leukocytes followed by 30 min of incubation in the dark on ice. Subsequently, the leukocytes were washed with PBS supplemented with 0.2% BSA and incubated with a secondary rabbit anti-goat (RAG) antibody conjugated with fluorescein isothiocynate (FITC) (Nordic Immunological Laboratories, Tilburg, the Netherlands) for another 30 min in the dark on ice. The LDL receptor expression by monocytes and neutrophils was measured using the same protocol, but with an anti-LDL receptor antibody (catalogue number AF2148, R&D Systems) followed by a secondary RAG antibody conjugated with FITC (Nordic Immunological Laboratories). All samples were measured on a XL flow cytometer (Beckman Coulter). Monocytes and neutrophils were identified in the side scatter versus CD45 dot plot.
Statistics
Differences between groups were tested using the unpaired t test when comparing two unrelated groups. One-way ANOVA with post-hoc analysis was used when multiple groups were compared. Graphpad 5.0 (Prism) and PASW 18.0 (SPSS, IBM) were used for the statistical analyses. A p value of less than 0.05 (two-sided) was regarded as statistical significant.
RESULTS
C3b-opsonization of LDL and acLDL
To determine whether both native LDL and acLDL were able to induce complement activation with subsequent C3b opsonization LDL and acLDL were incubated with NHS. This resulted in C3b opsonization of LDL and acLDL as established by ELISA (Fig. 1A). C3b opsonization of acLDL occurred already at NHS concentrations as low as 0.3%, whereas concentrations of at least 2.5% were necessary to induce C3b-opsonization of LDL. Addition of EDTA to NHS prevented C3b opsonization of LDL and acLDL (Fig. 1B).
FIGURE 1.
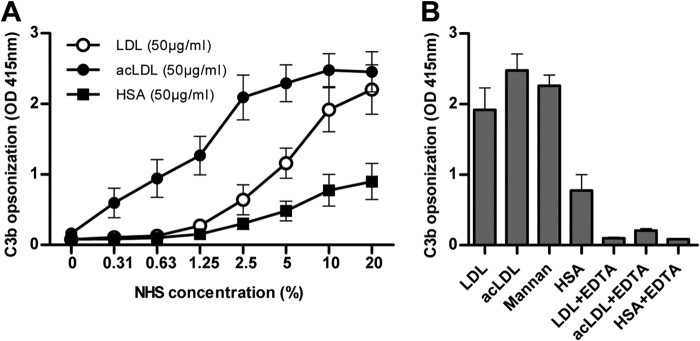
C3b opsonization of acLDL occurred already at very low concentrations of normal human serum (NHS), but concentrations of at least 2.5% were necessary to establish C3b opsonization of LDL (A). C3b opsonization occurred on both LDL and acLDL upon incubation with 10% NHS. Mannan served as a positive control, whereas human serum albumin (HSA) served as a negative control. The addition of EDTA to NHS prevented complement activation and C3b opsonization of LDL and acLDL completely (B). Every figure represents the mean ± S.E. of at least three experiments.
Binding of Opsonized LDL and acLDL to CR1
The capability of CR1 to bind opsonized LDL and acLDL was tested. For this purpose a cell model with CHO-CR1 and control CHO cells were used. Binding of opsonized LDL to CHO-CR1 was significantly higher compared with CHO (11.6 ± 1.1 a.u. versus 5.5 ± 0.3 a.u., p < 0.05). Similar results were found for opsonized acLDL (8.3 ± 1.4 a.u. versus 4.8 ± 0.8 a.u., p < 0.05) (Fig. 2A). This binding was dose-dependently reduced when LDL and acLDL were incubated with different ratios of HIS and NHS (Fig. 2, B and C).
FIGURE 2.
