

Background: Activated sugars required to form rare oligosaccharide subunits of Bacillus cereus biofilm-polysaccharide are not known.
Results: A new nucleotide sugar, UDP-fucosamine-4N-(2)-oxoglutarate (UDP-Yelosamine), is formed from UDP-4-keto-6-deoxy-d-GlcNAc by an aminotransferase and a carboxylate-amine ligase.
Conclusion: Both enzymes are promiscuous and convert other UDP-4-keto amino-sugars to their UDP-4-amino-2-oxoglutarate derivatives.
Significance: New insights into metabolic routes involved in the synthesis of bacterial biofilm polysaccharide have been uncovered.
Keywords: Bacterial Metabolism, Biofilm, Carbohydrate Biosynthesis, Enzyme Catalysis, Glycobiology
Abstract
Surface glycan switching is often observed when micro-organisms transition between different biotic and abiotic niches, including biofilms, although the advantages of this switching to the organism are not well understood. Bacillus cereus grown in a biofilm-inducing medium has been shown to synthesize an unusual cell wall polysaccharide composed of the repeating subunit →6)Gal(α1–2)(2-R-hydroxyglutar-5-ylamido)Fuc2NAc4N(α1–6)GlcNAc(β1→, where galactose is linked to the hydroxyglutarate moiety of FucNAc-4-amido-(2)-hydroxyglutarate. The molecular mechanism involved in attaching 2-hydroxyglutarate to 4-amino-FucNAc has not been determined. Here, we show two genes in B. cereus ATCC 14579 encoding enzymes involved in the synthesis of UDP-FucNAc-4-amido-(2)-oxoglutarate (UDP-Yelosamine), a modified UDP-sugar not previously reported to exist. Using mass spectrometry and real time NMR spectroscopy, we show that Bc5273 encodes a C4″-aminotransferase (herein referred to as Pat) that, in the presence of pyridoxal phosphate, transfers the primary amino group of l-Glu to C-4″ of UDP-4-keto-6-deoxy-d-GlcNAc to form UDP-4-amino-FucNAc and 2-oxoglutarate. Pat also converts 4-keto-xylose, 4-keto-glucose, and 4-keto-2-acetamido-altrose to their corresponding UDP-4-amino-sugars. Bc5272 encodes a carboxylate-amine ligase (herein referred as Pyl) that, in the presence of ATP and Mg(II), adds 2-oxoglutarate to the 4-amino moiety of UDP-4-amino-FucNAc to form UDP-Yelosamine and ADP. Pyl is also able to ligate 2-oxoglutarate to other 4-amino-sugar derivatives to form UDP-Yelose, UDP-Solosamine, and UDP-Aravonose. Characterizing the metabolic pathways involved in the formation of modified nucleotide sugars provides a basis for understanding some of the mechanisms used by bacteria to modify or alter their cell surface polysaccharides in response to changing growth and environmental challenges.
Introduction
Switching, altering, or adding cell surface glycan structures is a strategy adopted by numerous microbial pathogens during interactions with their host. Knowledge of the molecular mechanisms that allow these modifications to occur may lead to the development of new therapeutic agents that limit or prevent infection. Bacillus cereus provides a model system for studying such “surface glycan switching” phenomena. B. cereus is a food-borne, spore-forming pathogenic bacterium that is present in soil (1), dust (2), water, plants, and the intestinal tract of insects and mammals (3). The bacterium is also likely transiently present in other parts of an insect's body and in decaying organic matter (4). The ubiquitous nature of this bacterium, combined with its ability to exist as a planktonic vegetative cell, a spore, or as a biofilm community, requires that this bacteria is able to rapidly alter its cellular biochemistry as well as cell surface features to adapt to new environments. Leoff et al. (5) reported that B. cereus ATCC 14579 grown by shaking in a nutrient-rich medium provided the identification of an oligosaccharide structure consisting of secondary cell wall polysaccharides (SCWP)2 linked via the 6-phosphate of GlcNAc peptidoglycan residues. Subsequently, Candela et al. (6) described a second SCWP with a repeating sequence of →6)Gal(α1–2)(2-R-hydroxyglutar-5-ylamido)Fuc2NAc4N(α1–6)GlcNAc(β1→. This SCWP was detected in the planktonic phase and also in late stage biofilms when the bacterium was grown in a selective nutrient medium rather than a nutrient-rich medium (6). Candela et al. (6) suggested that the formation of the SCWP is regulated by both growth conditions and the growth phase of the bacterium. However, the mechanisms controlling the production of this unusual SCWP are not known nor is it known how a 2-hydroxyglutaryl moiety is incorporated into the repeating oligosaccharide subunit of the polysaccharide. As far as we are aware, the only other example of hydroxyglutarate being used in this manner is in the O-specific polysaccharide of the lipopolysaccharide produced by Flexibacter maritimus which is a Gram-negative pathogen of fish (7).
