

Background: The transcription factor Hes3 regulates the growth of neural and brain cancer stem cells.
Results: Hes3 regulates growth, gene expression, evoked insulin release in cultured insulinoma cells, and sensitivity to streptozotocin in vivo.
Conclusion: Hes3 is a novel regulator of cellular functions of importance in diabetes.
Significance: Introducing Hes3 and its regulators in diabetes research may provide new opportunities for the design of novel therapeutics.
Keywords: Beta Cell (B-cell), Cell Culture, Islet, Neural Stem Cell (NSC), Neurodegenerative Disease, Neuroprogenitor Cell, Notch Pathway, Pancreas, Parkinson Disease, Signal Transduction
Abstract
The transcription factor Hes3 is a component of a signaling pathway that supports the growth of neural stem cells with profound consequences in neurodegenerative disease models. Here we explored whether Hes3 also regulates pancreatic islet cells. We showed that Hes3 is expressed in human and rodent pancreatic islets. In mouse islets it co-localizes with alpha and beta cell markers. We employed the mouse insulinoma cell line MIN6 to perform in vitro characterization and functional studies in conditions known to modulate Hes3 based upon our previous work using neural stem cell cultures. In these conditions, cells showed elevated Hes3 expression and nuclear localization, grew efficiently, and showed higher evoked insulin release responses, compared with serum-containing conditions. They also exhibited higher expression of the transcription factor Pdx1 and insulin. Furthermore, they were responsive to pharmacological treatments with the GLP-1 analog Exendin-4, which increased nuclear Hes3 localization. We employed a transfection approach to address specific functions of Hes3. Hes3 RNA interference opposed cell growth and affected gene expression as revealed by DNA microarrays. Western blotting and PCR approaches specifically showed that Hes3 RNA interference opposes the expression of Pdx1 and insulin. Hes3 overexpression (using a Hes3-GFP fusion construct) confirmed a role of Hes3 in regulating Pdx1 expression. Hes3 RNA interference reduced evoked insulin release. Mice lacking Hes3 exhibited increased islet damage by streptozotocin. These data suggest roles of Hes3 in pancreatic islet function.
Introduction
The basic helix-loop-helix transcription factor Hes3 is a member of the Hes/Hey gene family that regulate developmental processes in progenitor cells from various tissues (1–7). Family members such as Hes1 and Hes5 are direct transcriptional targets of the cleaved intracellular domain of the Notch receptor (8). Hes3 stands out within this family as an indirect target of a non-canonical branch of the Notch signaling pathway (9). Specifically, in rodent neural stem cell (NSC)3 cultures, activation of the Notch receptor by soluble forms of the Delta4 and Jagged1 ligands induces the PI 3-kinase-dependent phosphorylation of Akt, mammalian target of rapamycin, STAT3, on serine residue 727, and subsequent induction of Hes3 transcription leading to increased cell survival and growth (10). Another activator of the Akt/mammalian target of rapamycin/STAT3-serine pathway, insulin, also induces Hes3 transcription and promotes cell growth (11). Hes3 is a functional mediator of this pathway in normal and cancerous tissues. NSC cultures from the subventricular zone of adult Hes3 null mice can be established but they are non-responsive to treatments that normally promote Hes3 expression and increase their number such as Delta4 and insulin (11). Inhibition of Hes3 expression by RNA interference in cultures of primary human brain cancer stem cells opposes their growth (12).
Hes3 has two forms: Hes3a and Hes3b (13). Hes3a cannot bind DNA but can still form heterodimers with other basic helix-loop-helix factors. Hes3b can both bind DNA and form heterodimers. The expression of another member of the Hes/Hey gene family, Hes1, and of other basic helix-loop-helix factors exhibit an oscillatory pattern (2). Oscillatory expression of the basic helix-loop-helix Ascl1 characterizes the self-renewing state, whereas sustained expression of specific genes results in fate determination, suggesting oscillatory versus sustained expression patterns are means of regulating cell fate.
Several studies support a role of Hes3 and its regulators in a number of normal and cancerous tissues, and in various regenerative processes. Macrophage inhibitory factor induces Hes3 expression and promotes NSC/progenitor cell proliferation and maintenance (14). Delta4, alone or in combination with other treatments such as basic fibroblast growth factor and epidermal growth factor (EGF), increases the number of endogenous progenitors in several areas of the adult brain (10, 11, 15–17). Delta4 induces Hes3 expression and promotes the acquisition of the definitive NSC fate from iPS-derived primitive NSCs (18). When Hes3 is “knocked out” from the Hes1:Hes5 double-mutant mouse line, neuroepithelial cells prematurely differentiate into neurons during embryonic development (19). A phosphomimetic STAT3-serine construct promotes prostate tumorigenesis independently of the JAK-STAT pathway (20), which involves the phosphorylation of STAT3 on tyrosine 705 (21). Notch-dependent STAT3-serine phosphorylation contributes to the growth of embryonic stem cell-derived NSCs following induction of Hoxb1 expression (22). The anti-tumor efficacy of a small molecule inhibitor of γ-secretase, an enzyme involved in Notch receptor activation (3), can be predicted by the level of expression of Hes3 in breast cancer xenograft models (23).
Here, using a mouse insulinoma cell line (MIN6) and observations in isolated and dissociated/cultured mouse and human islets, we addressed possible functions of Hes3, which may be of interest to the field of diabetes. We showed that Hes3 is expressed in mouse and human pancreatic islets and that genetic manipulation of Hes3 in MIN6 cells affects gene expression; key genes regulated include insulin and pancreatic and duodenal homeobox 1 (Pdx1), a transcription factor involved in pancreatic development and diabetes (24). In addition, Hes3 regulates the cell number and evoked insulin release. Using a Hes3 null mouse strain where the Hes3 gene was replaced by the reporter lacZ gene (25), we confirmed Hes3 expression in the adult pancreatic islet and induction following streptozotocin (STZ)-induced damage, and showed that in the absence of Hes3, STZ-induced damage is more pronounced, as indicated by reduced beta cell number and increased blood glucose levels in vivo. Taken together, these findings suggest that Hes3 may have important roles in pancreatic islet function.
EXPERIMENTAL PROCEDURES
Cell Culture
MIN6 cells were grown in either serum containing medium (DMEM (Invitrogen, 61965-026), 15% fetal calf serum (Biochrom Superior, F0615), 70 μm 2-mercaptoethanol (Sigma, M6250), 100 μg/ml of Pen-Strep (Invitrogen, 15140-122)), or serum-free N2 medium (DMEM-F12 (Sigma, D8062), 100 μg/ml of apo-transferrin (Sigma, T2036), 20 nm progesterone (Sigma, P8783), 100 μm putrescine (Sigma, P5780), 30 nm selenite (Sigma, S5261), and 100 μg/ml of Pen-Strep)). To examine changes in the properties of MIN6 cells under different culture conditions, cells were first maintained in serum containing medium for 5 days. Cells were then passaged and grown under serum-free conditions for another 5 days. The cells were then passaged again, back into serum containing medium (“RC” for return condition) for an additional 5 days prior to analysis. Serum and no serum controls were run in parallel in which the cells were maintained in either serum containing or serum-free conditions for the final 10 days of the experiment. For Exendin-4 treatments, cells were seeded at 50,000 cells/24-well plate well and treated with different concentrations of Exendin-4 (Biotrend, BP0111) beginning at 24 h after plating for different times, as indicated. For the sustained treatments, Exendin-4 was added every 2 days during medium changes, and cells were fixed and immunostained at day 5. In this paper, we denote serum-containing medium as “CC” (common conditions) and serum-free medium as “DC” (defined conditions).
JAK inhibitor treatments were performed on MIN6 cells seeded at 50,000 cells/well in a 24-well plate in DC. The inhibitor (Calbiochem 420097) was added to the culture medium daily from day 1 to 5; cell fixation and visualization was on day 5.
DNA Microarray
RNA was extracted from MIN6 cells grown in T75 flasks under the above mentioned culture conditions. For experiments using Hes3 siRNA, cells were transfected at 24 h after seeding. RNA integrity number for each sample was checked using Agilent Bioanalyzer (Agilent Michigan). Quadruplicate samples from a single experiment were run in DNA microarray equipment (Affymetrix). Volcano plots, heat maps, and statistics were generated using the R-2.15.2 software.
RNA Isolation and Reverse Transcriptase PCR
RNA was extracted from whole pancreas or MIN6 cells using the High Pure RNA isolation kit (Roche Applied Science, 11828665001) and reverse transcribed using Promega M-MLV reverse transcriptase (Promega, M170B). PCR was performed for total Hes3, Hes3a, Hes3b, Pdx1, insulin, and GAPDH (for primer list, please see below) using Dream Taq Green DNA polymerase (Thermo Scientific, EP0711).
Western Blotting
MIN6 cells were grown in 6-well plates for 5 days, then lysed with 500 μl of CytoBuster Protein Extraction Reagent (Novagen, 71009-3) containing protease inhibitors (Sigma, P8340). Samples were resolved by Western blotting using standard techniques.
Immunohistochemistry/Immunocytochemistry
Mouse pancreata were fixed by transcardial perfusion with 4% PFA followed by post-fixation overnight at 4 °C. Human isolated islets were fixed with 4% PFA for 30 min. The tissue was cryoprotected with 30% sucrose and embedded in OCT compound (Sakura, 4583). 12–14-μm sections were cut and mounted on glass slides. MIN6 cells were fixed in 4% PFA for 30 min. Both the sections and cells were permeabilized in 0.1% Triton X-100 in PBS and blocked in 5% powdered milk and 0.1% Triton X-100 in PBS prior to incubation with primary antibody. Images were taken using a laser confocal microscope (LEICA, Germany).