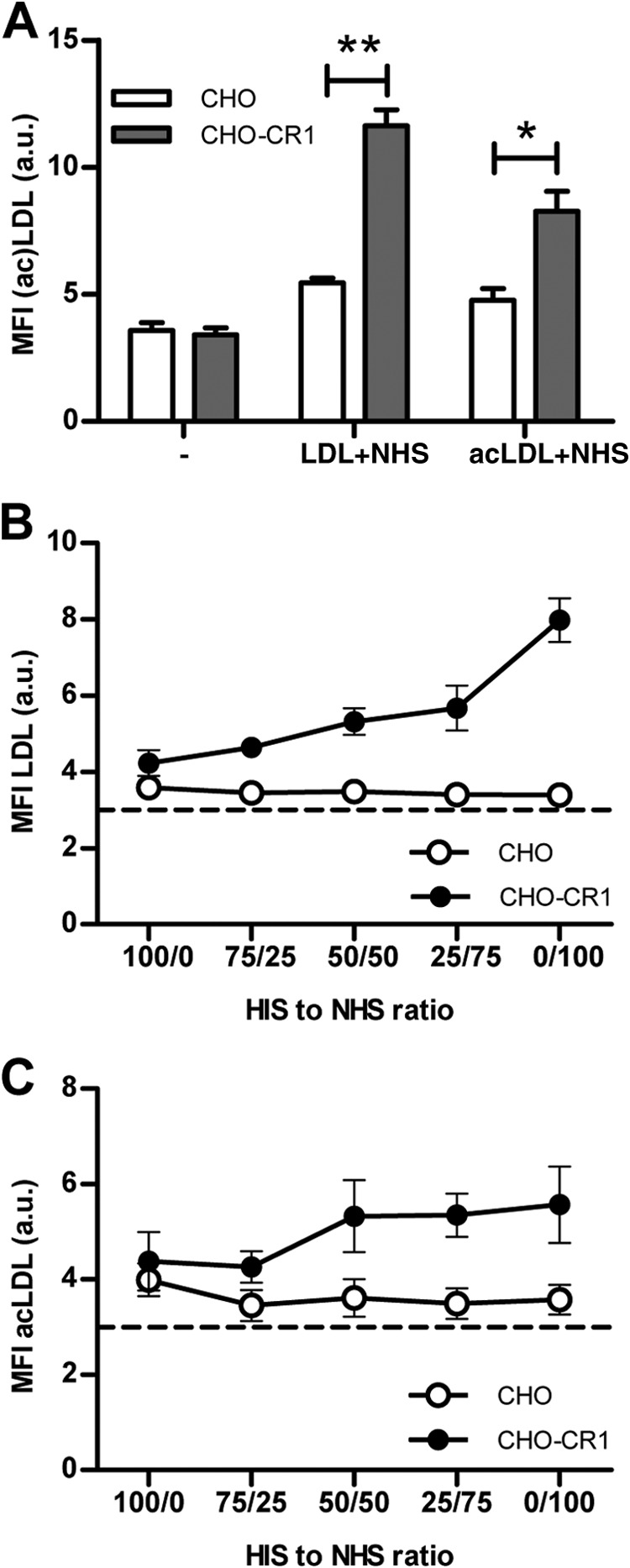
CHO-CR1 bound significantly more LDL (10 μg/ml) and acLDL (10 μg/ml) than CHO when LDL and acLDL were first incubated with normal human serum (NHS) (A). LDL and acLDL binding to CHO-CR1 was diminished when heat inactivated serum (HIS) was used to pre-incubate LDL and acLDL instead of NHS (B and C). The dotted horizontal lines in the figures represent the autofluorescent background signal. *, p < 0.05; **, p < 0.01.
Binding of non-opsonized LDL and acLDL to CHO and CHO-CR1 was similar between CHO and CHO-CR1 (Fig. 3A). Binding of LDL to CHO via the LDL receptor was demonstrated by a reduction in LDL binding to CHO by pre-incubation of CHO with an LDL-receptor blocking antibody (Fig. 3B). Binding of labeled LDL could be reduced to a similar extent by inducing competition for LDL-receptor binding between labeled and non-labeled LDL. Both, increasing concentrations of NHS or non-labeled LDL, reduced the binding of labeled LDL to CHO in a similar way as the addition of LDL-receptor blocking antibodies (Fig. 3, C and D). These data demonstrate that the addition of NHS to induce complement activation in the experiments from Fig. 3A effectively reduced the binding of labeled LDL to the LDL-receptor on CHO-cells.
