

Background: PTF1a is an essential transcription factor for pancreas development and function. Mechanisms regulating PTF1a degradation are unknown.
Results: TRIP12 interacts with PTF1a. Its E3 ubiquitin ligase activity decreases protein stability of PTF1a.
Conclusion: PTF1a is a new target of TRIP12. TRIP12 promotes proteasomal degradation of PTF1a and regulates PTF1a activities.
Significance: TRIP12/PTF1a interaction could contribute to the regulation of pancreatic acinar cell homeostasis.
Keywords: Basic Helix-Loop-Helix Transcription Factor (bHLH), E3 Ubiquitin Ligase, Pancreas, Pancreatic Cancer, Proteasome, Protein Degradation, Protein-Protein Interaction, Transcription Factor, Ubiquitylation (Ubiquitination), Yeast Two-Hybrid
Abstract
Pancreas transcription factor 1a (PTF1a) plays a crucial role in the early development of the pancreas and in the maintenance of the acinar cell phenotype. Several transcriptional mechanisms regulating expression of PTF1a have been identified. However, regulation of PTF1a protein stability and degradation is still unexplored. Here, we report that inhibition of proteasome leads to elevated levels of PTF1a and to the existence of polyubiquitinated forms of PTF1a. We used the Sos recruitment system, an alternative two-hybrid system method to detect protein-protein interactions in the cytoplasm and to map the interactome of PTF1a. We identified TRIP12 (thyroid hormone receptor-interacting protein 12), an E3 ubiquitin-protein ligase as a new partner of PTF1a. We confirmed PTF1a/TRIP12 interaction in acinar cell lines and in co-transfected HEK-293T cells. The protein stability of PTF1a is significantly increased upon decreased expression of TRIP12. It is reduced upon overexpression of TRIP12 but not a catalytically inactive TRIP12-C1959A mutant. We identified a region of TRIP12 required for interaction and identified lysine 312 of PTF1a as essential for proteasomal degradation. We also demonstrate that TRIP12 down-regulates PTF1a transcriptional and antiproliferative activities. Our data suggest that an increase in TRIP12 expression can play a part in PTF1a down-regulation and indicate that PTF1a/TRIP12 functional interaction may regulate pancreatic epithelial cell homeostasis.
Introduction
Ubiquitination is recognized to be a fundamental mechanism regulating degradation of most intracellular proteins. The process of ubiquitination involves the sequential transfer of activated ubiquitin between an ubiquitin-activating enzyme (E1), an ubiquitin-conjugating enzyme (E2), and an ubiquitin-protein ligase (E3) (1). E3s are of particular importance because they confer substrate specificity to the system by interacting directly with substrate proteins and thereby directing the transfer of ubiquitin. E3s modify protein substrates by either monoubiquitination or sequential attachment of ubiquitin molecules to form polyubiquitin chains. Generally, polyubiquitinated proteins are rapidly degraded by the 26 S proteasome. Two types of E3s are commonly found: the RING (really interesting new gene) finger domain-containing E3s type and the HECT (−homologous to E6-associated protein C terminus) type. HECT-E3s are characterized by the HECT domain, a C-terminal region of ∼350 amino acids in length with significant similarity to the C terminus of E6-AP. Most HECT ligases contain one or more protein-protein or protein-lipid interaction domains located within the N terminus of the protein (2). Unlike the RING-finger E3s, which lack a catalytic site, the HECT E3s directly catalyze substrate ubiquitination.
TRIP12 is a 1,992-residue protein that was first isolated and characterized as a thyroid hormone receptor-interacting protein (3). More recent data implicated TRIP12 in a broad range of physiological processes and described its importance for mouse embryogenesis (4). TRIP12 displays a HECT domain, a WW (tryptophan-tryptophan) protein-protein interaction motif and an ARM domain (armadillo/β-catenin-like repeats). TRIP12 shows homology to yeast UFD4 and was identified as an E3 enzyme for the ubiquitin fusion degradation pathway in human cells (5). TRIP12 was recently designated as ULF (ubiquitin ligase for ARF) because it promotes lysine-independent ubiquitination and degradation of the tumor suppressor ARF in normal human fibroblast cells (6). TRIP12 was also shown to regulate the neddylation pathway (7) by directing the ubiquitination of APP-BP1 (amyloid β precursor protein binding protein 1), a component of the NEDD8 (neural precursor cell expressed developmentally down-regulated 8)-activating enzyme E1 (8). TRIP12 also regulates BAF57 (BGR1/BRM-associated factor 57) turnover and is therefore described as a sensor of SWI/SNF complex integrity (9). Lately, TRIP12 was demonstrated to control the histone ubiquitination after DNA breakage (10).
Pancreas-specific transcription factor 1a (PTF1a)3 is a member of the basic helix-loop-helix family of transcriptional regulatory proteins (11). It plays a crucial role in the early development of the pancreas (11, 12) and thereafter in the differentiation of acinar cells (13, 14). In the normal adult pancreas, its expression is restricted to acinar cells, predominantly localized in the nucleus where it directs, within the PTF1 complex, the transcriptional activation of exocrine-specific genes such as secretory digestive enzymes (13, 15). In the active transcription complex PTF1, PTF1a heterodimerizes with an E-protein by its HLH domain and interacts with the Suppressor of Hairless RBP-J or its paralogue, RBP-L, via its C-terminal region. In addition to the stabilization of acinar phenotype, PTF1a imposes a very low proliferative index to acinar cells through the transcriptional induction of p21/Cdkn1a, the down-regulation of cyclin D2 mRNA level, and the increase of p27 protein level (16). PTF1a was also identified as a major player in cerebellar neurogenesis (17–19). Its role in specifying GABAergic neurons was also established in the dorsal spinal cord (20). In retina, PTF1a determines retinal cell fates and promotes formation of GABAergic neurons (21, 22).
Down-regulation of PTF1a expression is observed in pancreatic pathologies. The majority of human pancreatic adenocarcinomas lack PTF1a expression. PTF1a protein level is also decreased in acute and chronic pancreatitis (23, 24). Recently, gene-associated sequences and transcriptional mechanisms regulating spatial and temporal expression of PTF1a were identified (25, 26). Functional modulation of PTF1a transcriptional activity by interaction with HES (Hairy/Enhancer of Split) family of transcriptional repressors or recruitment of the transcriptional co-activators p/CAF (p300/cAMP-response element-binding protein-binding protein-associated factor) was reported (27, 28). However, despite a clearly established role for PTF1a in pancreas development and acinar cell function, mechanisms regulating PTF1a degradation are still unknown.
In this study, we report that TRIP12 is a new partner of PTF1a. We demonstrate that TRIP12 targets PTF1a degradation through an ubiquitin-degradation pathway and also regulates PTF1a functions.
EXPERIMENTAL PROCEDURES
Yeast Two-hybrid Screening
Yeast two-hybrid system CytoTrap (Stratagene), also designated Sos recruitment system, which was adapted to identify PTF1a-interacting proteins was used. pSos vector (Stratagene), was modified using the Gateway vector conversion system (Invitrogen). PTF1a cDNA was transferred from the pENTR/FLAG-PTF1a vector into pSos vector by recombination using Gateway cloning technology (Invitrogen). pSos-PTF1a was used as bait to screen a human testis cDNA library (catalogue no. 975205, Agilent) constructed into the pMyr vector. After co-transfection into Saccharomyces cerevisiae strain cdc25H, 5 × 106 clones were screened, and several positives were identified. All clones were PCR-amplified. Their sequences were identified by comparison with the GenBankTM repertoire. All yeast transformations were done by the standard lithium acetate method. The CytoTrap screening was performed as previously described (29). Positive clones were grown, plasmids were isolated using Wizard Plus SV Miniprep kit (Promega), and sequencing was carried out using the services of Genome Express. Identification of inserts was done by database searches at the NCBI BLAST Network Service.