Detection of β-Galactosidase Enzymatic Activity
Tissue sections were washed 3 × 5 min in LacZ wash buffer (PBS, 0.01% sodium deoxycholate, Sigma D6750, 0.002% Nonidet P-40, Roche 11754599001, and 2 mm MgCl2, Sigma 630063) and incubated overnight in LacZ staining buffer (LacZ buffer, 5 mm potassium ferrocyanide, Sigma, 736716; 5 mm potassium ferricyanide, Sigma, 702587) containing 2.45 mm 5-bromo-4-chloro-3-indoxy-β-d-galactopyranoside (X-Gal, ROTH, R0404). Sections were then visualized using brightfield microscopy.
Cell Proliferation
MIN6 cell proliferation was determined 4 days after the final cell passage by incubating the cells with 10 μm EdU for 8 h followed by visualization using the Click-IT EdU Alexa Fluor 594 Imaging Kit (Invitrogen, C10339).
Cell Viability Assay
Adherent MIN6 cells were cultured on sterile glass coverslips in CC, DC, and RC for 5 days. Cells were washed briefly with PBS and incubated with 4 μm Eth-D solution (LIVE/DEAD Viability/Cytotoxicity Kit for mammalian cells, Invitrogen, L-3224) for 30 min. Cells were fixed with 4% PFA and imaged.
Insulin Secretion Assay (with Variable KCl)
Combined glucose/KCl stimulated insulin production assay was performed on MIN6 cells as described in Ref. 26 using a Rat Insulin ELISA kit (Mercodia, 10-1250-01) and detected with a microplate reader (TECAN). Glucose concentrations were as follows: resting, 0 mm; stimulated, 25 mm. KCl concentrations were as follows: resting, 5 mm; stimulated, 55 mm.
Insulin Secretion Assay (with Non-variable KCl)
For these experiments, cells were maintained in a low KCl concentration (5 mm), which was not altered between resting and stimulated states. Glucose concentrations were as follows: resting, 2.8 mm; stimulated, 20 mm.
Hes3 Knockdown and Overexpression
MIN6 cells were plated at 50,000 cells/well in a 24-well plate. 24 h later, cells were transfected with Hes3 siRNA (2 μm final concentration in the culture medium) or scrambled control siRNA (Santa Cruz, sc-37942 and sc-37007) using DharmaFECT4 (Thermo Scientific, T-2004-001) as described by the manufacturer. Cells were assayed 12–24 h after transfection for changes in proliferation and in gene expression as determined by gene array analysis, immunocytochemistry, and Western blot. For Hes3 overexpression, MIN6 cells were transfected using Lipofectamine 2000 as described by the manufacturer with either pcDNA3.1 containing a C terminus GFP-tagged Hes3a gene or with an empty vector. Cells were cultured up to day 5, immunostained, and imaged using laser confocal microscopy.
Mouse Models of Diabetes
For high dose streptozotocin (Sigma, S0130) experiments, 8-week-old C57Bl/6J mice were injected intraperitoneally with a single dose of 150 mg/kg of streptozotocin or with PBS (vehicle control). For low dose streptozotocin experiments, 8-week-old C57Bl/6J mice were injected intraperitoneally with 5 daily doses of 50 mg/kg of streptozotocin or with PBS (vehicle control). Pancreata were extracted 4 weeks later.
Db/Db and Ob/Ob Mice
Pancreata were harvested from 10-month-old mice homozygous for the diabetes spontaneous mutations, Lepr db or Lep ob. C57Bl/6J mice served as control. C57BLKS/J mice were used as controls.
Total Body Irradiation of Mice
8-Week-old C57Bl/6J mice were administered a single 4-gray dose of irradiation for 3 min and 41 s. Pancreata were extracted 5 days after irradiation.
In Vivo Blood Glucose Measurements
One droplet of blood from a tail clip incision was placed on a glucose test strip and read using a glucometer (ACCU-CHEK Aviva, Roche Applied Science).
In Vivo Intraperitoneal Glucose Tolerance Test
Mice were fasted for 16 h (overnight) prior to the experiment. Glucose levels were measured 0, 15, 30, 60, 90, and 120 min after intraperitoneal injection of a 20% glucose solution (2 g of glucose/kg of body weight) (27).
Mouse Pancreatic Islet Isolation
Mice were euthanized and injected with Working Enzyme solution (Dissociation buffer, Collagenase, Sigma, C7657) through the bile duct to perfuse the pancreas. Pancreas tissue was incubated for 20 min at 37 °C on a rocking platform at 100 rpm for dissociation. Dissociated tissue was washed with Quenching buffer (Hank's balanced salt solution, Invitrogen, 14025092; 10% FBS, Biochrom) and filtered. Different gradients of Ficoll (Sigma, F637) were applied to collect the islets. Islets were resuspended in Islet Culture Medium (CMRL 1066, Invitrogen, PL000676; 10% FBS Biochrom; PenStrep, Invitrogen). In this medium, hand-picking of islets was performed. Following this, islets were placed in the appropriate culture medium for experimentation as described under “Results.”
Human Pancreatic Islets
Human isolated islets (4 preparations) were obtained by the Diabetes Research Group, King's College London, UK.
Dissociation/Culture of Human Pancreatic Islets
Human pancreatic islets were washed 3 times with PBS at room temperature and dissociated using 0.05% trypsin/EDTA (Invitrogen, 25300) for 2 min at 37 °C. Cells were then seeded at a density of 50,000 cells per well in a 24-well plate and cultured either in Islet Culture Medium (as described in mouse pancreatic islet isolation) or N2 medium for 48–72 h. Cells were fixed with 4% PFA, immunostained, and imaged.
Statistical Analysis
Results shown are the mean ± S.D. or S.E., as noted in the figure legends. Experiments were performed in triplicate, unless otherwise noted. Asterisks identify experimental groups that were significantly different (p value 0.05) from control groups by the Student's t test (paired, two-sided) (Microsoft Excel).
Antibodies Used
Primary antibodies used were: anti-HES3 1:100 (Santa Cruz, M-135, sc-25393), anti-Pdx1 1:1000 (Abcam, ab47383), anti-insulin 1:200 (GeneTex, GT X27842), anti-glucagon 1:200 (GeneTex, K79bB10), anti-somatostatin 1:200 (GeneTex, YC7), and anti-NKX6.1 1:200 (R&D Systems, AF5857). Secondary antibodies used were: donkey anti-rabbit IgG (H+L) 594 (Dianova, 711-505-152), donkey anti-goat IgG (H+L) 488 (Dianova, 705-486-147), donkey anti-guinea pig IgG (H+L) 488 (Dianova, 706-486-148), donkey anti-guinea pig IgG (H+L) 649 (Dianova, 706-496-148), donkey anti-mouse IgG (H+L) 649 (Dianova, 715-496-150), and donkey anti-rat IgG (H+L) 649 (Dianova, 712-496-150). All secondary antibodies were diluted 1:500 in blocking buffer and incubated at room temperature for 1–2 h. DAPI is applied at 1:10,000 dilution for 15 min for nuclear staining. Images were taken using a laser confocal microscope (LEICA, Germany).
PCR Primers
The primers used were: MuHes3a-F, AAGCTCCCTGCCATAGCGGA; MuHes3a-R, ATGCGTGCACGGCGCTTCTT; MuHes3b-F, ACATCACAGCATGGGCACCGAGCCCACATC; MuHes3b-R, GTTGATGCGTGCACGGCGCTTCTTC; MuHes3-F, ATCTCCAAGCCTCTGATGGAGAA; MuHes3-R, AGCTTTCGTTTCCGTATCTGATG; HuHes3-F, GAGAAGCCTTCAGAACTCCTTGC; HuHes3-R, CTGCCGACCTCATCTCCGCG; MuPdx1-F, CCACCCCAGTTTACAAGCTC; MuPdx1-R, TGTAGGCAGTACGGGTCCTC; HuPdx1-F, CTTTCTCCTCCTCCTCCTTCTA; HuPdx1-R, GGTCATACTGGCTCGTGAATAG; MuInsulin-F, GACCAGCTATAATCAGAGACC; MuInsulin-R, AGTTGCAGTAGTTCTCCAGCTG; MuHes1-F, TCTCTCCTTGGTCCTGGAATA; Mu-Hes1-R, CTTCGCCTCTTCTCCATGATAG; MuGAPDH-F, CTGGAGAAACCTGCCAAGTA; MuGAPDH-R, TGTTGCTGTAGCCGTATTCA; HuGAPDH-F, GATTCCACCCATGGCAAATTC; and HuGAPDH-R, GTCATGAGTCCTTCCACGATAC.