FIGURE 3.

Non-opsonized LDL and acLDL binding to CHO cells was investigated by flow cytometry. CHO and CHO-CR1 bound LDL (10 μg/ml) in a similar proportion, but binding of acLDL (10 μg/ml) was less clear (A). Binding of labeled LDL was reduced when CHO were pre-incubated with increasing concentrations of anti-human LDL receptor antibodies (B). Comparable reductions in LDL binding to CHO were observed when labeled (fluorescent) LDL (10 μg/ml) was combined with increasing concentrations of native (non-labeled) LDL (C) or normal human serum (NHS) (D) due to competitive binding between labeled and non-labeled LDL. The dotted horizontal lines represent the autofluorescent background signal.
Binding of Human IgM, C1q, MBL, and Properdin to LDL and acLDL
The binding of several complement factors to LDL and acLDL was tested to explore, which complement factors may be involved in CR1-bound LDL and acLDL. Therefore, binding of purified forms of IgM, C1q, MBL, and properdin to LDL- and acLDL-coated plates was established by ELISA. IgM bound to acLDL dose dependently, but not to LDL when the lipoproteins were incubated with IgM (Fig. 4, A and B). C1q bound to both LDL and acLDL, but binding to acLDL was more than 2-fold higher when compared with LDL or to the positive control IgM (Fig. 4C). Binding of C1q to acLDL was already observed at concentrations of 0.08 μg/ml, whereas C1q binding to LDL or natural IgM were observed at concentrations above 1 μg/ml (Fig. 4D). MBL did not bind to either LDL or acLDL, whereas MBL effectively bound to mannan, which served as a positive control for MBL binding (Fig. 4, E and F). Properdin bound to acLDL, but not to LDL (Fig. 4, G and H). Altogether these results imply that opsonization of either LDL or acLDL may involve different pathways of complement activation.
FIGURE 4.

Binding of different complement factors to LDL and acLDL was investigated by ELISA. Isolated natural IgM (10 μg/ml) bound to acLDL, but not to LDL (A) and a clear dose response curve observed with natural IgM binding to acLDL (B). Isolated C1q (5 μg/ml) bound to LDL to a similar extent as the positive control natural IgM, whereas C1q was highly bound by acLDL (C) with a clear dose response curve (D). Isolated mannose binding lectin (MBL) (0.13 μg/ml) did not bind to either LDL or acLDL, in contrast to mannan, which served as a positive control (E), which was also observed with higher concentrations of MBL (F). Properdin (10 μg/ml) highly bound acLDL, but not LDL (G). The binding of properdin to acLDL was already observed at very low concentrations (H). Every figure represents the mean ± S.E. of at least three experiments.
Involvement of Different Complement Pathways in the Binding of LDL and acLDL to CR1
The involvement of the classical and lectin pathways in C3b opsonization of LDL and acLDL were investigated. For this purpose C3b opsonization of LDL and acLDL was detected using a C3b-ELISA on LDL- or acLDL-coated plates incubated with NHS substituted with different concentrations of either anti-C1q or d-mannose. C3b opsonization of LDL was almost completely diminished by anti-C1q at an anti-C1q concentration of 5 μg/ml or higher. C3b opsonization of acLDL was only gradually reduced by anti-C1q with a maximum decrease in C3b opsonization of acLDL of 36% at an anti-C1q concentration of 10 μg/ml (Fig. 5A). d-Mannose was able to block C3b opsonization of LDL at a maximum concentration of 400 mm in a similar way as for mannan. In contrast, d-mannose did not affect C3b opsonization of acLDL (Fig. 5B).
FIGURE 5.