Cell Culture
HEK-293T cells, HEK-293FT cells, and AR42J cells (clone B13) provided by Prof. Timo Otonkoski (University of Helsinki, Helsinki, Finland) with the permission of Dr. Itaru Kojima (Gunma University, Maebashi, Japan) derived from an azaserine-induced pancreatic acinar tumor and acinar pancreatic 266.6 cells (derived from a mouse tumor induced with an elastase I/SV40 (simian virus 40) T-antigen fusion gene) were used. The cells were cultured in DMEM 4.5 g/liter d-glucose (Invitrogen) supplemented with 10% (v/v) fetal bovine serum, nonessential amino acids, 50 IU/ml penicillin, and 50 μg/ml streptomycin in a 5% CO2 and 95% humidified atmosphere at 37 °C.
Plasmids
Full-length human TRIP12 cDNA (accession number D28476) was obtained from the Kazusa DNA Research Institute (30). TRIP12 cDNA was subcloned into Gateway® pcDNATM-DEST47 vector that contains a GFP tag (pDEST47) (Invitrogen). Full-length human PTF1a cDNA was subcloned into pcDNA3.1 vector. For the different deletion mutants of TRIP12, DNA sequences corresponding to different regions were amplified by PCR with appropriate primers (Table 1) from the above constructs and subcloned into pcDNA™-DEST47 vector. The TRIP12-C1959A mutant with a cysteine to alanine substitution at the conserved cysteine 1959 and the PTF1a mutants with a lysine to alanine substitution on lysine 290, 312, or both were generated by introducing a point mutation using QuikChange kit (Stratagene) according to the manufacturer's instructions. Plasmids pRK5-HA-Ub-WT, pRK5-HA-Ub-K48O, and pRK5-HAUb-K63O were obtained from Addgene. pLKO shRNA control (SHC202) and pLKO ShTRIP12 (TRC1TRCN0000022374) lentiviral plasmids were purchased from Sigma-Aldrich. Vectors were expanded in competent Escherichia coli TOP10 bacterial strain (Invitrogen) and purified using the Maxiprep kit (Qiagen).
TABLE 1.
Primers used for the generation of cDNA fragments
Point mutations inserted in primers used to generate mutants of Ptf1a and TRIP12 are in bold.
| Constructs | Forward primers | Reverse primers |
|---|---|---|
| Ptf1a | ATggACgCggTgTTgCTggAgCACTTCCC | TCAggACACAAACTCAAATggTggTTCg |
| TRIP12 | ggggACAAgTTTgTACAAAAAAgCAggCTTCgAAggAgATAgAACCATgTCCAACCggCCTAATAAC | ggggACCACTTTgTACAAgAAAgCTgggTTgTAggAAAgATggAACgACTg |
| TRIP12 (1–445) | CCCCAgAAAACTCAACAgCgCATCTTCCTTCAACAACATAg | ggggACCACTTTgTACAAgAAAgCTgggTTgTATCCTTgTAgTAgCTgCTgggC |
| TRIP12 (446–1551) | ggggACAAgTTTgTACAAAAAAgCAggCTTCgAAggAgATAgAACCATgTTgCAAgCCAgTgATgAAAgT | ggggACCACTTTgTACAAgAAAgCTgggTTgTAATAAAAAAgCATTTgCCgggT |
| TRIP12 (1552–1992) | ggggACAAgTTTgTACAAAAAAgCAggCTTCgAAggAgATAgAACCATggTAACTgCATTTgATCgggAC | ggggACCACTTTgTACAAgAAAgCTgggTTgTAggAAAgATggAACgACTg |
| PTF1a-K290A | ggACTgATgAAAAACAACTCgCggAACAAAATATTATCCg | CggATAATATTTTgTTCCgCgAgTTgTTTTTCATCAgTCC |
| PTF1a-K312A | CCCCAgAAAACTCAACAgCgCATCTTCCTTCAACAACATAg | CTATgTTgTTgAAggAAgATgCgCTgTTgAgTTTTCTgggg |
| TRIP12-C1959A | gCCCTCTgTAATgACTgCTgTgAACTATCTTAAgTTgCCgg | CCggCAACTTAAgATAgTTCACAgCAgTCATTACAgAgggC |
Lentiviral Vector Production
All replication defective, self-inactivating lentiviral vectors were generated in a BSL-3 facility (Vectorology platform, INSERM U1037, Toulouse, France) as previously described by Torrisani et al. (31). Briefly, transient transfection of HEK-293FT cells with packaging and lentiviral vector plasmids were performed using LENTI-Smart INT kit (InvivoGen) following the manufacturer's recommendations. All batches were verified replicative virus-free. Lentiviral vector concentrations were quantified by p24 ELISA (Innotest, Ingen, Paris).
RNA Extraction and Quantitative RT-PCR Analysis
Total RNA was isolated from 266.6 cell lines with TRIzol® reagent (Invitrogen) according to the supplier's instructions. One μg of total RNA was reverse transcribed into cDNA using RevertAid H minus reverse transcriptase kit (Thermo-Scientific) according to the manufacturer's recommendations. Duplicate quantitative RT-PCR assays were carried out in a StepOnePlusTM real time PCR system (Applied Biosystems) with SsoFastTM EvaGreen® supermix Mix (Bio-Rad) and specific primers (Table 2). Relative amounts of mRNA were calculated by the comparative threshold cycle (CT) method as 2-ΔCT, where ΔCT = CT TRIP12 mRNA-CT RPS16 mRNA.
TABLE 2.
Primers used for mRNA expression analysis
| Targets | Forward primers | Reverse primers |
|---|---|---|
| TRIP12 | GTGCCTCCAATGGAAGTTGT | CCGGCTGTCAATCCTGTTAT |
| PTF1a | GAAGGTTATCATCTGCCATCG | TCAAAGGGTGGTTCGTTCTC |
| RPS16 | AATGGGCTCATCAAGGTGAACGGA | TATCCACACCAGCAAATCGCTCCT |
Transfection, Transduction, Western Blot, and Immunoprecipitation
Briefly, HEK-293T, 266.6 cells were seeded at a 38,000 cells/cm2 density and upon reaching 40–50% confluence were transfected with 5–10 μg of plasmid of interest. Transfection was performed using the FuGENE 6® (Roche) or Lipofectamine® 2000 (Invitrogen) reagents according to the manufacturers' protocols. After 36 h, total cellular protein was prepared according to our routinely used protocol. The cells were lysed in 10 mm HEPES (pH 7.9) buffer containing 10 mm KCl, 0.1 mm EDTA, 0.1 mm EGTA, 1 mm dithiothreitol, 2% Complete protease inhibitor mixture® (Roche, 1 tablet/ml), 1% soybean trypsin inhibitor, 1 mm Na3VO4, and 1% Nonidet P-40®. Proteins were separated by SDS-PAGE followed by Western blot assays. The protein expression signal was detected with Pierce SuperSignal Western blotting substrate. Loading control was performed after incubation with an anti-GAPDH antibody. Antibodies used for experiments are indicated in Table 3.
TABLE 3.
Antibodies used for experimental procedures
The rabbit polyclonal anti-PTF1a directed against the C-terminal KSFDNIENEPPFEFVS peptide of PTF1a is a custom antibody generated by Eurogentec.
| Antibodies against | Species | Usesa | Dilutions | Suppliers |
|---|---|---|---|---|
| PTF1a | Rabbit polyclonal | WB | 1:2000 | Eurogentec |
| IP | 10 μl/assay | |||
| IHC | 1:500 | |||
| TRIP12 | Rabbit polyclonal | IHC | 1:200 | Bethyl Laboratories |
| IP | 4 μg/500 μg of protein | |||
| Ubiquitin | Mouse monoclonal | WB | 1:2000 | Cell Signaling Technology |
| GFP | Mouse monoclonal | WB | 1:200 | Santa Cruz Biotechnology |
| IP | 4 μg/500 μg of protein | |||
| GAPDH | Rabbit polyclonal | WB | 1:1000 | Santa Cruz Biotechnology |
| HA | Rabbit monoclonal | WB | 1:1000 | Cell Signaling |
a WB, Western blot; IP, immunoprecipitation; IHC, immunohistochemistry.