RESULTS
Hes3 Is Expressed in Adult Pancreatic Islets
Hes3 immunolabeling of isolated adult human pancreatic islets showed expression of Hes3 (counterstained for glucagon, insulin, Pdx1, somatostatin, and Nkx6.1) (Fig. 1A). Western blot and PCR analysis confirmed Hes3 expression in protein and RNA extracts from isolated human (hu) and mouse (mu) pancreatic islets (Fig. 1, B and C). Isolated adult human pancreatic islets can be placed in culture for research or transplantation purposes (28). Typically, these cells are cultured in serum-containing media. However, in NSC cultures, serum inclusion in the culture medium opposes the nuclear localization of Hes3, and this may suppress transcriptional functions (9). For this purpose, we cultured dissociated human islet cells in both commonly used serum-containing (“CC”) and serum-free, defined (“DC”) conditions. We point out that these two media compositions differ in various aspects, as detailed under “Experimental Procedures,” not only in terms of the presence or absence of serum. Under both conditions, cells grew efficiently and the cultures maintained expression of Pdx1, Nkx6.1, and insulin (Fig. 1, D and E). The incidence of cells with nuclear Hes3, the intensity of Hes3 label, the incidence of cells expressing insulin, and the incidence of cells co-expressing Hes3 and insulin was higher in DC than in CC (Fig. 1, F–I).
FIGURE 1.

Hes3 is expressed in adult pancreatic islets and dissociated cultures of pancreatic islet cells. A, confocal immunohistochemical images of sections from isolated human pancreatic islets show nuclear Hes3 expression in cells co-expressing glucagon, insulin, Pdx1, somatostatin, and Nkx6.1 (image widths: 246 μm). B, Western blot analysis shows expression of Hes3, along with Pdx1, in isolated human and mouse pancreatic islets. C, PCR analysis from isolated human pancreatic islets shows Hes3, Pdx1, and insulin expression. D, cultures from dissociated human pancreatic islets in CC and DC exhibit Pdx1 and Nkx6.1 expression (image widths: 387 μm). E, cultures from dissociated human pancreatic islets in CC and DC exhibit insulin and Hes3 expression (image widths: 157 μm). F–I, quantification of the percentage of cells with nuclear Hes3 staining, Hes3 immunolabel intensity, percentage of cells expressing insulin, and percentage of cells expressing both Hes3 and insulin, in cultures from dissociated human pancreatic islets in CC and DC (signal intensity was measured using the Fiji image processing software. Data are mean from 500+ cells; error bars represent S.D. A total of 4 human islet preparations were used to generate these data.) J, confocal immunohistochemical images of sections from the adult mouse pancreas show that Hes3 is expressed in the nucleus of pancreatic islet cells co-expressing glucagon, insulin, Pdx1, somatostatin, and Nkx6.1 (image widths: 246 μm; insets show cell nuclei stained with DAPI). K, quantification of the percentage of cells in the islet that express Hes3, the percentage of Pdx1 positive cells in the islet that express Hes3, the percentage of Pdx1 positive cells that co-express insulin and Hes3, and the percentage of glucagon positive cells that express Hes3 in 4-, 8-, and 15-week-old mice (n = 3; errors bars represent S.E.). L, immunohistochemical images of sections from the postnatal mouse pancreas show that Hes3 immunoreactivity appears postnatally (P8, postnatal day 8, etc.). The main images show Hes3 in white and insets are co-stainings of Hes3 with insulin and glucagon (image widths: 246 μm). M, Hes3 mRNA expression in cDNA from total pancreas from mice of different ages (postnatal days 3 to 21) (data are shown for both isoforms of Hes3: Hes3a and Hes3b).
Hes3 immunolabeling of the adult mouse pancreas showed that Hes3 is expressed in a significant proportion of beta and alpha cells (Fig. 1, J-K). (Insets are images of the cell nuclei stained with DAPI.) Hes3 immunofluorescence appeared postnatally at approximately day 8 and gradually increased until approximately day 20 (Fig. 1L). (Insets show merged channel images.) PCR analysis of total adult mouse pancreas confirmed the expression of both Hes3 isoforms (Hes3a and Hes3b) (Fig. 1M).
Hes3 Is Expressed in Cultured MIN6 Cells under Defined Growth Conditions
To address functional roles of molecular mechanisms in pancreatic islet cells, the mouse insulinoma cell line MIN6 is a common tool that allows the assessment of cell growth and glucose-stimulated insulin secretion (29). MIN6 cells are typically cultured in CC. We also cultured them in DC to assess whether, like with the human cells, these conditions promote nuclear Hes3 expression. In some experiments, following growth in DC, we passaged cells back in serum-containing conditions (“RC”) to assess the reversibility of the cell state.
Cells grew efficiently in all three conditions (Fig. 2A). (Percent of cells that incorporate EdU following an 8-h EdU pulse: CC: 47.7 ± 8.4%; DC: 15.0 ± 2.1%; RC: 49.8 ± 9.7%.) In CC and RC, no cells exhibited nuclear localization of Hes3. In contrast, in DC, ∼15% of cells exhibited strong nuclear Hes3 localization at each point in time (Fig. 2B). In DC, cells also exhibited a higher incidence of Pdx1 expression. Cells in all three conditions exhibited low cell death rates, as assessed by ethidium bromide incorporation. (Percent of cells that incorporate ethidium bromide during a 30-min pulse before cell fixation: CC: 6.2 ± 0.3%; DC: 4.4 ± 0.4%; RC: 5.5 ± 0.2%.) Gene expression differed between CC and DC, and between DC and RC, as revealed by DNA microarray analysis (Fig. 2C). In DC, protein expression of Pdx1 and insulin were higher than in CC or RC (Fig. 2D).
FIGURE 2.

Hes3 is expressed in cultured MIN6 cells under defined conditions. A, cells propagate under all three culture conditions as shown by EdU incorporation (representative immunocytochemistry images shown; insets represent DAPI staining) (image width: 387 μm). B, DC promote nuclear Hes3 localization and increase nuclear Pdx1 localization. Nuclear Hes3 and Pdx1 co-localize. (representative immunocytochemistry images shown) (image widths: 450 μm). C, culture conditions affect gene expression profiles as revealed by Affymetrix DNA microarrays. D, culture conditions affect Pdx1 expression, and insulin content (Western blotting images shown); DC increase Pdx1 expression and insulin content; the effects are reversed by re-plating the cells in RC (GAPDH is used as the housekeeping gene). E, released insulin measurements by ELISA under the three different conditions show that the cells are responsive to glucose challenge in all conditions (n = 3; errors bars represent S.E. R, resting state; S, stimulated state). F, stimulation index measurements (expressed as % of the value in CC) by ELISA under the three different conditions show that the cells are responsive to glucose challenge in all conditions (n = 3; errors bars represent S.E.).
When challenged with a combination of glucose and KCl following a period of glucose starvation (26), cells in all three conditions responded by releasing insulin (Fig. 2, E and F). In DC, the stimulation index was elevated compared with CC and RC.
MIN6 Cells Cultured in DC Respond to Pharmacological Stimulation by Exendin-4
The GLP-1 analog Exendin-4 activates signal transduction pathways that stimulate insulin release (30). Exendin-4 operates via Akt/Irs2 and mammalian target of rapamycin (31–34), signaling components that in NSCs lead to Hes3 induction (10).
Treatment with 0, 20, and 50 nm Exendin-4 for 4 h, 6 h, 12 h, 24 h, or 5 days did not show a measurable effect on Hes3 expression/subcellular localization as assessed by immunocytochemistry (data not shown). However, treatment with 200 nm Exendin-4 over the same time course up to 24 h showed an increase in Hes3 expression (Fig. 3, A–C). In contrast, none of these treatments resulted in nuclear Pdx1 localization (Fig. 3, A and B, and data not shown).
FIGURE 3.

MIN6 cells respond to pharmacological stimulation. A, treatment with Exendin-4 (200 nm) induces the time-dependent expression of Hes3. Some of the Hes3+ cells co-express Pdx1 (image widths: 387 μm). B, details from A show Hes3/Pdx1 co-expression. C, quantification of the effects of Exendin-4 (200 nm in DC) on the percentage of cells with nuclear Hes3 expression, at different time points after treatment (n = 3; errors bars represent S.D.). D, Exendin-4 (200 nm for 5 days in DC) increases the nuclear localization of Pdx1 and Hes3; Pdx1 and Hes3 co-localize (representative immunocytochemistry images are shown) (image widths: 450 μm). E, quantification of the effects of Exendin-4 (200 nm for 5 days in DC) on the percentage of cells with nuclear Pdx1 and nuclear Hes3, compared with controls (n = 3; errors bars represent S.E.). F, Exendin-4 (200 nm for 5 days in CC) increases the nuclear localization of Pdx1 and Hes3; Pdx1 and Hes3 co-localize (representative immunocytochemistry images shown) (image widths: 450 μm). G, quantification of the effects of Exendin-4 (200 nm for 5 days in CC) on the percentage of cells with nuclear Pdx1 and nuclear Hes3, compared with controls (n = 3; errors bars represent S.E.).
Treatment with 200 nm Exendin-4 for 5 days significantly increased nuclear Hes3 localization (Fig. 3, D and E). This treatment, as well as 50 and 100 nm Exendin-4 for 5 days, also significantly increased nuclear Pdx1 expression.
In CC fewer than 3% of the cells exhibit nuclear Hes3 or Pdx1 expression. A 200 nm Exendin-4 treatment for 5 days induced a significant increase in the percentage of cells expressing nuclear Hes3 and nuclear Pdx1; nuclear Hes3 and Pdx1 colocalized in the same cells (Fig. 3, F and G).
Hes3 Regulates MIN6 Cell Number and Gene Expression
Hes3 RNA interference reduced cell number in DC; in contrast, it had no significant effect in CC (Fig. 4, A and B, and data not shown). Hes3 RNA interference in DC induced significant changes in gene expression as revealed by an Affymetrix DNA microarray experiment; Hes3 knockdown in CC had a smaller effect on gene expression (Fig. 4, C and D; Table 1).