C3b opsonization of LDL was highly reduced by the addition of anti-C1q to 10% NHS, whereas C3b opsonization of acLDL was only modestly reduced by antiC1q (20 μg/ml). The dose response pattern of anti-C1q and its reduction on C3b deposition on LDL is comparable to its control IgM, whereas increasing concentrations of anti-C1q showed a more linear reduction in C3b opsonization of acLDL (A). The addition of d-mannose to 10% NHS reduced C3b deposition on LDL, but not on acLDL. The reduction in C3b opsonization of LDL by d-mannose was similar to its control mannan (B). Binding of LDL to CHO-CR1 was significantly reduced by the addition of d-mannose (100 mm), MgEGTA (20 mm) or EDTA (20 mm) to normal human serum (NHS). Anti-C1q (20 μg/ml) to NHS resulted in a non-significant reduction in LDL binding to CHO-CR1 (C). AcLDL binding to CHO-CR1 was only reduced by the addition of EDTA to NHS, but not by anti-C1q, d-mannose, or MgEGTA (D). Every figure represents the relative mean ± S.E. of at least three experiments. *, p < 0.05; **, p < 0.01.
In addition, we conducted experiments to test, which complement pathways were involved in LDL and acLDL binding to CHO-CR1. For this purpose we incubated CHO-CR1 with fluorescent labeled LDL and acLDL, which were incubated with NHS substituted with either anti-C1q, d-mannose, MgEGTA or EDTA. By blocking the classical pathway with anti-C1q LDL binding to CHO-CR1 was slightly decreased. When the lectin pathway was blocked with d-mannose LDL binding to CHO-CR1 was significantly decreased. LDL binding to CHO-CR1 could also be inhibited with MgEGTA, which blocks both the classical and lectin pathway but not the alternative pathway, and EDTA, which blocks all three complement pathways (Fig. 5C). AcLDL binding to CHO-CR1 was significantly decreased by the addition of EDTA and slightly affected by anti-C1q, but not by d-mannose or MgEGTA (Fig. 5D).
CR1 Up-regulation on Leukocytes and Binding of apo B-containing Lipoproteins
Blood from different volunteers (n = 6) was used to investigate whether up-regulation of CR1 would result in increased binding of apo B containing lipoproteins on leukocytes. Activation of complement and leukocytes was induced by incubation of whole blood with LPS. This resulted in a significant increase in CR1 expression on monocytes and neutrophils when compared with baseline conditions at T0 and with control experiments using NaCl without LPS (Fig. 6, A and B). A concomitant increased binding of apo B by monocytes and neutrophils upon incubation with LPS was observed (Fig. 6, C and D). No significant differences in LDL receptor expression on monocytes or neutrophils were observed after incubation with either NaCl or LPS (data not shown).
FIGURE 6.

Human monocytes and neutrophils were incubated with LPS resulting in an increased CR1 expression (A and B) and concomitant increased binding of apolipoprotein (apo) B containing lipoproteins (C and D) when compared with baseline condition T0 and incubation with 0. 9% NaCl. Every figure represents the mean ± S.E. of six separate experiments. *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
DISCUSSION
The current experiments show for the first time that opsonized LDL and acLDL can bind to CR1. LDL and acLDL can activate the complement system through different pathways with subsequent C3b opsonization of these lipoproteins followed by binding to CR1. Recently, it was demonstrated in vitro that complement activation occurred when epithelial cells were incubated with oxLDL in NHS supplemented cell cultures. This resulted in increased C3b levels, which could be prevented by using complement inactive serum (HIS) instead of NHS, which is in line with our results (11).
Enzymatically modified LDL (E-LDL) activates the C1 complex via the classical pathway (31). Our data suggest that LDL binds to CR1 primarily via C3b formed by activation of the classical pathway, whereas acLDL binds to CR1 via both the classical and alternative pathways. We were able to demonstrate C1q binding to native LDL in a similar proportion as to IgM, but only with high concentrations of purified C1q. In contrast, a previous study showed that reconstitution of purified C1q to 10% C1q-depleted serum, resulted only in C3b deposition on modified forms of LDL, but not on native LDL (11). We observed that only sufficiently high enough NHS concentrations lead to C3b-opsonization of native LDL.
We also observed a reduction in CR1-bound LDL with decreased C3b opsonization in the presence of high concentrations of d-mannose. These results were unexpected since binding of MBL to LDL or acLDL could not be established in our hands, which was in line with published data (32). In contrast, a different study demonstrated binding of MBL to acLDL, but reconstitution of MBL-deficient serum with purified MBL did not result in complement activation on acLDL (12). Therefore, the exact contribution of MBL and the lectin pathway to the binding of LDL to CR1 needs to be explored further.