For immunoprecipitation experiments, cells were lysed in 1 ml of 50 mm Tris/HCl (pH 8.0) immunoprecipitation buffer containing 120 mm NaCl, 0.5% Nonidet P-40®, protease inhibitor mixture®. Lysates were incubated overnight with appropriate antibodies and protein G-agarose beads (Roche). The immune complexes were pelleted by centrifugation washed in lysis buffer, and the proteins were recovered by boiling the beads in SDS sample buffer, fractionated by SDS-PAGE, and identified by Western blot analysis with appropriate antibodies (Table 3).
266.6 cells were seeded at a density of 104 cells/well of a 48-well dish. After 24 h, cells were incubated with 150 ng of p24 of lentiviral vectors in the presence of protamine sulfate (4 μg/ml) for 12 h. Transduced cells were selected for 3 weeks using puromycin (5 μg/ml; InvivoGen). TRIP12 and PTF1a protein expression was measured by Western blot analysis using radioimmune precipitation assay buffer supplemented with 1% Soybean Trypsin Inhibitor and Complete protease inhibitor mixture (Roche).
Proteasome Inhibition Experiments
Thirty-six hours after pcDNA-PTF1a transfection, HEK-293T cells were treated with proteasome inhibitors MG132 (20 μm) or lactacystin (5 μm). Control cells were treated with an equal amount of DMSO. In addition to the proteasome inhibitor, a 4–16-h treatment with 50 μg/ml of cycloheximide (CHX) was performed when indicated. All protein lysates were analyzed by Western blot using the appropriate antibodies (Table 3).
Stability of PTF1a in Mammalian Cells
To examine the effect of TRIP12 overexpression on PTF1a degradation, HEK-293T cells (3.106 cells) were co-transfected with 4 μg of pcDNA3.1-PTF1a and 10 μg of TRIP12 wild-type or TRIP12-C1959A plasmids or control plasmids. Stability of PTF1a was monitored in HEK-293T cells after inhibition of protein synthesis by CHX. Within 24–36 h after transfection of the pcDNA3.1-PTF1a plasmid, CHX (50 μg/ml) was added, and the CHX treatment was terminated at the indicated time points. Equal amounts of protein from cell lysates were subjected to SDS-PAGE followed by Western blot and detection with the proper antibodies. Stability of PTF1A in 266.6 stable cell lines was determined by Western blot at the indicated time points after CHX treatment (10 μg/ml).
In Vivo Ubiquitination Assay
Total protein was extracted from cells using modified radioimmune precipitation assay buffer (50 mm Tris/HCl, pH 8.0, 150 mm NaCl, 0.5% Nonidet P-40®, 0.5% sodium deoxycholate, 1% SDS) containing protease inhibitor mixture). Cells were lysed and heated at 95 °C during 10 min to dissociate contaminant proteins associated with PTF1a. Cell lysates were then diluted 10-fold with radioimmune precipitation assay buffer (50 mm Tris/HCl, pH 8.0, 150 mm NaCl, 0.5% Nonidet P-40®, 0.5% sodium deoxycholate, 0.1% SDS), and immunoprecipitation was performed with anti-PTF1a antibody.
Reporter Gene and Proliferation Assays
6XA26-Luc is a reporter construct containing a hexamer of A element of the rat elastase-1 promoter (kindly provided by F. X. Real, CNIO, Madrid, Spain) (16). pRL-SV40 containing the Renilla LUC coding sequence driven by the SV40 promoter was used as an internal control for normalization. For transactivation assays, HEK-293T cells were co-transfected with 0.1 μg of plasmid expressing PTF1a and 0.1 μg of plasmid expressing RBP-L, HEB (kindly provided by R. J. MacDonald and G. Swift (University of Texas Southwestern Medical Center, Dallas, TX)) and RLSV40 (as a transfection control). To determine the co-repression induced by TRIP12, 0.1 μg of PTF1a and RBP-L/HEB expression vectors or the corresponding empty vectors were co-transfected with increasing amounts of TRIP12 or TRIP12-C1959A plasmids (0.1–0.5 μg) and 0.1 μg of 6XA26-Luc. Luciferase activities were measured 48 h after transfection using the dual luciferase reporter assay system (Promega). Firefly and Renilla luciferase luminescence were measured using a luminometer. Proliferation rate was measured by counting the cells at day 6 after HEK-293T cells transfection of PTF1a in the presence or not of TRIP12 or TRIP12-C1959A.
Bioluminescence Resonance Energy Transfer (BRET)
BRET was performed with cellular extracts. HEK293T cells were transfected separately with a BRET donor (PTF1a-HRluc vector) or a BRET acceptor (TRIP12-YFP vector). After 48 h, total cellular proteins were extracted with CelLytic NuCLEAR extraction kit (Sigma-Aldrich) according to the manufacturer's instructions. A total of 3 μg of lysate containing a BRET donor was mixed with 0–40 μg of lysate containing a BRET acceptor and incubated during 30 min at 4 °C. Coelentherazin-h (Promega) was added to a final concentration of 5 μm in lysis buffer, just before reading. Readings were done using a LB 941 Tristar reader (Berthold France SA, Thoiry, France), with signal detection in the 470–490 nm (donor) and 520–540 nm (acceptor). BRET signal was determined by calculating the ratio of light emitted by YFP over light emitted by Rluc. The net BRET values were obtained by subtracting the BRET background signal corresponding to Rluc construct alone. These values are expressed in NET milli BRET units. All experiments were performed at least three times with comparable results.
Statistical Analysis
In vitro data were analyzed by two-tailed, paired Student's t test, and statistical significance was accepted at a confidence interval of p value <0.05 using a multiple statistics software package. Mean values are given ± S.E. The number of independent experiments is indicated in the text and/or figure legends.
RESULTS
PTF1a Is Degraded through the Ubiquitin-Proteasome Pathway
PTF1a protein sequence contains one region rich in proline, glutamic acid, serine, and threonine or putative PEST sequence (amino acids 33–55), suggesting the presence of a degradation signal. Because most cellular proteins are degraded by the ubiquitin-proteasome pathway, we examined the possible involvement of the proteasome in the degradation of PTF1a. We treated the AR42J acinar cell line endogenously expressing PTF1a and HEK-293T cells transiently expressing PTF1a with the proteasome inhibitor MG132. Our results showed that the level of PTF1a is significantly increased after MG132 treatment, suggesting that the protein is degraded by the proteasome in both cell lines (Fig. 1A). To exclude the possibility that the accumulation of PTF1a was the result of newly synthesized protein, we pretreated the two acinar cell lines AR42J and 266.6 for 1 h with CHX, a protein synthesis inhibitor, followed by administration of MG132. As shown in Fig. 1B, the level of PTF1a is also increased in these conditions. We then confirmed the involvement of the proteasome in PTF1a degradation by using lactacystin, another specific proteasome inhibitor (Fig. 1C) (32). Because proteins destined to proteasome degradation are often ubiquitinated, we investigated whether PTF1a undergoes ubiquitination. Upon MG132 treatment, the polyubiquitinated forms of PTF1a were detected after immunoprecipitation of PTF1a in cell extracts and immunoblotting with anti-ubiquitin or anti-PTF1a antibody (Fig. 1D). Taken together, all these data showed that degradation of PTF1a is mediated by an ubiquitin-dependent pathway.
FIGURE 1.