FIGURE 4.
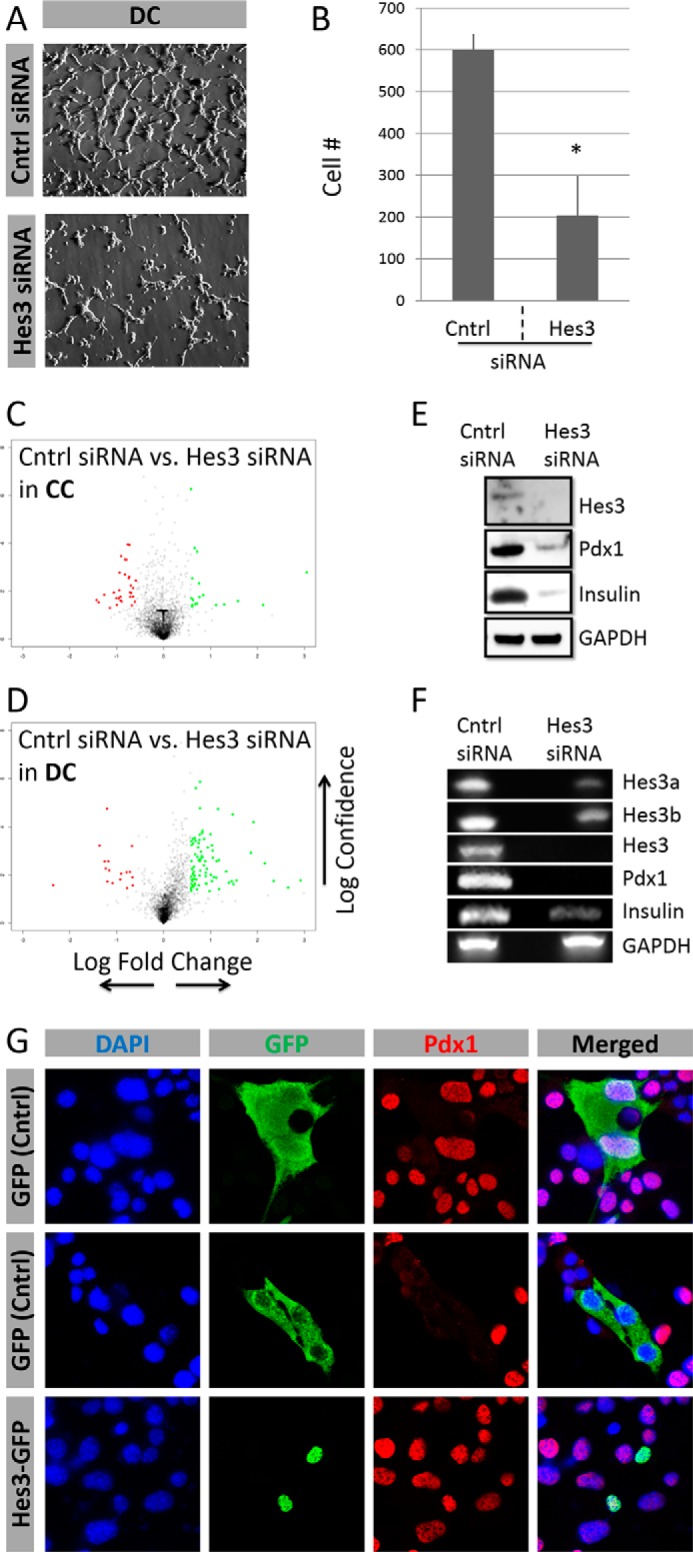
Hes3 regulates cell number and gene expression in MIN6 cell cultures. A, Hes3 interference by siRNA in DC opposes their growth (representative brightfield images shown) (image width: 450 μm). B, quantification of cell number following Hes3 interference in DC. Data are presented as number of cells per field of view (n = 3; errors bars represent S.D.). C and D, Hes3 interference by siRNA in cells cultured in CC and DC induces gene expression profile changes (2 days after interference). Data are presented as a “volcano” plot generated from Affymetrix DNA microarrays. The y axis represents statistical confidence (from quadruplicate samples) and the x axis represents fold-change (logarithmic scale). Red dots represent genes whose expression is reduced by Hes3 siRNA and green dots those whose expression increases. Gray dots represent genes whose expression changes less than 2-fold. E, Hes3 interference opposes Hes3, Pdx1, and insulin protein expression. (Western blotting data are shown; GAPDH was used as the housekeeping gene.) F, Hes3 interference opposes Hes3a Hes3b, total Hes3, Pdx1, and insulin gene transcription (PCR data shown; GAPDH was used as the housekeeping gene). G, Hes3 overexpression induces nuclear Pdx1 localization in DC. In DC, at each time point, ∼15% of cells exhibit nuclear Hes3 localization. Transient overexpression of a control GFP plasmid has no effect on nuclear Pdx1 localization. The top two rows show examples of GFP-labeled cells with nuclear and non-nuclear Pdx1. In contrast, overexpression of a Hes3-GFP fusion plasmid results in 100% of labeled cells exhibiting nuclear Pdx1 (representative immunocytochemistry images shown) (image width: 246 μm).
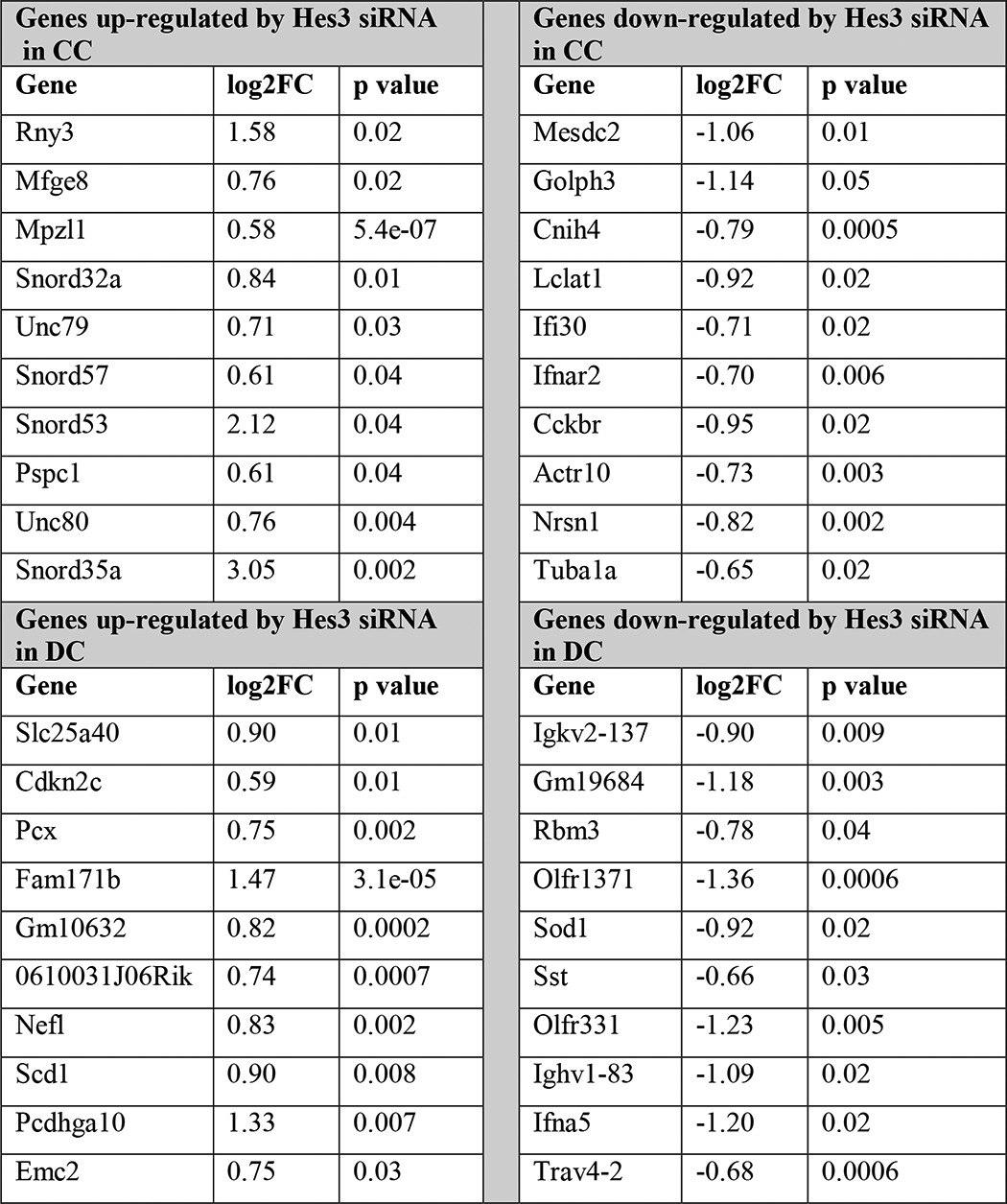
TABLE 1.
Gene expression regulation by Hes3 knockdown in MIN6 cells
MIN6 cells were passaged into separate plates and cultured in CC or DC for 1 day prior to transfection. Cells were transfected with scrambled control siRNA orHes3 siRNA, and total RNA was collected after 4 days in vitro. Gene expression profiles were determined using an Affymetrix DNA microarray (data are from a single experiment with quadruplicate samples; top 10 log2FC values are presented for each category).