Here, we report on a new metabolic pathway (see Fig. 1) in which two enzymes are required to convert UDP-4-keto-6-deoxy-2-N-acetylamido-hexose to UDP-FucNAc-4N-(2)-oxoglutarate (herein referred as UDP-Yelosamine). The first enzyme is a C4″-aminotransferase (Pat), which catalyzes the transfer of the primary amino group of l-Glu to C-4 of UDP-4-keto-6-deoxy-d-GlcNAc to form UDP-4-amino-FucNAc and 2-oxoglutarate (2OG). The second enzyme (Pyl) is a carboxylate-amine ligase that catalyzes the attachment of 2OG to the 4-amino group of the modified nucleotide-sugar. Amino acid sequence homology suggests that this ATP-dependent enzyme is a member of the ATP-Grasp family of proteins (8, 9).
FIGURE 1.

Proposed pathway for the formation of UDP-Yelosamine in B. cereus ATCC 14579. A UDP-GlcNAc 4,6-dehydratase converts UDP-GlcNAc to UDP-4-keto-6-deoxy-GlcNAc. Note: at steady state, the UDP-4-keto-sugar form (K) is converted nonenzymatically to a hydrated form Y. The enzyme encoded by Bc5273 (herein referred as Pat) is a l-Glu:UDP-4-keto-sugar C4″-aminotransferase and yields 2OG and UDP-d-FucNAc-4N. Pat activity requires a cofactor PLP and mediates C4″-transaminase exchange between the amino moiety (NH2) of l-glutamate and the keto (=O) group of UDP-4-keto-sugar to give a UDP-4-amino-sugar (A) and 2OG. Subsequently, Bc5272 encodes a carboxylate-amine ligase (herein referred to as Pyl) that, in the presence of ATP and Mg(II), adds 2OG to the 4-amino moiety of UDP-4-amino-FucNAc to form UDP-Yelosamine and ADP. Pyl is an ATP-dependent ligase, and we proposed (see brackets) ATP is required to activate 2OG to a phospho-intermediate derivative.
EXPERIMENTAL PROCEDURES
Bacterial Strain and Growth
Stock of wild type B. cereus ATCC 14579 was stored in 30% glycerol at −80 °C, streaked onto BHI agar (BD Biosciences), and grown for 18 h at 30 °C. The medium (agar or liquid) used was Luria Bertani (LB per liter: 10 g of tryptone, 5 g of yeast extract, 10 g of NaCl). Escherichia coli DH10B (Invitrogen) cells were used for cloning, and Rosetta2, a BL21(De3) strain derivative (Novagen), was used to generate recombinant proteins.
Cloning of Bc5272 (Pyl) and Bc5273 (Pat) from B. cereus ATCC 14579
A single colony of B. cereus ATCC 14579 grown on LB-agar was suspended in 50 μl of sterile water and then heated for 5 min at 96 °C. The suspension was centrifuged (13,000 × g, 2 min), and a portion of the supernatant (5 μl) was used as the source of genomic DNA. The PCR primer sets used to amplify the coding region of each gene included at their 5′ a 15-nucleotide extension with sequence homology to the cloning site of the pET28a plasmid. The primers are as follows: SY86, AAGCTTGCGGCCGCACTCGAGCAC; SY87, CATGgTATATCTCCTTCTTAAAGTTAAAC; SY91, GTGCGGCCGCAAGCTTATCTTTATAAATAAAATATCCAATTTTATC; SY97, GAAGGAGATATAcCATGAAAAAAAGAGTTCTAATTCTAGG; SY92, GTGCGGCCGCAAGCTTTCTATTGTTTAAAATCTCTTTAAAAG; and SY98, GAAGGAGATATACCATGATTAATAATACAATAAGAAATATCC. Individual genes (Bc5272 or Bc5273) were PCR-amplified using high fidelity Pyrococcus DNA polymerase (0.4 units of Phusion Hot Start II; New England Biolabs). The reaction (20 μl) included buffer, dNTPs (0.4 μl 10 mm), B. cereus genomic DNA (5 μl), and PCR primer sets (1 μl each of 10 μm). The PCR thermocycle conditions were 1× 98 °C denaturation cycle for 30 s followed by 25× cycles (each of 8 s denaturation at 98 °C; 25 s annealing at 50 °C; and 30 s elongation at 72 °C) and finally 4 °C. A similar PCR was used to amplify the expression plasmid (pET28) using a specific inverse-PCR primer set (SY86 and SY87 located near the NcoI and HindIII sites, respectively) with a 25-s annealing cycle at 58 °C and a 3-min elongation at 72 °C. After PCR, a portion (4 μl each) of the amplified plasmid and insert were mixed, digested with 10 units of DpnI (15 min, 37 °C), and then transformed into DH10B competent cells. Positive clones were selected on LB agar containing kanamycin (50 μg/ml), and clones were verified by PCR and by DNA sequencing. Plasmids harboring specific genes were sequenced, and the DNA sequences were deposited in GenBankTM (with respective accession numbers KM486797 and KM486798). The recombinant DNA coding sequence was cloned to yield a recombinant protein fused at the C terminus to a short peptide linker of 6 histidines (His6).