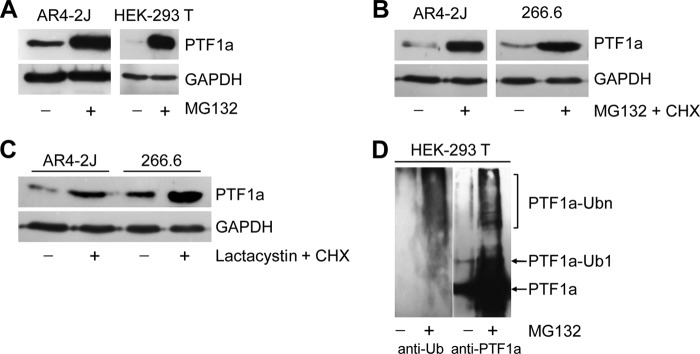
PTF1a is degraded by the ubiquitin-proteasome system. A, acinar AR4–2J cells expressing endogenous PTF1a and HEK-293T cells transfected with PTF1a vector were treated or not with MG132 (5 μm) for 10 and 16 h, respectively. Cell lysates were analyzed by Western blot using an anti-PTF1a antibody. B, acinar AR4–2J and 266.6 cells were treated with the protein synthesis inhibitor CHX (50 μg/ml) in the presence of MG132 (5 μm) for 4 h, and cell lysates were analyzed by Western blot using an anti-PTF1a antibody. C, acinar AR4–2J and 266.6 cells were treated with CHX in the presence or absence of the proteasome inhibitor lactacystin for 4 h, and cell lysates were analyzed by Western blot using anti-PTF1a antibody. D, HEK-293T cells were transfected with PTF1a expression vector and treated or not with MG132 for 10 h. Cell lysates were immunoprecipitated with anti-PTF1a antibody and immunoblotted with anti-PTF1a antibody (right panel) or anti-ubiquitin antibody (left panel); mono- and polyubiquitinated forms of PTF1a are indicated. Ub or Ubn, ubiquitin.
PTF1a Is a New Target of the E3 Ubiquitin Protein Ligase TRIP12
To determine candidate molecular mediators of PTF1a proteolytic degradation, we mapped its interactome using the Sos recruitment system, an alternative method developed in our group for detecting protein-protein interactions in the cytoplasm instead of the nucleus as the conventional two-hybrid systems (29). Moreover, the Sos recruitment system is particularly adapted to nuclear proteins that often interact with transcriptional factors and that nonspecifically activate the classical Gal4 two-hybrid system. The results of the yeast double-hybrid screening yielded nine proteins interacting with PTF1a, including RBPJ, which belongs to the PTF1 complex (Table 4). Among them, TRIP12, recently characterized as an E3 ubiquitin-protein ligase, was identified as a new partner of PTF1a. The physical interaction between TRIP12 and PTF1a was examined by reciprocal co-immunoprecipitation experiments in HEK-293T cells co-transfected with PTF1a and GFP-tagged TRIP12 expression vectors (Fig. 2A) and in 266.6 cells expressing endogenous proteins (Fig. 2B). TRIP12 co-immunoprecipitates with PTF1a in both cell lines, thus confirming that these two proteins belong to the same protein complex.
TABLE 4.
Proteins interacting with PTF1a identified from the yeast two-hybrid screening
| Proteins identified | UniProtKB | |
|---|---|---|
| RPGRIP1 | Retinitis pigmentosa GTPase regulator-interacting protein 1 | Q96KN7 |
| CCNDBP1 | Cyclin D-type binding-protein 1 | O95273 |
| TRIP12 | Thyroid hormone receptor interactor 12 | Q14669 |
| ZW10 | Centromere/kinetochore protein zw10 homologue | O43264 |
| SPATA7 | Spermatogenesis-associated 7 | Q9P0W8 |
| RBPJ | Recombination signal binding protein for immunoglobulin kappa J regiona | Q06330 |
| AAMP | Angio-associated, migratory cell protein | Q13685 |
| CCDC27 | Coiled-coil domain containing 27 | Q2M243 |
| TCF4 | Transcription factor 4 | P15884 |
a Previous name: recombining binding protein suppressor of hairless (Drosophila).
FIGURE 2.

TRIP12 interacts with PTF1a and regulates PTF1a stability. A, co-immunoprecipitation of exogenous TRIP12 with exogenous PTF1a. HEK-293T cells were co-transfected with GFP-tagged TRIP12 and PTF1a expressing vectors. Cell lysates were immunoprecipitated or not (Input) with either nonspecific anti IgG, anti-GFP antibody, or anti-PTF1a antibody. The immunoprecipitates and lysates were analyzed by SDS-PAGE and subsequent immunoblotting with anti-PTF1a or anti-GFP antibodies. B, interaction between endogenous PTF1a and endogenous TRIP12. Acinar 266.6 cell lysates were immunoprecipitated or not (Input) with anti-TRIP12 antibody or with nonspecific IgG antibody and subsequently immunoblotted with anti-PTF1a or anti-TRIP12 antibodies C, TRIP12 and PTF1a protein levels in ShTRIP12 and ShControl 266.6 stable cell lines were measured by Western blot using appropriate antibodies. The level of GAPDH was used as loading control. D, TRIP12 and PTF1a mRNA level in ShTRIP12 and ShControl 266.6 stable cell lines was measured by quantitative RT-PCR using specific primers. The level of ribosomal protein S16 was used for normalization. The graph represents the means (± S.E.) of three different experiments. The results are expressed as percentages of levels in ShControl 266.6 cell line. E, TRIP12/PTF1a interaction in TRIP12 depleted 266.6 cells. Cell lysates from ShControl and ShTRIP12 stable 266.6 cell lines were immunoprecipitated or not (Input) with an anti-TRIP12 antibody. As control, cell lysate from ShControl 266.6 cell line was immunoprecipitated with a nonspecific IgG. Immunoprecipitated proteins were visualized by Western blot analysis using TRIP12 and PTF1a antibodies. The GAPDH level was used to verify equal amount of loaded protein in the Input lanes. F, PTF1a protein stability in TRIP12-depleted 266.6 cells. ShTRIP12 and ShControl 266.6 stable cell lines were treated with 10 μg/ml CHX for the indicated time. PTF1a protein level was analyzed by Western blot using an anti-PTF1a antibody. Relative amounts of PTF1a were calculated after normalizing to GAPDH protein level. Stability of PTF1a was determined from three separate experiments. Black squares represent ShControl 266.6 cell line, and white circles represent 266.6 ShTRIP12 cell line. IP, immunoprecipitation.
TRIP12 Is a Key Regulator of PTF1a Stability
Because TRIP12 and PTF1a belongs to the same protein complex, we reasoned that PTF1a can be a substrate of TRIP12. We therefore examined whether the protein stability of PTF1a is affected when decreasing expression of TRIP12 by a ShRNA strategy. We verified that a stable expression of ShTRIP12 in 266.6 cells significantly diminished TRIP12 expression at protein and mRNA levels (Fig. 2, C and D, respectively). We observed an increased PTF1a protein expression in response to TRIP12 deletion (Fig. 2C), confirming the important role of TRIP12 on PTF1a protein expression. In addition, we confirmed the specificity of TRIP12/PTF1a interaction by observing a decreased PTF1a amount in anti-TRIP12 immunoprecipitates obtained from TRIP12-depleted cells (Fig. 2E). This increased expression of PTF1a in TRIP12-depleted cells can be explained by an increased stability of PTF1a protein (Fig. 2F). A major signal for proteasomal degradation is lysine 48-linked polyubiquitin chains on target proteins for degradation. We thus examined linkage specificity of TRIP12-mediated ubiquitin chain assembly by performing in vivo ubiquitination. First, wild-type HA-ubiquitin was co-expressed with PTF1a and TRIP12 in HEK-293T. Analysis of ubiquitination with anti-HA antibody showed that PTF1a is polyubiquitinated to much higher levels in the presence of TRIP12 (Fig. 3A). To determine which type of ubiquitin linkage is involved in the ubiquitination of PTF1a, we examined the ability of HA-ubiquitin mutants to ubiquitinate PTF1a in the presence or not of TRIP12. PTF1a and various ubiquitin mutants were co-expressed in the presence or not of TRIP12. Lys48-only (K48O) and Lys63-only (K63O) ubiquitin mutants promote the proteasome-linked Lys48 and the proteasome-independent Lys63 linkage of cellular proteins, respectively. The K48R (lysine to arginine mutation at position 48) disrupted Lys48 ubiquitin linkage, leading to accumulation of ubiquitinated proteins (Fig. 3B). Our results demonstrated that ubiquitin molecules are linked to PTF1a through Lys48 and Lys63. Nevertheless overexpression of TRIP12 enhanced the assembly of ubiquitin to PTF1a through Lys48 chains, thus providing a signal to the proteasome, but had no effect on Lys63-linked ubiquitination (Fig. 3C). Regulation of PTF1a ubiquitination by TRIP12 was also impaired using a K48R mutant (Fig. 3C).