We studied the importance of the alternative pathway in these processes in addition to the classical and lectin pathways. Properdin was able to bind acLDL, but not native LDL. Moreover, MgEGTA, which blocks selectively the classical and lectin pathway but not the alternative pathway, did not affect acLDL binding to CHO-CR1, whereas it significantly reduced LDL binding to CHO-CR1. These results illustrate the importance of the alternative pathway in the binding of acLDL to CR1. It was already described that oxLDL is able to increase factor B in vitro, which is a stimulator of the alternative pathway (11).
Fig. 7 provides a schematic model, summarizing the complement pathways and its complement factors involved in LDL and acLDL binding to CR1. This model proposes that the classical pathway is involved in both LDL and acLDL binding to CR1, whereas the alternative pathway via properdin also regulates acLDL binding to CR1. The exact contribution of MBL and the lectin pathway in LDL binding to CR1 remains unclear. C4b may also be involved, but in our model we only studied C3b as the ligand to bind (ac)LDL to CR1 since C3b has been the most extensively studied complement factor in this respect.
FIGURE 7.

Schematic overview of the mechanism of CR1-bound LDL and acLDL. Binding of LDL to CR1 is mediated via the classical pathway. C1q binds first to LDL by C3b opsonization, which serves as the ligand for CR1. The role of MBL and the lectin pathway in CR1-bound LDL remains unclear since we were unable to show MBL binding to LDL despite an effect of alienating the lectin pathway. Binding of acLDL to CR1 is mediated via the classical and alternative pathway. AcLDL can be bound by both C1q and properdin (P), which are able to initiate the classical and alternative pathway, respectively. In addition, IgM, which is able to bind C1q, can also bind to acLDL. The LDL and modified LDL binding capacity of CR1 may provide an additional mechanism for cellular bound apolipoprotein B-containing lipoproteins.
We used flow cytometry to quantify LDL and acLDL binding to CHO and CHO-CR1, but comparable results were obtained by using immunofluorescence microscopy (data not shown). It should be noted that all studies presented here were done in vitro and ex vivo and it is still not clear whether LDL and modified forms of LDL are able to induce complement activation in vivo. However, using shotgun proteomics it has been demonstrated that C3 and C4 are present on HDL in vivo (8). In our experiments, C3b was not present on freshly isolated LDL or acLDL and addition of NHS was necessary to initiate C3b opsonization of LDL and acLDL. The absence of C3b on freshly isolated LDL from serum may be explained by avid binding of opsonized LDL- to CR1-expressing cells like endothelial cells, leukocytes, and erythrocytes in the cellular blood compartment (15, 21–23). Our experiments with human leukocytes provided indirect evidence that CR1 contributes to the binding of apo B containing lipoproteins on human leukocytes. The increased binding of apo B containing lipoproteins to monocytes and neutrophils could not be attributed to an up-regulation of the LDL receptor since the LDL receptor expression remained unchanged during these experiments. Others have described an increase in the number of LDL receptors on macrophages during LPS stimulation, but those incubations lasted 24 h (33).
Our data suggest a previously undescribed cellular binding mechanism for LDL and modified LDL via CR1, which is ubiquitously distributed throughout the human body. CR1 was first isolated from human erythrocytes where CR1 facilitates immune adherence of immune complexes and microorganisms to erythrocytes (18–20, 34). CR1 is also present on various inflammatory cells. Monocytes, macrophages, and neutrophils are able to internalize CR1 as a defense mechanism for phagocytosis of CR1-bound immune complexes in contrast to erythrocytes (17).