Hes3 RNA interference reduced the expression of Pdx1 and insulin in DC as assessed by Western blotting and PCR approaches (Fig. 4, E and F). Transient overexpression with a vector with the GFP gene fused to Hes3 revealed that all (100%) of the transfected cells exhibited Pdx1 expression. In contrast, cells transfected with the control GFP vector did not have a greater incidence of Pdx1 expression than non-transfected controls (∼15%) (Fig. 4G).
Hes3 Regulates Evoked Insulin Release in MIN6 Cells
Hes3 RNA interference reduced insulin content, the amount of released insulin, and stimulation index as assessed by ELISA measurements of insulin (Fig. 5, A–C), using a stimulation paradigm that includes both glucose and KCl. Using a different stimulation paradigm that utilizes the same concentration of low KCl and different glucose concentrations in DC, in both the resting and stimulated states, we did not observe any significant stimulated insulin release (insulin content: scrambled control siRNA/resting, 28.684 ng ± 1.553; control siRNA/stimulated: 25.974 ng ± 1.805; Hes3 siRNA/resting, 22.541 ng ± 2.059; Hes3 siRNA/stimulated, 18.193 ng ± 1.893; stimulation index: control siRNA: 0.824 ± 0.24; Hes3 siRNA: 1.082 ± 0.62).
FIGURE 5.
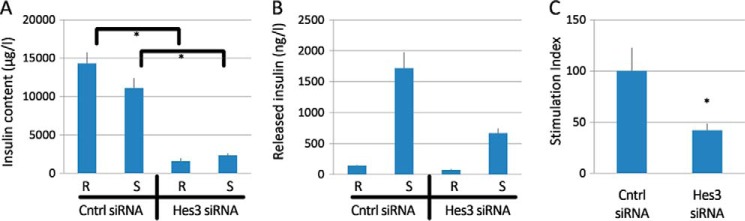
Hes3 regulates responses to challenge by glucose/KCl in MIN6 cell cultures. A–C, insulin content, released insulin, and stimulation index measurements by ELISA (stimulation index is expressed as % of control siRNA) following transfection with either a control siRNA or Hes3 siRNA show that opposing Hes3 expression reduces the responsiveness to stimulation in MIN6 cells cultured in DC (n = 3; errors bars represent S.E. R, resting state; S, stimulated state).
Hes3 Null Mice Exhibit Grossly Normal Pancreatic Islet Marker Expression and Blood Glucose Values
Hes3 null mice have no obvious phenotype under normal conditions (25). Indeed, islet morphology and biomarker expression did not reveal any obvious differences from wild-type mice (Fig. 6A). PCR analysis for Pdx1 and insulin also revealed levels similar to wild-type controls (Fig. 6B). Another member of the Hes/Hey gene family, Hes1, exhibited a marked increase in RNA levels in islets from the Hes3 null mice.
FIGURE 6.
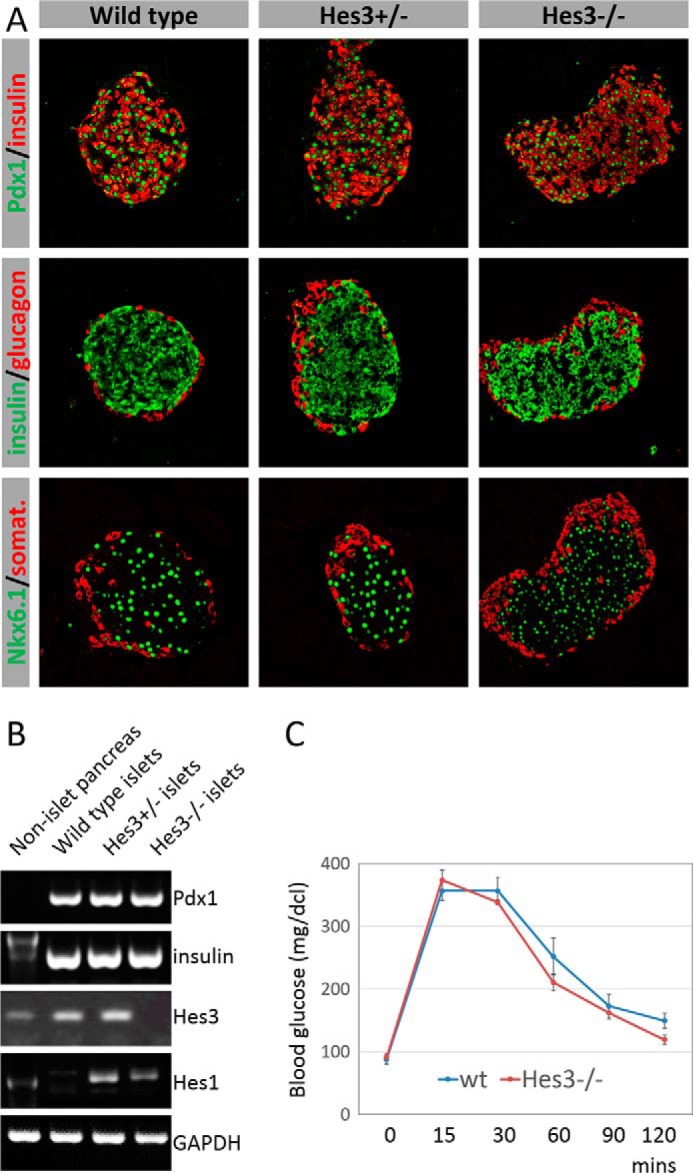
Hes3 null mice exhibit grossly normal pancreatic islet marker expression and blood glucose values. A, immunohistochemical detection of pancreatic islet markers in sections from whole pancreas (image width: 387 μm). B, PCR analysis of Pdx1, insulin, Hes3, and Hes1 expression in isolated islets from wild-type, Hes3 heterozygous, and Hes3 null; pancreatic tissues after islet removal was also used. C, glucose tolerance test scores are shown for wild-type and Hes3 null mice (blood glucose was measured at different time points within a 120-min period. Data represent mean ± S.E. from 7 wild-type and 5 Hes3 null mice).
Fasting blood glucose levels and glucose tolerance test scores were also normal (Fig. 6C). Likewise, total body weight was also similar between wild-type and Hes3 null mice (wild-type, 22.6 g ± 0.3; Hes3−/−, 20.8 ± 0.3).
Hes3 Null Mice Exhibit Increased STZ Sensitivity
Following STZ damage (using either high or low dose STZ protocols), activation of the Hes3a promoter was significantly enhanced, as assessed by X-Gal staining (Fig. 7, A and B). When wild-type mice are subjected to the low STZ damage protocol, blood glucose levels increase ∼4–5 days after the last STZ injection, relative to vehicle-treated animals (Fig. 7, C and D). In contrast, blood glucose levels in Hes3 null mice increased within 2 days from the last injection. Hes3 null mice reached diabetic blood glucose levels (>200 mg/dcl) earlier than wild-type controls (Fig. 7E). Immunohistochemical analysis of the pancreata of wild-type and Hes3 null mice 5 days after the last STZ injection revealed a decrease in the percentage of insulin+ and Nkx6.1+ cells and an increase in the relative number of glucagon+ cells in the pancreatic islets (Fig. 7, F–H).
FIGURE 7.

Hes3 null mice exhibit a more pronounced effect following STZ-induced damage. A, X-Gal staining of pancreatic islets from Hes3 null mice following damage by the high STZ protocol at different time points after STZ injection (image width: 223 μm). B, X-Gal staining of pancreatic islets from Hes3 null mice following damage by the low STZ protocol. Images were taken 10 days after vehicle or STZ injection regimen completion (image width: 446 μm). C, blood glucose levels in wild-type and Hes3 null mice after control (vehicle) injections. Injections were given on five consecutive days starting at day 0. D, blood glucose levels in wild-type and Hes3 null mice after low dose STZ injections. Injections were given on five consecutive days starting at day 0 (data are mean ± S.E. from 8 to 11 mice per group). E, quantification of the percentage of mice that have blood glucose levels over 200 mg/dl (i.e. diabetic mice) over the course of 10 days (data represent the experiment described in D). F, immunohistochemical detection of insulin and glucagon in wild-type and Hes3 null mice treated with low STZ (images were taken at day 10). Insets show merged channels together with DAPI staining (image width: 267 μm). G, immunohistochemical detection of Nkx6.1 in wild-type and Hes3 null mice treated with low STZ (images were taken at day 10). Insets show merged channels together with DAPI staining (image width: 267 μm). H, quantification of the percentage of insulin+, glucagon+, and Nkx6.1+ cells per islet as shown in F and G. Data are mean ± S.D. from 30 islets per group.
DISCUSSION
Pancreatic islets demonstrate remarkable plasticity, being able to alter mass depending on systemic insulin demand (35). Understanding this mechanism will help us design new therapeutic strategies for diabetes. We previously elucidated a signal transduction pathway that plays important roles regulating the plasticity of a different cell type, the NSC (9). A key component is the transcription factor Hes3 (10). Several inputs into Hes3 increase its expression and nuclear localization, and promote the growth of NSCs in vitro and in vivo (36, 37). In vivo, they increase the number of Hes3+ cells and confer powerful neuroprotection in animal models of ischemic stroke and Parkinson disease, likely, in part through the release of trophic factors from Hes3+ cells (10, 11, 15, 17, 38).