FIGURE 3.
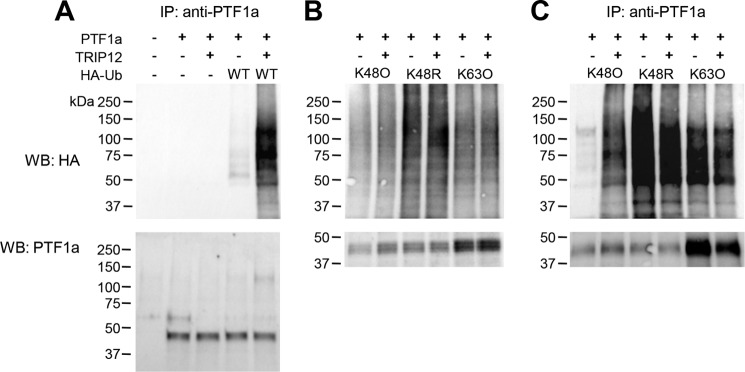
TRIP12 mediates Lys48-linked polyubiquitination of PTF1a. A, HEK-293T cells were co-transfected with PTF1a, WT HA-ubiquitin, and GFP-tagged TRIP12 expression plasmids as indicated. After MG132 exposure for 2 h, cell lysates were denatured and immunoprecipitated using anti-PTF1a antibodies, and equal amounts of PTF1a were immunoblotted with anti-HA antibody. B, HEK-293T cells were transfected with plasmids encoding HA-ubiquitin mutated at individual lysines except Lys48 (K48O), Lys63 (K63O), or mutated at Lys48 (K48R) as indicated. After MG132 exposure for 8 h, 50 μg of denatured cell lysates were loaded, HA-ubiquitinated proteins were detected with anti-HA antibody and PTF1a with anti-PTF1a antibody. C, HEK-293T cells were co-transfected with PTF1a, HA-ubiquitin mutants, and TRIP12 expression plasmids as indicated. After MG132 exposure for 8 h, cell lysates were denatured and immunoprecipitated using anti-PTF1a antibody. PTF1a was detected by immunoblotting with anti-HA or PTF1a antibodies. Standards molecular weights are indicated. IP, immunoprecipitation; WB, Western blot.
Protein Stability of PTF1a Is Linked to E3 Ligase Activity of TRIP12
HEK-293T cells were transfected with PTF1a and GFP-tagged TRIP12 expression vectors, and PTF1a levels were monitored after inhibiting new protein synthesis with cycloheximide. The half-life of PTF1a is shortened by 2.4 ± 0.2-fold (mean ± S.E., n = 3, p < 0.001) by over-expression of TRIP12 confirming that PTF1a is a target protein of TRIP12 (Fig. 4A).
FIGURE 4.

TRIP12 destabilizes and induces proteasome-dependent degradation of PTF1a. A, HEK-293T cells were treated with 50 μg/ml CHX for the indicated time after transfection with PTF1a and TRIP12-GFP. Fifty μg of cell lysates were analyzed by Western blot using an anti-PTF1a antibody, relative amounts of PTF1a were calculated after normalizing to GAPDH, and the half-life of PTF1a was determined from three separate experiments. Black squares, PTF1a; white circles, co-expression of PTF1a and TRIP12. B, sequence alignment of HECT domains from several E3 ubiquitin ligases, cysteine 1959 of TRIP12 is conserved. C, HEK-293T cells transfected with PTF1a and TRIP12 wild type, TRIP12-C1959A, or HECT domain expression vectors were analyzed by immunoblotting with anti-PTF1a antibody after treatment with CHX (50 μg/ml) as indicated. Relative amounts of PTF1a were calculated after normalizing to GAPDH, and the stability of PTF1a is represented from three separate experiments. White circles, co-expression of PTF1a and TRIP12 wild type; white squares, PTF1a and TRIP12-C1959A; black triangles, PTF1a and HECT domain. D, HEK-293T cells were co-transfected with GFP-tagged TRIP12-C1959A mutant and PTF1a expression vectors. Cell lysates were immunoprecipitated or not (Input) with anti-PTF1a antibody, anti-GFP antibody or nonspecific IgG antibody. The immunoprecipitates and cell lysates were analyzed by Western blot using anti-GFP or anti-PTF1a antibodies. E, HEK-293T cells were transfected with PTF1a expression vector in the presence or not of GFP-tagged TRIP12 construct and treated with CHX (50 μg/ml) in the presence or not of MG132 (5 μm) for 4 h, and cell lysates were analyzed by Western blot using an anti-PTF1a antibody. IP, immunoprecipitation.
E3 ligases of the HECT domain family share a region of homology at the C terminus, which contains a conserved cysteine that catalytically transfers ubiquitin to its substrate (33). To evaluate whether this catalytic cysteine is necessary for PTF1a degradation by TRIP12, we generated a construct in which the catalytically cysteine is mutated to alanine. According to the alignment of C-terminal sequences of various HECT domains, we chose Cys1959 as a mutation residue and constructed TRIP12-C1959A (Fig. 4B). We co-transfected HEK-293T cells with PTF1a with either GFP-tagged TRIP12 or GFP-tagged TRIP12-C1959A. As shown in Fig. 4C, the protein stability of PTF1a is unaffected by the co-expression of TRIP12-C1959A. We verified that this effect does not result from an impaired interaction of TRIP12-C1959A and PTF1a. The results of co-immunoprecipitation experiments showed that TRIP12-C1959A is able to interact with PTF1a (Fig. 4D). Taken together, these results demonstrated that protein stability of PTF1a is linked to E3 ligase activity of TRIP12. Consistent with this conclusion, the increase of PTF1a degradation induced by TRIP12 co-expression could be rescued by the proteasome inhibitor MG132 (Fig. 4E).
We also tested whether the TRIP12-HECT domain alone can affect the stability of PTF1a. As shown in Fig. 4C, PTF1a is not destabilized by TRIP12-HECT domain, suggesting that the catalytic domain is essential but not sufficient for PTF1a degradation and that regions outside of HECT domain are required to destabilize PTF1a. To identify the regions of TRIP12 required for interaction with PTF1a, we generated GFP-tagged deletion mutants of TRIP12 (Fig. 5A) and performed PTF1a-TRIP12 domain co-immunoprecipitation assays. As shown in Fig. 5B, PTF1a interacts with the TRIP12 mutant harboring the ARM and WWE protein-interacting domains (named TRP12 446–1551). The N-terminal region and the HECT domain of the E3 ligase are not involved in PTF1a recognition. To confirm further the interaction between PTF1a and the TRIP12 (446–1551) mutant, we performed a BRET assay, which allows the real time detection of protein-protein interactions. As shown in Fig. 5C, the BRET signal confirmed that PTF1a and the TRIP12 mutant containing the ARM and WWE protein-interacting domains belong to the same complex.
FIGURE 5.