The expression of CR1 by inflammatory cells is highly variable within subjects. As reported previously, an increase in CR1 expression by monocytes and neutrophils occurs upon the ingestion of a fat meal (35). On average, monocytes and neutrophils express 20,000 to 140,000 CR1 molecules per cell (36, 37). Erythrocytes express only ∼300–800 CR1 molecules per erythrocyte, but most of the total CR1 in the circulation is present on erythrocytes since they form the majority of the cellular blood compartment (38). The liver clears immune complexes and microorganisms bound to erythrocytes at high rates (18). Something similar may occur with CR1-mediated erythrocyte-bound LDL, which may be an atheroprotective mechanism (15, 22). In line with this mechanism, it has been suggested that erythrocytes contribute to reverse cholesterol transport (39). We have shown recently that the presence of apo B on erythrocytes is associated with a lower carotid intima media thickness and decreased prevalence of cardiovascular disease in humans (22, 23).
In conclusion, CR1 is able to bind opsonized native LDL and acLDL. Binding of LDL to CR1 is mediated via the classical pathway, whereas binding of acLDL is mediated via both the classical and alternative pathways.
Acknowledgments
This study is dedicated to the memory of our dear friend Prof. Dr. Hans van Dijk. We thank Prof. Dr. J.P. Atkinson and Dr. M.K. Liszewski (Washington University School of Medicine, St Louis) and Dr. S. Rooijakkers (University Medical Center Utrecht, Utrecht, the Netherlands) for kindly providing the appropriate CHO cells.
Footnotes
- CRP
- C-reactive protein
- MBL
- mannose-binding lectin
- LDL
- low density lipoprotein
- CR
- complement receptor
- LPS
- lipopolysaccharide
- acLDL
- acetylated LDL
- NHS
- normal human serum.
REFERENCES
- 1. Oksjoki R., Kovanen P. T., Pentikäinen M. O. (2003) Role of complement activation in atherosclerosis. Curr. Opin. Lipidol. 14, 477–482 [DOI] [PubMed] [Google Scholar]
- 2. Ricklin D., Hajishengallis G., Yang K., Lambris J. D. (2010) Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kojima M., Presanis J. S., Sim R. B. (2003) The mannose-binding lectin (MBL) route for activation of complement. Adv. Exp. Med. Biol. 535, 229–250 [DOI] [PubMed] [Google Scholar]
- 4. Walport M. J. (2001) Complement. Second of two parts. N. Engl. J. Med. 344, 1140–1144 [DOI] [PubMed] [Google Scholar]
- 5. Walport M. J. (2001) Complement. First of two parts. N. Engl. J. Med. 344, 1058–1066 [DOI] [PubMed] [Google Scholar]
- 6. Bhakdi S., Torzewski M., Klouche M., Hemmes M. (1999) Complement and atherogenesis: binding of CRP to degraded, nonoxidized LDL enhances complement activation. Arterioscler. Thromb. Vasc. Biol. 19, 2348–2354 [DOI] [PubMed] [Google Scholar]
- 7. van Oostrom A. J., Alipour A., Plokker T. W., Sniderman A. D., Cabezas M. C. (2007) The metabolic syndrome in relation to complement component 3 and postprandial lipemia in patients from an outpatient lipid clinic and healthy volunteers. Atherosclerosis. 190, 167–173 [DOI] [PubMed] [Google Scholar]
- 8. Vaisar T., Pennathur S., Green P. S., Gharib S. A., Hoofnagle A. N., Cheung M. C., Byun J., Vuletic S., Kassim S., Singh P., Chea H., Knopp R. H., Brunzell J., Geary R., Chait A., Zhao X. Q., Elkon K., Marcovina S., Ridker P., Oram J. F., Heinecke J. W. (2007) Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J. Clin. Invest. 117, 746–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alipour A., van Oostrom A. J., Van Wijk J. P., Verseyden C., Plokker H. W., Jukema J. W., Rabelink A. J., Castro Cabezas M. (2009) Mannose binding lectin deficiency and triglyceride-rich lipoprotein metabolism in normolipidemic subjects. Atherosclerosis 206, 444–450 [DOI] [PubMed] [Google Scholar]