The choice of culture conditions is key to the appropriate modeling of a signaling pathway in vitro. Defined culture conditions that omit unknown factors improve reproducibility and are demonstrably able to model aspects of the biology of NSCs in vivo (39). Although cells in tissues are exposed to several serum components within their microenvironment, these precise conditions that include active cytokines, membrane-bound receptors, and extracellular matrix components are very difficult to model in vitro by simply including serum. Regardless of the culture method, conditions in vitro are non-physiological. Here, conditions were chosen based on our understanding of how to maintain the operation of the Hes3 signaling pathway, allowing us to model this aspect of pancreatic islet cells. Key features of these conditions are the absence of serum, the pre-coating of culture plates with polyornithine and fibronectin, and the use of low oxygen (5%) incubators (40). The ability of MIN6 cells as well as human primary dissociated islet cells to efficiently grow in serum-free conditions may also be of interest to drug discovery efforts where the inclusion of serum together with its many unknown components may render data interpretation difficult. By applying our understanding of NSC signal transduction to pancreatic islet cell systems, we demonstrate that Hes3 is expressed in the mouse and human pancreatic islet, and that it regulates several functions in vitro, including cell growth and evoked insulin release (Fig. 8A).
FIGURE 8.
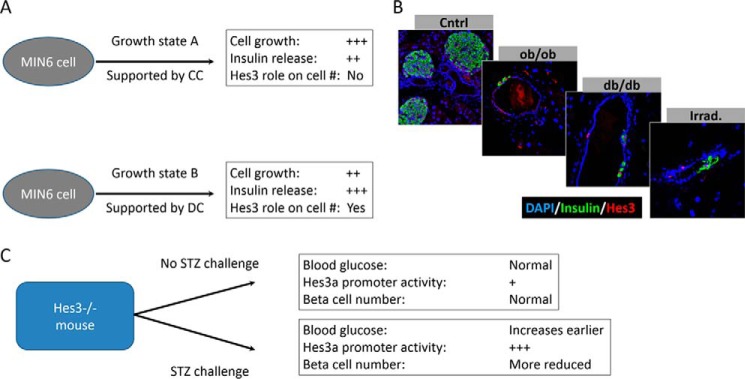
Roles of Hes3 in pancreatic islet function. A, MIN6 cells can be cultured under different culture conditions, in which the role of Hes3 varies. B, potentially relevant signaling pathways that may intercept with Hes3 include the leptin signaling system and irradiation-induced stresses. In genetic models of leptin signaling disruption (ob/ob and db/db mouse strains), Hes3 induction in the pancreatic duct is observed. Likewise in wild-type mice subjected to total body irradiation. C, Hes3 null mice appear normal with no obvious phenotype. Upon challenge with STZ, however, phenotypes are revealed, including earlier increases in blood glucose levels and a decreased number of beta cells in the islet, relative to wild-type controls.
There may be a large number of genes downstream of Hes3 that affect pancreatic islet function. A DNA microarray approach revealed that Hes3 in CC positively regulates Cckbr (cholecystokinin B receptor), a gene involved in pancreatic islet neogenesis (41–44) (Table 1). It may be of interest to address a potential role of Hes3 in neogenesis, possibly intercepting with gastrin signaling through its receptor (Cckbr), in future studies.
The DNA microarray data also show that, among the top genes regulated by Hes3 knockdown, small nucleolar RNAs appear preferentially and negatively regulated by Hes3 in CC. It is difficult to make absolute statements on the subcellular localization of Hes3 using a polyclonal antibody; however, it is possible that because in the two culture conditions Hes3 localization appears very different (CC, cytoplasmic; DC, nuclear), data from these two culture conditions may preferentially represent passive repressor versus transcriptional functions of Hes3. It is also possible that the apparent subcellular localization differences in Hes3 represent different forms of the gene (Hes3a versus Hes3b) and future studies may address the distinct functions of these two forms. This could be especially important because, as we show, both forms are present in total pancreas tissue and the levels of the transcripts appear to be differentially regulated during postnatal development. In light of recent work showing that oscillatory versus sustained expression of Hes/Hey and other related genes contributes to cell fate specification (7), it will be useful to address, in future studies, the regulation of the expression of each Hes3 form.
The repressor activity of Hes3 renders it a difficult gene to study, being a candidate for transcriptional squelching. Indeed, stable transfection of MIN6 cells with full-length Hes3b-GFP results in GFP+ cells that appear healthy for weeks but form colonies of only 1, 2, or 3 cells (data not shown). In this paper, we rely mostly on Hes3 RNA interference, and inferences from Hes3 overexpression (positive regulation of Pdx1) are corroborated by Hes3 siRNA experiments.
To identify additional putative mediators of Hes3 manipulation, a more focused approach based on work performed in other cell types, including NSCs, was also applied. The Hes/Hey gene family comprises members with widely different functions in NSCs. For example, whereas Hes3 expression is consistent with self-renewal, Hes1, through cytoplasmic interactions with JAK and STAT3, promotes STAT3-tyrosine phosphorylation and subsequent differentiation to the glial fate (45). Hes1 and Hes3 also represent two distinct branches of the Notch signaling pathway (Hes1, canonical; Hes3, STAT3-Ser/Hes3 signaling axis). As passive repressors that can interact with other gene family members, it is possible that these genes mutually oppose each other's expression, and this may contribute to cell state and fate choices. Supporting such a possibility, we show that in isolated primary pancreatic islets from Hes3 null mice, the expression of Hes1, a gene with roles in pancreatic development (1, 46), is markedly increased. It is possible that other Hes/Hey genes and other gene families may also be regulated, and this will be important to address in subsequent studies to identify genes that may compensate for the absence of Hes3 in the context of development and/or the response to damage. This may explain our finding that in the Hes3 null-isolated islets, Pdx1 expression appears normal. In contrast, in MIN6 cells subjected to Hes3 interference, an acute manipulation that may not allow for compensatory mechanisms to operate, Pdx1 expression is markedly reduced.
The involvement of JAK in Hes3 regulation deserves additional study. In NSCs, JAK inhibition promotes Hes3 expression, especially when combined with cytokine treatments that activate both the Akt pathway (which leads to STAT3-serine phosphorylation and Hes3 transcription) and the JAK-STAT pathway (which suppresses Hes3 transcription), likely because it promotes the pro-Hes3 function, whereas inhibiting the anti-Hes3 function (10). In fact, JAK inhibition can convert a pro-differentiation cytokine (ciliary neurotrophic factor) into a mitogen that maintains the self-renewal state (10, 12). In MIN6 cells (in CC), leptin induces the activation of MAPK and JAK and promotes cell proliferation; whereas MAPK inhibition experiments show that this kinase family mediates proliferation, it is not clear if JAK also contributes (47). Our observations using MIN6 cells show no effect of JAK inhibition of the subcellular localization of Hes3 or Pdx1 (control treatment: 19.8 ± 1.3% of cells with nuclear Hes3, 94.1 ± 1.5% of cells with nuclear Pdx1; 200 nm JAK inhibitor for 5 days: 22.6 ± 1.9% of cells with nuclear Hes3, 96.4 ± 1.6% of cells with nuclear Pdx1). However, in primary cultured fetal mouse pancreatic progenitors, JAK inhibition increases cell yield (10). This could be due to the fact that, as a transformed cell line, MIN6 cells exhibit skewed JAK or downstream signaling (48).
A potential clue pointing toward a role of other signaling pathways, including the JAK pathway in vivo comes from our observations that in mice with disrupted leptin signaling (ob/ob and db/db mouse strains), we observe Hes3 expression outside the pancreatic islet, and specifically in the ductal epithelium (Fig. 8B). In contrast, we have never observed such immunolabeling in wild-type mice. Another indication comes from mice subjected to total body irradiation, which also exhibit Hes3 expression in the duct. These mouse models may serve to identify additional upstream and downstream regulators and mediators of Hes3. These observations raise an intriguing question: is the function of Hes3 to stimulate fast proliferation or to induce cells to partake in the regeneration process by enhanced survival and cell fate choices? The latter possibility would not necessarily require a fast proliferation mechanism. The fact that in DC, MIN6 cells incorporate less EdU than in CC may support a role in survival over proliferation, although many other differences in these culture conditions may account for the EdU incorporation differences. Our results show that under certain growth conditions that support Hes3 expression, it plays important roles in regulating their cell number. It will be of importance to address whether the subcellular localization or another aspect of Hes3 signaling such as specific interactions with other proteins or DNA sequences drive this process. Of course, other differences between the culture conditions may be responsible for this. Hes3 knockdown experiments support a role of Hes3 in cell number regulation, but this may be due to effects on survival and not the regulation of cell number. Additional experiments with, preferably, primary cell systems that allow the measurement of proliferative activity and cell cycle duration, will be required to address this point. In the meantime, our previous work with NSCs showed that treatment with soluble Notch receptor ligands that induce Hes3 expression and increase cell number do so not by changing cell cycle duration but by greatly reducing the probability of cell death (10).