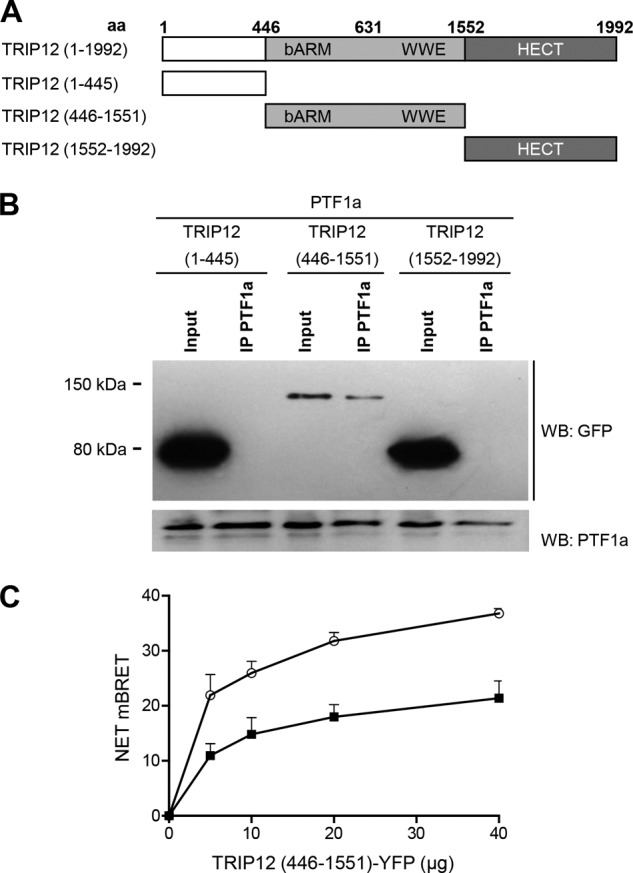
The N-terminal region and the HECT domain of TRIP12 are not involved in PTF1a recognition. A, wild-type and truncated TRIP12 proteins. aa, amino acid. All constructs were GFP-tagged. B, HEK-293T cells were co-transfected with PTF1a and vectors for GFP-tagged truncated TRIP12 proteins as indicated. Cell lysates were immunoprecipitated or not (Input) with an anti-PTF1a antibody then analyzed by Western blot using anti-GFP or anti-PTF1a antibodies. A negative control consisting of anti-rabbit IgG beads alone led to no band (not shown). C, HEK-293T cells were transfected separately with the BRET donor (C- or N-terminally labeled PTF1a-HRluc vector) or the BRET acceptor (C-terminal labeled TRIP12 (446–1551) mutant-YFP vector). After 48 h, total cellular proteins were extracted. A total of 3 μg of lysate containing the BRET donor was mixed with variable amounts (0–40 μg) of lysate containing the BRET acceptor. White circles, C-terminal labeled PTF1a-HRluc vector; black squares, N-terminal labeled PTF1a-HRluc vector. The Rluc substrate coelenterazin-h was added 15 min before reading. The data represent the net BRET values expressed in NET milli BRET units. WB, Western blot.
Lysine 312 Residue Is Essential for PTF1a Degradation
E3 ligases catalyze the last step in the conjugation process, namely the covalent attachment of ubiquitin to an epsilon-NH2 group of an internal lysine residue in the substrate to generate a covalent isopeptide group. PTF1a contains eight conserved lysine residues; two are located in the basic helix-loop-helix domain, whereas the other six are in the C-terminal region of the protein. Because the C-terminal sequence of p53 also contains six lysine residues that are targets for ubiquitination, we performed an alignment of C-terminal sequences of PTF1a and p53 (34). We found that lysine 290 and lysine 312 of PTF1a are in an amino acid environment similar to that of two lysine residues of p53 and reasoned that they could be potential sites of modification by ubiquitin (Fig. 6A). To investigate their role, we generated K290A, K312A, and K290A/K312A lysine to alanine mutants of PTF1a.
FIGURE 6.

Lysine 312 of PTF1a is required for ubiquitination and proteasome degradation. A, alignment of human PTF1a (amino acids 251–348) and p53 (amino acids 305–389) sequences. An asterisk indicates conserved amino acid residues, and a colon shows amino acid residues with similar properties. Gray boxes indicate lysine residues of PTF1a and p53 proteins located in similar amino acid environment. B, HEK-293T cells were co-transfected with GFP-tagged TRIP12 and PTF1a mutant expression vectors as indicated. Cell lysates were immunoprecipitated or not with an anti-PTF1a antibody or a nonspecific IgG antibody. The immunoprecipitates and lysates were analyzed by Western blot with anti-GFP or anti-PTF1a antibodies. C, HEK-293T cells transfected with wild-type PTF1a or PTF1a-K290A, PTF1a-K312A, and PTF1a-K290A/K312A mutants were treated or not with MG132 (5 μm) for 4 h. Cell lysates were analyzed by Western blot using an anti-PTF1a antibody, and mono- and polyubiquitinated forms of PTF1a are indicated. D, HEK-293T cells transfected with TRIP12 and the indicated PTF1a wild type and K290A, K312A, and K290A/K312A mutants were analyzed by immunoblotting with an anti-PTF1a antibody after treating with CHX (50 μg/ml) as indicated (left panel). Relative amounts of PTF1a were calculated after normalizing to GAPDH, and half-life of PTF1a was determined from three separate experiments in the right panel. White circles, co-expression of TRIP12 and PTF1a wild type; black squares, TRIP12 and PTF1a-K290A; black triangles, TRIP12 and PTF1a-K312A; white diamonds, TRIP12 and PTF1a-K290A/K312A.
We first verified that lysine to alanine mutations neither modified PTF1a expression nor interaction with TRIP12 (Fig. 6B). Upon MG132 treatment, an accumulation of K290A mutant is similar to that of wild-type protein, whereas K312A and double K290A/K312A mutant protein levels are not modified, thus indicating that lysine 312 is essential for proteasome degradation (Fig. 6C). Moreover, the absence of molecular species with higher mass suggested that mutation of lysine 312 completely inhibits ubiquitination of PTF1a and thus represents a unique site of ubiquitination. Accordingly, the half-life of K290A mutant was similar to that of wild-type protein, whereas K312A and double K290A/K312A mutation stabilized PTF1a. Indeed, the half-life of the proteins is extended from 2.5 ± 0.4 h to 6.7 ± 0.6 h (mean ± S.E., n = 3, p < 0.01), thus demonstrating that lysine 312 is crucial for the degradation of PTF1a (Fig. 6D).
TRIP12 Regulates PTF1a Transcriptional and Antiproliferative Activities
The function of PTF1a is to control acinar proliferation and differentiation through antiproliferative and transcriptional activities. We examined whether, consistent with the degradation of PTF1a, TRIP12 inhibits these activities. We co-transfected HEK-293T cells with increasing amounts of TRIP12 or corresponding empty vector in combination with expression vectors for each component of the PTF1 complex (PTF1a, RBP-L, and HEB). To assess PTF1a transcription activity, we measured the activity of a luciferase reporter under the control of a synthetic promoter containing six multimerized A boxes from the rat Elastase 1 gene (16). As seen in Fig. 7A, co-expression of TRIP12 with PTF1a reduces the luciferase activity by 80% in a dose-dependent manner. Expression of TRIP12 alone does not activate PTF1-responsive promoter (data not shown). Transfection with the TRIP12 mutant lacking E3 ligase activity fails to inhibit PTF1a transcriptional activity (Fig. 7A). We also examined whether TRIP12 affects PTF1a antiproliferative activity. As shown in Fig. 7B, PTF1a significantly decreases HEK-293T cell proliferation by ∼44%. This effect is reversed by concomitant expression of TRIP12. Again the importance of TRIP12 ubiquitin ligase activity in regulating PTF1a function was demonstrated with the absence of effect of the TRIP12-C1959A mutant.
FIGURE 7.