- 10. Gauvreau D., Roy C., Tom F. Q., Lu H., Miegueu P., Richard D., Song W. C., Stover C., Cianflone K. (2012) A new effector of lipid metabolism: complement factor properdin. Mol. Immunol. 51, 73–81 [DOI] [PubMed] [Google Scholar]
- 11. Ebrahimi K. B., Fijalkowski N., Cano M., Handa J. T. (2013) Decreased membrane complement regulators in the retinal pigmented epithelium contributes to age-related macular degeneration. J. Pathol. 229, 729–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fraser D. A., Tenner A. J. (2010) Innate immune proteins C1q and mannan-binding lectin enhance clearance of atherogenic lipoproteins by human monocytes and macrophages. J. Immunol. 185, 3932–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goldstein J. L., Brown M. S. (2009) The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 29, 431–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mitra S., Goyal T., Mehta J. L. (2011) Oxidized LDL, LOX-1 and atherosclerosis. Cardiovasc. Drugs Ther. 25, 419–429 [DOI] [PubMed] [Google Scholar]
- 15. Bovenberg S. A., Alipour A., Elte J. W., Rietveld A. P., Janssen J. W., van de Geijn G. J., Njo T. N., van Mechelen R., Hervas S. M., Cabezas M. C. (2010) Cell-mediated lipoprotein transport: A novel anti-atherogenic concept. Atheroscler. Suppl. 11, 25–29 [DOI] [PubMed] [Google Scholar]
- 16. Langeggen H., Berge K. E., Johnson E., Hetland G. (2002) Human umbilical vein endothelial cells express complement receptor 1 (CD35) and complement receptor 4 (CD11c/CD18) in vitro. Inflammation 26, 103–110 [DOI] [PubMed] [Google Scholar]
- 17. Fearon D. T., Wong W. W. (1983) Complement ligand-receptor interactions that mediate biological responses. Annu. Rev. Immunol. 1, 243–271 [DOI] [PubMed] [Google Scholar]
- 18. Cornacoff J. B., Hebert L. A., Smead W. L., VanAman M. E., Birmingham D. J., Waxman F. J. (1983) Primate erythrocyte-immune complex-clearing mechanism. J. Clin. Invest. 71, 236–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Waxman F. J., Hebert L. A., Cornacoff J. B., VanAman M. E., Smead W. L., Kraut E. H., Birmingham D. J., Taguiam J. M. (1984) Complement depletion accelerates the clearance of immune complexes from the circulation of primates. J. Clin. Invest. 74, 1329–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nelson R. A., Jr. (1953) The Immune-Adherence Phenomenon. Science 118, 733–737 [DOI] [PubMed] [Google Scholar]
- 21. Alipour A., van Oostrom A. J., Izraeljan A., Verseyden C., Collins J. M., Frayn K. N., Plokker T. W., Elte J. W., Castro Cabezas M. (2008) Leukocyte activation by triglyceride-rich lipoproteins. Arterioscler. Thromb. Vasc. Biol. 28, 792–797 [DOI] [PubMed] [Google Scholar]
- 22. Bovenberg S. A., Klop B., Alipour A., Martinez-Hervas S., Westzaan A., van de Geijn G. J., Janssen H. W., Njo T., Birnie E., van Mechelen R., Rietveld A. P., Elte J. W., Castro Cabezas M. (2012) Erythrocyte-associated apolipoprotein B and its relationship with clinical and subclinical atherosclerosis. Eur. J. Clin. Invest. 42, 365–370 [DOI] [PubMed] [Google Scholar]
- 23. Klop B., van de Geijn G. J., Bovenberg S. A., van der Meulen N., Elte J. W., Birnie E., Njo T. L., Janssen H. W., van Miltenburg A., Jukema J. W., Cabezas M. C. (2013) Erythrocyte-Bound Apolipoprotein B in Relation to Atherosclerosis, Serum Lipids and ABO Blood Group. PLoS One 8, e75573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Makrides S. C., Scesney S. M., Ford P. J., Evans K. S., Carson G. R., Marsh H. C., Jr. (1992) Cell surface expression of the C3b/C4b receptor (CR1) protects Chinese hamster ovary cells from lysis by human complement. J. Biol. Chem. 267, 24754–24761 [PubMed] [Google Scholar]