Understanding the signal transduction pathways upstream and downstream of Hes3 may help identify new treatments that affect pancreatic function. GLP-1 analogs activate the Akt pathway which, in NSCs, leads to phosphorylation of STAT3-serine, Hes3 transcription, and improved cell survival (10). In MIN6 cells, where Exendin-4 protects against pro-apoptotic insults (49), high concentrations of Exendin-4 significantly increased nuclear Hes3 expression in CC and DC. Although in this context it is not clear if changes in Hes3 expression/localization are responsible for the protective effects, it will be of interest to address this issue, especially with primary cell systems. In CC, we observe a very low incidence of nuclear Hes3 localization. Exendin-4, however, induces nuclear Hes3 expression, suggesting that it activates a signaling pathway that overrides nuclear exclusion of Hes3, worthy of additional research. In both CC and DC we observed more pronounced effects after sustained exposure to relatively high Exendin-4 concentrations. It is conceivable that a negative feedback mechanism, downstream of Exendin-4, may limit the extent of the action of Exendin-4 and that repeat cycles of activation may be necessary for effects to become obvious. For example, in NSCs, Notch ligand activation induces the phosphorylation of STAT3-serine within ∼20 min. However, the phosphorylation abruptly decreases within another 20 min, returning to baseline, also suggesting a negative feedback system. The reduction is observed despite the continued presence of ligand, suggesting a refractory period following receptor activation. Such a system could conceivably involve kinases such as LKB1, which is phosphorylated at approximately the time that STAT3-serine phosphorylation is down-regulated (10). However, the functional consequence of this modification on LKB1 (or potentially others) in NSCs has not been determined. Regardless, it will be valuable to determine whether Hes3 is a functional mediator of GLP-1 signaling and to identify negative feedback mechanisms whose inhibition may provide the basis for new therapeutics that will enhance the efficacy of GLP-1 analogs. Clues to such mechanisms may be provided by our observations of extra-islet Hes3 expression in mice that are subjected to irradiation or that have disrupted leptin signaling.
It will be valuable to investigate further the relationship between Hes3 and Pdx1. In cells lacking Pdx1, Exendin-4 fails to promote cell proliferation (50) and future studies may address if Hes3 is involved in this effect. In this work, we demonstrate that in MIN6 cells, Hes3 regulates Pdx1 expression. Previous work employing chromatin immunoprecipitation approaches also on MIN6 cells demonstrated that Pdx1 binds to the Hes3 promoter (51). Our observations may be uncovering aspects of this reciprocity. In the acute treatment, Hes3 translocation is a more sensitive (and earlier) response than Pdx1 expression or translocation. In contrast, in the sustained Exendin-4 experiments, the tables are reversed. Here, nuclear Pdx1 changes appear at lower concentrations than the ones needed to observe Hes3 changes. These observations may serve as assays to investigate the relationship between Hes3 and Pdx1 further.
The Hes3 null mouse line is a remarkable one as it has no evident phenotypes, but the biology behind Hes3 suggests that its absence should impact the health of the animal (11). It may be that the answer to this riddle is that Hes3 is not essential under normal conditions but highly convenient to have in instances of damage and stress, either because Hes3 absence can be compensated for under normal conditions by other genes, or because Hes3 function is primarily allocated to damage situations (Fig. 8C). Here, we employed STZ damage models to this mouse strain, revealing new phenotypes in line with this hypothesis. Whereas in normal conditions, glucose tolerance test results are indistinguishable between Hes3 null mice and wild-type controls, following STZ damage we observe an induction of Hes3a promoter activation (the mouse strain only allows the assessment of Hes3a promoter activity), a greater deficit in beta cells, and an earlier diabetic phenotype. Following low STZ damage, the percentage of alpha cells in the Hes3 null mice was greater than in the wild-type controls; it will be of great interest to address whether this reflects the fact that alpha cells are not affected by the absence of Hes3, despite the fact that a proportion of them express this transcription factor, or whether alpha to beta cell conversion may be somehow impacted by the absence of Hes3. These initial results suggest a role of Hes3 in the sensitivity of pancreatic beta cells to STZ and challenge future experiments designed to assess the role of Hes3 in different paradigms of damage and regeneration.
Acknowledgment
We thank Dr. Jun-ichi Miyazaki for permission to use MIN6 cells in this work and Dr. Ryoichiro Kageyama for providing the Hes3 null mouse strain.
This work was supported by Boehringer Ingelheim and in part by the Helmholtz Alliance ICEMED-Imaging and Curing Environmental Metabolic Diseases, through the Initiative and Network Fund of Helmholtz Association Grant 051_40001 and Deutsche Forschungsgemeinschaft Grant SFB 655, “Cells into tissues,” Project A24. Ingo Uphues and Michael Mark are employees of Boehringer Ingelheim and thus contributed to study design, data analysis, and preparation of the manuscript.
- NSC
- neural stem cell
- STZ
- streptozotocin
- PFA
- paraformaldehyde
- CC
- common condition
- DC
- defined condition
- RC
- return condition
- EdU
- 5-ethynyl-2′-deoxyuridine.
REFERENCES
- 1. Kageyama R., Ohtsuka T., Kobayashi T. (2007) The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development 134, 1243–1251 [DOI] [PubMed] [Google Scholar]
- 2. Imayoshi I., Isomura A., Harima Y., Kawaguchi K., Kori H., Miyachi H., Fujiwara T., Ishidate F., Kageyama R. (2013) Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science 342, 1203–1208 [DOI] [PubMed] [Google Scholar]
- 3. Artavanis-Tsakonas S., Rand M. D., Lake R. J. (1999) Notch signaling: cell fate control and signal integration in development. Science 284, 770–776 [DOI] [PubMed] [Google Scholar]
- 4. Imayoshi I., Kageyama R. (2011) The role of Notch signaling in adult neurogenesis. Mol. Neurobiol. 44, 7–12 [DOI] [PubMed] [Google Scholar]
- 5. Ueo T., Imayoshi I., Kobayashi T., Ohtsuka T., Seno H., Nakase H., Chiba T., Kageyama R. (2012) The role of Hes genes in intestinal development, homeostasis and tumor formation. Development 139, 1071–1082 [DOI] [PubMed] [Google Scholar]
- 6. Hirata H., Yoshiura S., Ohtsuka T., Bessho Y., Harada T., Yoshikawa K., Kageyama R. (2002) Oscillatory expression of the bHLH factor Hes1 regulated by a negative feedback loop. Science 298, 840–843 [DOI] [PubMed] [Google Scholar]
- 7. Kageyama R., Niwa Y., Shimojo H., Kobayashi T., Ohtsuka T. (2010) Ultradian oscillations in Notch signaling regulate dynamic biological events. Curr. Top. Dev. Biol. 92, 311–331 [DOI] [PubMed] [Google Scholar]
- 8. Guruharsha K. G., Kankel M. W., Artavanis-Tsakonas S. (2012) The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 13, 654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Poser S. W., Park D. M., Androutsellis-Theotokis A. (2013) The STAT3-Ser/Hes3 signaling axis: an emerging regulator of endogenous regeneration and cancer growth. Front Physiol. 4, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Androutsellis-Theotokis A., Leker R. R., Soldner F., Hoeppner D. J., Ravin R., Poser S. W., Rueger M. A., Bae S. K., Kittappa R., McKay R. D. (2006) Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 442, 823–826 [DOI] [PubMed] [Google Scholar]
- 11. Androutsellis-Theotokis A., Rueger M. A., Mkhikian H., Korb E., McKay R. D. (2008) Signaling pathways controlling neural stem cells slow progressive brain disease. Cold Spring Harb. Symp. Quant. Biol. 73, 403–410 [DOI] [PubMed] [Google Scholar]
- 12. Park D. M., Jung J., Masjkur J., Makrogkikas S., Ebermann D., Saha S., Rogliano R., Paolillo N., Pacioni S., McKay R. D., Poser S., Androutsellis-Theotokis A. (2013) Hes3 regulates cell number in cultures from glioblastoma multiforme with stem cell characteristics. Sci. Rep. 3, 1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hirata H., Ohtsuka T., Bessho Y., Kageyama R. (2000) Generation of structurally and functionally distinct factors from the basic helix-loop-helix gene Hes3 by alternative first exons. J. Biol. Chem. 275, 19083–19089 [DOI] [PubMed] [Google Scholar]
- 14. Ohta S., Misawa A., Fukaya R., Inoue S., Kanemura Y., Okano H., Kawakami Y., Toda M. (2012) Macrophage migration inhibitory factor (MIF) promotes cell survival and proliferation of neural stem/progenitor cells. J. Cell Sci. 125, 3210–3220 [DOI] [PubMed] [Google Scholar]
- 15. Androutsellis-Theotokis A., Rueger M. A., Park D. M., Mkhikian H., Korb E., Poser S. W., Walbridge S., Munasinghe J., Koretsky A. P., Lonser R. R., McKay R. D. (2009) Targeting neural precursors in the adult brain rescues injured dopamine neurons. Proc. Natl. Acad. Sci. U.S.A. 106, 13570–13575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oya S., Yoshikawa G., Takai K., Tanaka J., Higashiyama S., Saito N., Kirino T., Kawahara N. (2008) Region-specific proliferative response of neural progenitors to exogenous stimulation by growth factors following ischemia. Neuroreport. 19, 805–809 [DOI] [PubMed] [Google Scholar]