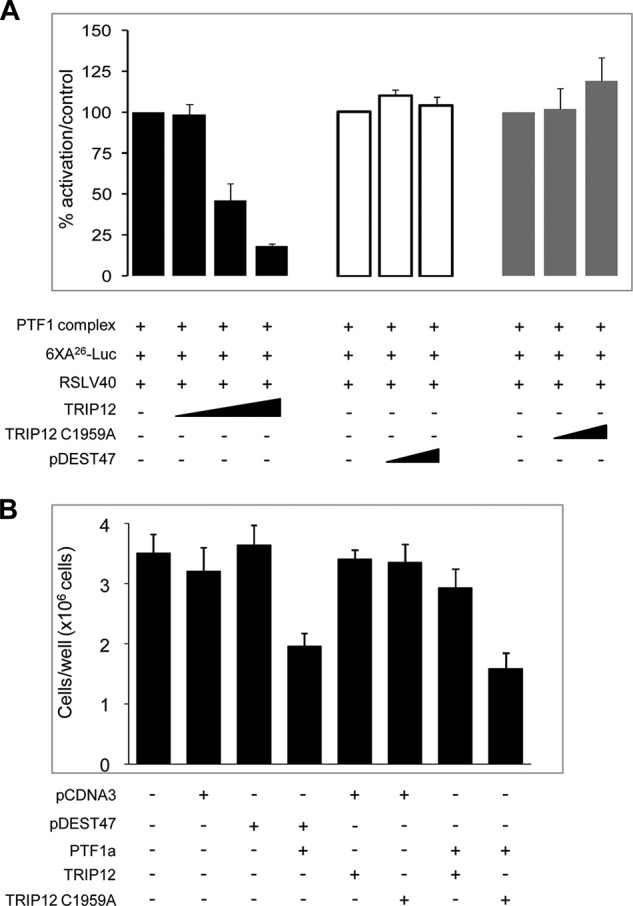
TRIP12 reverses gene activation and antiproliferative activity of PTF1a. A, HEK-293T cells were transiently co-transfected with PTF1 complex (composed of PTF1a, RBPL, and HEB), the 6XA26-luc reporter construct that contains an hexamer of PTF1-responsive promoter and RSLV40 vector, in the presence of increasing amounts of TRIP12 (100, 200, and 500 ng/well) (black bars), TRIP12-C1959A (200 and 500 ng/well) (gray bars) or control pDEST47 plasmid (200 and 500 ng/well) (white bars). The RSLV40 vector was included as an internal control for normalization. The results are expressed as percentages of activation (means ± S.E., n = 3), and each experiment is performed in triplicate. No significant differences on the transcriptional activity are observed when TRIP12 or TRIP-C1959A constructs were transfected in the absence of PTF1a (data not shown). B, HEK-293T cells were co-transfected with PTF1a and with TRIP12 or TRIP12-C1959A or their corresponding controls plasmids (as indicated), stopped, and counted 6 days post-transfection. The results of experiments performed in triplicate are represented as counted cells per well and expressed in means ± S.E. (n = 3).
DISCUSSION
In this study, we provide evidence that PTF1a, an essential pancreatic transcription factor, is a new physiological target of TRIP12. We identified TRIP12 by using an optimized yeast two-hybrid system, and our study strongly supports the crucial role of this E3 ubiquitin ligase in the proteolysis and inhibition of PTF1a activities. Our data are the first to report mechanisms involved in PTF1a stability and degradation. To date, known mechanisms that regulate PTF1a function are transcriptional mechanisms. They control PTF1a levels and post-translational acetylation by p/CAF, the acetyltransferase cofactor that potentiates PTF1a transcriptional activity (28). Rbms3, a RNA binding protein, was recently suggested to stimulate the translation of PTF1a mRNA by binding to its 3′-UTR (35). In contrast, miR-18a was shown to target repression of PTF1a by binding to its 3′-UTR (36). Moreover, four genetic mutations leading to the expression of a truncated protein lacking C-terminal region associated with the absence of pancreatic and cerebellar development in newborn infants were identified (19, 37, 38).
We report that TRIP12 is a novel interacting partner of PTF1a. Although we cannot conclude from our experiments that the interaction between PTF1a and TRIP12 is direct, our mapping experiments indicate that a region of TRIP12 sequence containing the armadillo repeats and the WWE domain mediates the association with PTF1a. Interestingly, the WWE domain was reported as required for the interaction with APP-BP1 (8). Although the HECT domain of TRIP12 was shown to interact with and to ubiquitinate substrates of the ubiquitin fusion degradation pathway, we report neither binding nor catalytic activity toward PTF1a of this domain alone (5). Thus, the possibility that several intramolecular interactions and/or a specific active conformations involving other regions in the case of PTF1a degradation exists.
The C-terminal domain of PTF1a is essential for transcriptional activity of PTF1a because it recruits the mammalian Suppressor of Hairless RBP (recombination signal-binding protein 1)-Jκ or its paralogue RBP-L (RBP-like) into the PTF1a complex binding to the promoter of acinar genes (15). The C-terminal region of PTF1a is also responsible for the antiproliferative activity of the protein (16). We demonstrate here that the conserved lysine residue located in this region, Lys312, is crucial for PTF1a degradation by the proteasome pathway. In contrast to p53, for which competition between acetylation and ubiquitination was documented at several lysine residues in the C terminus, Lys312 is not a target of p/CAF, whereas Lys200 located in basic helix-loop-helix region (Lys201 in human sequence) is the crucial lysine for acetylation (28, 39).
We demonstrate that PTF1a/TRIP12 interaction has functional consequences because it negatively regulates PTF1a expression, transcriptional, and antiproliferative activities. Altogether, the present study identifies a new physiological target of TRIP12, provides new insights into the mechanisms through which PTF1a is regulated, and suggests an important role for the PTF1a/TRIP12 interaction in vivo. Moreover, our results support the hypothesis that TRIP12 could play an important role in the regulation of pancreatic epithelial homeostasis.
Acknowledgments
We thank R. J. MacDonald and G. Swift (University of Texas Southwestern Medical Center, Dallas, TX) for the HEB and mouse Rbpsuh-L plasmids; FX Real (CNIO, Madrid, Spain) for the 6XA26-Luc reporter construct; R. F. Kikuno and O. Ohara (Kazusa DNA Research Institute, Chiba, Japan) for TRIP12 clone; and C. Lafargues (INSERM U1037, Toulouse, France) for technical assistance.
This work was supported by INSERM, Agence Nationale pour la Recherche, and Association pour la Recherche contre le Cancer.
- PTF1a
- pancreas transcription factor 1a
- CHX
- cycloheximide
- BRET
- bioluminescence resonance energy transfer.
REFERENCES
- 1. Hershko A., Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 2. Rotin D., Kumar S. (2009) Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10, 398–409 [DOI] [PubMed] [Google Scholar]
- 3. Lee J. W., Choi H. S., Gyuris J., Brent R., Moore D. D. (1995) Two classes of proteins dependent on either the presence or absence of thyroid hormone for interaction with the thyroid hormone receptor. Mol. Endocrinol. 9, 243–254 [DOI] [PubMed] [Google Scholar]
- 4. Kajiro M., Tsuchiya M., Kawabe Y., Furumai R., Iwasaki N., Hayashi Y., Katano M., Nakajima Y., Goto N., Watanabe T., Murayama A., Oishi H., Ema M., Takahashi S., Kishimoto H., Yanagisawa J. (2011) The E3 ubiquitin ligase activity of Trip12 is essential for mouse embryogenesis. PLoS One 6, e25871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park Y., Yoon S. K., Yoon J. B. (2009) The HECT domain of TRIP12 ubiquitinates substrates of the ubiquitin fusion degradation pathway. J. Biol. Chem. 284, 1540–1549 [DOI] [PubMed] [Google Scholar]
- 6. Chen D., Shan J., Zhu W. G., Qin J., Gu W. (2010) Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature 464, 624–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hochstrasser M. (1998) There's the rub: a novel ubiquitin-like modification linked to cell cycle regulation. Genes Dev. 12, 901–907 [DOI] [PubMed] [Google Scholar]
- 8. Park Y., Yoon S. K., Yoon J. B. (2008) TRIP12 functions as an E3 ubiquitin ligase of APP-BP1. Biochem. Biophys. Res. Commun. 374, 294–298 [DOI] [PubMed] [Google Scholar]
- 9. Keppler B. R., Archer T. K. (2010) Ubiquitin-dependent and ubiquitin-independent control of subunit stoichiometry in the SWI/SNF complex. J. Biol. Chem. 285, 35665–35674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gudjonsson T., Altmeyer M., Savic V., Toledo L., Dinant C., Grøfte M., Bartkova J., Poulsen M., Oka Y., Bekker-Jensen S., Mailand N., Neumann B., Heriche J. K., Shearer R., Saunders D., Bartek J., Lukas J., Lukas C. (2012) TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 150, 697–709 [DOI] [PubMed] [Google Scholar]
- 11. Krapp A., Knöfler M., Ledermann B., Bürki K., Berney C., Zoerkler N., Hagenbüchle O., Wellauer P. K. (1998) The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev. 12, 3752–3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kawaguchi Y., Cooper B., Gannon M., Ray M., MacDonald R. J., Wright C. V. (2002) The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat. Genet. 32, 128–134 [DOI] [PubMed] [Google Scholar]