- 25. Nickells M., Hauhart R., Krych M., Subramanian V. B., Geoghegan-Barek K., Marsh H. C., Jr., Atkinson J. P. (1998) Mapping epitopes for 20 monoclonal antibodies to CR1. Clin. Exp. Immunol. 112, 27–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Redgrave T. G., Roberts D. C., West C. E. (1975) Separation of plasma lipoproteins by density-gradient ultracentrifugation. Anal. Biochem. 65, 42–49 [DOI] [PubMed] [Google Scholar]
- 27. Van Berkel T. J., Nagelkerke J. F., Harkes L., Kruijt J. K. (1982) Processing of acetylated human low-density lipoprotein by parenchymal and non-parenchymal liver cells. Involvement of calmodulin? Biochem. J. 208, 493–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gaarkeuken H., Siezenga M. A., Zuidwijk K., van Kooten C., Rabelink T. J., Daha M. R., Berger S. P. (2008) Complement activation by tubular cells is mediated by properdin binding. Am. J. Physiol. Renal Physiol. 295, F1397–F1403 [DOI] [PubMed] [Google Scholar]
- 29. Roos A., Bouwman L. H., Munoz J., Zuiverloon T., Faber-Krol M. C., Fallaux-van den Houten F. C., Klar-Mohamad N., Hack C. E., Tilanus M. G., Daha M. R. (2003) Functional characterization of the lectin pathway of complement in human serum. Mol. Immunol. 39, 655–668 [DOI] [PubMed] [Google Scholar]
- 30. Nauta A. J., Trouw L. A., Daha M. R., Tijsma O., Nieuwland R., Schwaeble W. J., Gingras A. R., Mantovani A., Hack E. C., Roos A. (2002) Direct binding of C1q to apoptotic cells and cell blebs induces complement activation. Eur. J. Immunol. 32, 1726–1736 [DOI] [PubMed] [Google Scholar]
- 31. Biró A., Thielens N. M., Cervenák L., Prohászka Z., Füst G., Arlaud G. J. (2007) Modified low density lipoproteins differentially bind and activate the C1 complex of complement. Mol. Immunol. 44, 1169–1177 [DOI] [PubMed] [Google Scholar]
- 32. Faro J., Chen Y., Jhaveri P., Oza P., Spear G. T., Lint T. F., Gewurz H. (2008) L-ficolin binding and lectin pathway activation by acetylated low-density lipoprotein. Clin. Exp. Immunol. 151, 275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ye Q., Lei H., Fan Z., Zheng W., Zheng S. (2014) Difference in LDL receptor feedback regulation in macrophages and vascular smooth muscle cells: foam cell transformation under inflammatory stress. Inflammation 37, 555–565 [DOI] [PubMed] [Google Scholar]
- 34. Fearon D. T. (1979) Regulation of the amplification C3 convertase of human complement by an inhibitory protein isolated from human erythrocyte membrane. Proc. Natl. Acad. Sci. U.S.A. 76, 5867–5871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klop B., van de Geijn G. J., Njo T. L., Janssen H. W., Rietveld A. P., van Miltenburg A., Fernandez-Sender L., Elte J. W., Cabezas M. C. (2013) Leukocyte cell population data (volume conductivity scatter) in postprandial leukocyte activation. Int. J. Lab. Hematol. 6, 644–651 [DOI] [PubMed] [Google Scholar]
- 36. Quadri R. A., Schifferli J. A. (1992) Over-estimation of the number of complement receptor type 1 (CR1) on erythrocytes. Scand J. Immunol. 36, 125–130 [DOI] [PubMed] [Google Scholar]
- 37. Wilson J. G., Wong W. W., Schur P. H., Fearon D. T. (1982) Mode of inheritance of decreased C3b receptors on erythrocytes of patients with systemic lupus erythematosus. N. Engl. J. Med. 307, 981–986 [DOI] [PubMed] [Google Scholar]
- 38. Hinderling P. H. (1997) Red blood cells: a neglected compartment in pharmacokinetics and pharmacodynamics. Pharmacol. Rev. 49, 279–295 [PubMed] [Google Scholar]
- 39. Hung K. T., Berisha S. Z., Ritchey B. M., Santore J., Smith J. D. (2012) Red blood cells play a role in reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 32, 1460–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]