- 17. Pacioni S., Rueger M. A., Nisticò G., Bornstein S. R., Park D. M., McKay R. D., Androutsellis-Theotokis A. (2012) Fast, potent pharmacological expansion of endogenous hes3+/sox2+ cells in the adult mouse and rat hippocampus. PLoS One 7, e51630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Salewski R. P., Buttigieg J., Mitchell R. A., van der Kooy D., Nagy A., Fehlings M. G. (2013) The generation of definitive neural stem cells from PiggyBac transposon-induced pluripotent stem cells can be enhanced by induction of the NOTCH signaling pathway. Stem Cells Dev. 22, 383–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hatakeyama J., Bessho Y., Katoh K., Ookawara S., Fujioka M., Guillemot F., Kageyama R. (2004) Hes genes regulate size, shape and histogenesis of the nervous system by control of the timing of neural stem cell differentiation. Development 131, 5539–5550 [DOI] [PubMed] [Google Scholar]
- 20. Qin H. R., Kim H. J., Kim J. Y., Hurt E. M., Klarmann G. J., Kawasaki B. T., Duhagon Serrat M. A., Farrar W. L. (2008) Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 68, 7736–7741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Levy D. E., Darnell J. E., Jr. (2002) Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3, 651–662 [DOI] [PubMed] [Google Scholar]
- 22. Gouti M., Gavalas A. (2008) Hoxb1 controls cell fate specification and proliferative capacity of neural stem and progenitor cells. Stem Cells 26, 1985–1997 [DOI] [PubMed] [Google Scholar]
- 23. Zhang C. C., Pavlicek A., Zhang Q., Lira M. E., Painter C. L., Yan Z., Zheng X., Lee N. V., Ozeck M., Qiu M., Zong Q., Lappin P. B., Wong A., Rejto P. A., Smeal T., Christensen J. G. (2012) Biomarker and pharmacologic evaluation of the gamma-secretase inhibitor PF-03084014 in breast cancer models. Clin. Cancer Res. 18, 5008–5019 [DOI] [PubMed] [Google Scholar]
- 24. Fujimoto K., Polonsky K. S. (2009) Pdx1 and other factors that regulate pancreatic beta-cell survival. Diabetes Obes. Metab. 11, 30–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hirata H., Tomita K., Bessho Y., Kageyama R. (2001) Hes1 and Hes3 regulate maintenance of the isthmic organizer and development of the mid/hindbrain. EMBO J. 20, 4454–4466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ort T., Voronov S., Guo J., Zawalich K., Froehner S. C., Zawalich W., Solimena M. (2001) Dephosphorylation of β2-syntrophin and Ca2+/μ-calpain-mediated cleavage of ICA512 upon stimulation of insulin secretion. EMBO J. 20, 4013–4023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Phieler J., Chung K. J., Chatzigeorgiou A., Klotzsche-von Ameln A., Garcia-Martin R., Sprott D., Moisidou M., Tzanavari T., Ludwig B., Baraban E., Ehrhart-Bornstein M., Bornstein S. R., Mziaut H., Solimena M., Karalis K. P., Economopoulou M., Lambris J. D., Chavakis T. (2013) The complement anaphylatoxin C5a receptor contributes to obese adipose tissue inflammation and insulin resistance. J. Immunol. 191, 4367–4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ludwig B., Reichel A., Steffen A., Zimerman B., Schally A. V., Block N. L., Colton C. K., Ludwig S., Kersting S., Bonifacio E., Solimena M., Gendler Z., Rotem A., Barkai U., Bornstein S. R. (2013) Transplantation of human islets without immunosuppression. Proc. Natl. Acad. Sci. U.S.A. 110, 19054–19058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miyazaki J., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., Yamamura K. (1990) Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132 [DOI] [PubMed] [Google Scholar]
- 30. Göke R., Fehmann H. C., Linn T., Schmidt H., Krause M., Eng J., Göke B. (1993) Exendin-4 is a high potency agonist and truncated exendin-(9–39)-amide an antagonist at the glucagon-like peptide 1-(7–36)-amide receptor of insulin-secreting beta-cells. J. Biol. Chem. 268, 19650–19655 [PubMed] [Google Scholar]
- 31. Park S., Dong X., Fisher T. L., Dunn S., Omer A. K., Weir G., White M. F. (2006) Exendin-4 uses Irs2 signaling to mediate pancreatic beta cell growth and function. J. Biol. Chem. 281, 1159–1168 [DOI] [PubMed] [Google Scholar]
- 32. Norquay L. D., D'Aquino K. E., Opare-Addo L. M., Kuznetsova A., Haas M., Bluestone J. A., White M. F. (2009) Insulin receptor substrate-2 in beta-cells decreases diabetes in nonobese diabetic mice. Endocrinology 150, 4531–4540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van de Velde S., Hogan M. F., Montminy M. (2011) mTOR links incretin signaling to HIF induction in pancreatic beta cells. Proc. Natl. Acad. Sci. U.S.A. 108, 16876–16882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kwon G., Marshall C. A., Pappan K. L., Remedi M. S., McDaniel M. L. (2004) Signaling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes 53, S225–S232 [DOI] [PubMed] [Google Scholar]
- 35. Heit J. J., Karnik S. K., Kim S. K. (2006) Intrinsic regulators of pancreatic beta-cell proliferation. Annu. Rev. Cell Dev. Biol. 22, 311–338 [DOI] [PubMed] [Google Scholar]
- 36. Androutsellis-Theotokis A., Rueger M. A., Park D. M., Boyd J. D., Padmanabhan R., Campanati L., Stewart C. V., LeFranc Y., Plenz D., Walbridge S., Lonser R. R., McKay R. D. (2010) Angiogenic factors stimulate growth of adult neural stem cells. PLoS One 5, e9414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Androutsellis-Theotokis A., Walbridge S., Park D. M., Lonser R. R., McKay R. D. (2010) Cholera toxin regulates a signaling pathway critical for the expansion of neural stem cell cultures from the fetal and adult rodent brains. PLoS One 5, e10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Masjkur J., Rueger M. A., Bornstein S. R., McKay R., Androutsellis-Theotokis A. (2012) Neurovascular signals suggest a propagation mechanism for endogenous stem cell activation along blood vessels. CNS Neurol. Disord. Drug Targets 11, 805–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johe K. K., Hazel T. G., Muller T., Dugich-Djordjevic M. M., McKay R. D. (1996) Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev. 10, 3129–3140 [DOI] [PubMed] [Google Scholar]
- 40. Poser S. W., Androutsellis-Theotokis A. (2013) Growing neural stem cells from conventional and nonconventional regions of the adult rodent brain. J. Vis. Exp. 81, e50880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang T. C., Bonner-Weir S., Oates P. S., Chulak M., Simon B., Merlino G. T., Schmidt E. V., Brand S. J. (1993) Pancreatic gastrin stimulates islet differentiation of transforming growth factor α-induced ductular precursor cells. J. Clin. Invest. 92, 1349–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rooman I., Lardon J., Bouwens L. (2002) Gastrin stimulates beta-cell neogenesis and increases islet mass from transdifferentiated but not from normal exocrine pancreas tissue. Diabetes 51, 686–690 [DOI] [PubMed] [Google Scholar]
- 43. Suarez-Pinzon W. L., Lakey J. R., Brand S. J., Rabinovitch A. (2005) Combination therapy with epidermal growth factor and gastrin induces neogenesis of human islet β-cells from pancreatic duct cells and an increase in functional β-cell mass. J. Clin. Endocrinol. Metab. 90, 3401–3409 [DOI] [PubMed] [Google Scholar]
- 44. Gigoux V., Clerc P., Sanchez D., Coll M. G., Corominola H., Leung-Theung-Long S., Pénicaud L., Gomis R., Seva C., Fourmy D., Dufresne M. (2008) Reg genes are CCK2 receptor targets in ElasCCK2 mice pancreas. Regul. Pept. 146, 88–98 [DOI] [PubMed] [Google Scholar]
- 45. Kamakura S., Oishi K., Yoshimatsu T., Nakafuku M., Masuyama N., Gotoh Y. (2004) Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat. Cell Biol. 6, 547–554 [DOI] [PubMed] [Google Scholar]
- 46. Kopinke D., Brailsford M., Shea J. E., Leavitt R., Scaife C. L., Murtaugh L. C. (2011) Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development 138, 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tanabe K., Okuya S., Tanizawa Y., Matsutani A., Oka Y. (1997) Leptin induces proliferation of pancreatic beta cell line MIN6 through activation of mitogen-activated protein kinase. Biochem. Biophys. Res. Commun. 241, 765–768 [DOI] [PubMed] [Google Scholar]
- 48. Frank D. A. (2007) STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 251, 199–210 [DOI] [PubMed] [Google Scholar]
- 49. D'Amico E., Hui H., Khoury N., Di Mario U., Perfetti R. (2005) Pancreatic beta-cells expressing GLP-1 are resistant to the toxic effects of immunosuppressive drugs. J. Mol. Endocrinol. 34, 377–390 [DOI] [PubMed] [Google Scholar]
- 50. Li Y., Cao X., Li L. X., Brubaker P. L., Edlund H., Drucker D. J. (2005) Beta-cell Pdx1 expression is essential for the glucoregulatory, proliferative, and cytoprotective actions of glucagon-like peptide-1. Diabetes 54, 482–491 [DOI] [PubMed] [Google Scholar]
- 51. Sachdeva M. M., Claiborn K. C., Khoo C., Yang J., Groff D. N., Mirmira R. G., Stoffers D. A. (2009) Pdx1 (MODY4) regulates pancreatic beta cell susceptibility to ER stress. Proc. Natl. Acad. Sci. U.S.A. 106, 19090–19095 [DOI] [PMC free article] [PubMed] [Google Scholar]