- 13. Masui T., Swift G. H., Deering T., Shen C., Coats W. S., Long Q., Elsässer H. P., Magnuson M. A., MacDonald R. J. (2010) Replacement of Rbpj with Rbpjl in the PTF1 complex controls the final maturation of pancreatic acinar cells. Gastroenterology 139, 270–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dong P. D., Provost E., Leach S. D., Stainier D. Y. (2008) Graded levels of Ptf1a differentially regulate endocrine and exocrine fates in the developing pancreas. Genes Dev. 22, 1445–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beres T. M., Masui T., Swift G. H., Shi L., Henke R. M., MacDonald R. J. (2006) PTF1 is an organ-specific and Notch-independent basic helix-loop-helix complex containing the mammalian Suppressor of Hairless (RBP-J) or its paralogue, RBP-L. Mol. Cell. Biol. 26, 117–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rodolosse A., Chalaux E., Adell T., Hagège H., Skoudy A., Real F. X. (2004) PTF1α/p48 transcription factor couples proliferation and differentiation in the exocrine pancreas. Gastroenterology 127, 937–949; Correction (2004) Gastroenterology127, 1651 [DOI] [PubMed] [Google Scholar]
- 17. Hoshino M., Nakamura S., Mori K., Kawauchi T., Terao M., Nishimura Y. V., Fukuda A., Fuse T., Matsuo N., Sone M., Watanabe M., Bito H., Terashima T., Wright C. V., Kawaguchi Y., Nakao K., Nabeshima Y. (2005) Ptf1a, a bHLH transcriptional gene, defines GABAergic neuronal fates in cerebellum. Neuron 47, 201–213 [DOI] [PubMed] [Google Scholar]
- 18. Pascual M., Abasolo I., Mingorance-Le Meur A., Martínez A., Del Rio J. A., Wright C. V., Real F. X., Soriano E. (2007) Cerebellar GABAergic progenitors adopt an external granule cell-like phenotype in the absence of Ptf1a transcription factor expression. Proc. Natl. Acad. Sci. U.S.A. 104, 5193–5198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sellick G. S., Barker K. T., Stolte-Dijkstra I., Fleischmann C., Coleman R. J., Garrett C., Gloyn A. L., Edghill E. L., Hattersley A. T., Wellauer P. K., Goodwin G., Houlston R. S. (2004) Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat. Genet. 36, 1301–1305 [DOI] [PubMed] [Google Scholar]
- 20. Glasgow S. M., Henke R. M., Macdonald R. J., Wright C. V., Johnson J. E. (2005) Ptf1a determines GABAergic over glutamatergic neuronal cell fate in the spinal cord dorsal horn. Development 132, 5461–5469 [DOI] [PubMed] [Google Scholar]
- 21. Lelièvre E. C., Lek M., Boije H., Houille-Vernes L., Brajeul V., Slembrouck A., Roger J. E., Sahel J. A., Matter J. M., Sennlaub F., Hallböök F., Goureau O., Guillonneau X. (2011) Ptf1a/Rbpj complex inhibits ganglion cell fate and drives the specification of all horizontal cell subtypes in the chick retina. Dev. Biol. 358, 296–308 [DOI] [PubMed] [Google Scholar]
- 22. Dullin J. P., Locker M., Robach M., Henningfeld K. A., Parain K., Afelik S., Pieler T., Perron M. (2007) Ptf1a triggers GABAergic neuronal cell fates in the retina. BMC Dev. Biol. 7, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Adell T., Gómez-Cuadrado A., Skoudy A., Pettengill O. S., Longnecker D. S., Real F. X. (2000) Role of the basic helix-loop-helix transcription factor p48 in the differentiation phenotype of exocrine pancreas cancer cells. Cell Growth Differ. 11, 137–147 [PubMed] [Google Scholar]
- 24. Molero X., Adell T., Skoudy A., Padilla M. A., Gómez J. A., Chalaux E., Malagelada J. R., Real F. X. (2007) Pancreas transcription factor 1α expression is regulated in pancreatitis. Eur J. Clin. Invest. 37, 791–801 [DOI] [PubMed] [Google Scholar]
- 25. Masui T., Swift G. H., Hale M. A., Meredith D. M., Johnson J. E., Macdonald R. J. (2008) Transcriptional autoregulation controls pancreatic Ptf1a expression during development and adulthood. Mol. Cell. Biol. 28, 5458–5468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meredith D. M., Masui T., Swift G. H., MacDonald R. J., Johnson J. E. (2009) Multiple transcriptional mechanisms control Ptf1a levels during neural development including autoregulation by the PTF1-J complex. J. Neurosci. 29, 11139–11148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ghosh B., Leach S. D. (2006) Interactions between hairy/enhancer of split-related proteins and the pancreatic transcription factor Ptf1-p48 modulate function of the PTF1 transcriptional complex. Biochem. J. 393, 679–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rodolosse A., Campos M. L., Rooman I., Lichtenstein M., Real F. X. (2009) p/CAF modulates the activity of the transcription factor p48/Ptf1a involved in pancreatic acinar differentiation. Biochem. J. 418, 463–473 [DOI] [PubMed] [Google Scholar]
- 29. Thalappilly S., Suliman M., Gayet O., Soubeyran P., Hermant A., Lecine P., Iovanna J. L., Dusetti N. J. (2008) Identification of multi-SH3 domain-containing protein interactome in pancreatic cancer: a yeast two-hybrid approach. Proteomics 8, 3071–3081 [DOI] [PubMed] [Google Scholar]
- 30. Nakajima D., Saito K., Yamakawa H., Kikuno R. F., Nakayama M., Ohara R., Okazaki N., Koga H., Nagase T., Ohara O. (2005) Preparation of a set of expression-ready clones of mammalian long cDNAs encoding large proteins by the ORF trap cloning method. DNA Res. 12, 257–267 [DOI] [PubMed] [Google Scholar]
- 31. Torrisani J., Bournet B., du Rieu M. C., Bouisson M., Souque A., Escourrou J., Buscail L., Cordelier P. (2009) let-7 MicroRNA transfer in pancreatic cancer-derived cells inhibits in vitro cell proliferation but fails to alter tumor progression. Hum. Gene Ther. 20, 831–844 [DOI] [PubMed] [Google Scholar]
- 32. Kisselev A. F., Goldberg A. L. (2001) Proteasome inhibitors: from research tools to drug candidates. Chem. Biol. 8, 739–758 [DOI] [PubMed] [Google Scholar]
- 33. Huibregtse J. M., Scheffner M., Beaudenon S., Howley P. M. (1995) A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. U.S.A. 92, 2563–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rodriguez M. S., Desterro J. M., Lain S., Lane D. P., Hay R. T. (2000) Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol. Cell. Biol. 20, 8458–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu C. K., Lai Y. C., Chen H. R., Chiang M. K. (2012) Rbms3, an RNA-binding protein, mediates the expression of Ptf1a by binding to its 3′UTR during mouse pancreas development. DNA Cell Biol. 31, 1245–1251 [DOI] [PubMed] [Google Scholar]
- 36. Yang Y., Ding L., An Y., Zhang Z. W., Lang Y., Tai S., Guo F., Teng C. B. (2012) MiR-18a regulates expression of the pancreatic transcription factor Ptf1a in pancreatic progenitor and acinar cells. FEBS Lett. 586, 422–427 [DOI] [PubMed] [Google Scholar]
- 37. Al-Shammari M., Al-Husain M., Al-Kharfy T., Alkuraya F. S. (2011) A novel PTF1A mutation in a patient with severe pancreatic and cerebellar involvement. Clin. Genet. 80, 196–198 [DOI] [PubMed] [Google Scholar]
- 38. Tutak E., Satar M., Yapicioglu H., Altintas A., Narli N., Hergüner O., Bayram Y. (2009) A Turkish newborn infant with cerebellar agenesis/neonatal diabetes mellitus and PTF1A mutation. Genet. Couns. 20, 147–152 [PubMed] [Google Scholar]
- 39. Le Cam L., Linares L. K., Paul C., Julien E., Lacroix M., Hatchi E., Triboulet R., Bossis G., Shmueli A., Rodriguez M. S., Coux O., Sardet C. (2006) E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell 127, 775–788 [DOI] [PubMed] [Google Scholar]