Expression and Purification of Recombinant Pat and Pyl, Bc5273 and Bc5272
E. coli strains containing pET28a:Bc5273-His6 (herein referred as Pat), pET28a:Bc5272-His6 (herein referred as Pyl), or an empty plasmid (control) were cultured in separate flasks of LB medium (20 ml) supplemented with kanamycin (50 μg/ml) and chloramphenicol (35 μg/ml). The cells were grown for 16 h at 37 °C with shaking at 250 rpm. A portion of each culture (5 ml) was then transferred to fresh 250 ml of LB medium containing the same concentrations of antibiotics. The cultures were grown at 37 °C until the cell density reached A600 = 0.5 to 0.6. Recombinant gene expression was then induced by the addition of isopropyl β-d-1-thiogalactopyranoside to 0.5 mm, and the cells were grown for an additional 20 h at 18 °C with shaking at 250 rpm. The culture was centrifuged (6,000 × g for 10 min at 4 °C), and the cell pellet was suspended in 10 ml of lysis buffer (50 mm Tris-HCl, pH 8.0, 30% (v/v) glycerol, 50 mm KCl, and 10 mm β-mercaptoethanol). The cells were lysed with a Misonics S4000 sonicator for 12 cycles at 30% amplitude. The lysate was centrifuged (6,000 × g for 15 min at 4 °C), and the supernatant was supplemented with 2 mm β-mercaptoethanol, recentrifuged (20,000 × g for 30 min at 4 °C), and the supernatant (labeled S20) was flash-frozen in liquid nitrogen and kept at −80 °C. His-tagged proteins were isolated using fast-flow nickel-Sepharose (GE Healthcare, 2 ml of resin packed in a 1-cm inner diameter × 15-cm polypropylene column). Each column was equilibrated with buffer A (50 mm Tris-HCl, pH 8.0, 300 mm NaCl, 10% (v/v) glycerol). An aliquot of S20 (5 ml) was loaded onto the column, and unbound protein was washed with buffer A. His-tagged protein was eluted with buffer A containing increasing concentrations of imidazole (10–250 mm). The fractions containing the His-tagged Pat or Pyl activities were flash-frozen in liquid nitrogen and kept at −80 °C.
Large Enzymatic Preparation of Substrates
Substrates for Pat reaction specificity included UDP-4-keto-6-deoxy-d-GlcNAc (i.e. UDP-4-keto-HexNac), UDP-4-keto-xylose (i.e. UDP-4-keto-d-pentose), UDP-4-keto-6-deoxy-d-Glc (i.e. UDP-4-keto-hexose), and UDP-4-keto-6-deoxy-l-AltNAc. These nucleotide sugars were prepared by the following enzymatic reactions: UDP-4-keto-6-deoxy-d-GlcNAc was obtained by incubating 10 mm UDP-GlcNAc in 50 mm Tris-HCl, pH 8.0, with 3 μg of purified recombinant UDP-GlcNAc 4,6-dehydratase from B. cereus (NC_004722.1). UDP-4-keto-d-xylose was obtained by incubating 5 mm NAD+ and 5 mm UDP-GlcA in 50 mm sodium phosphate, pH 7.6, with 1 μg of recombinant RsU4kpxs from Ralstonia solanacearum (10). UDP-4-keto-6-deoxy-d-Glc was obtained by incubating 5 mm NAD+ and 10 mm UDP-Glc in 50 mm sodium phosphate, pH 7.6, with 13 μg of purified recombinant UG4,6-Dh from Botryotinia fuckeliana (11). UDP-4-keto-6-deoxy-l-AltNAc was prepared by incubating 5 mm UDP-GlcNAc in 50 mm Tris-HCl, pH 7.6, with the combined 1.5 μg of purified recombinant-Pen (EAO54187.1) and 8.5 μg of purified recombinant-Pal (EAO54188.1) from Bacillus thuringiensis. Following enzymatic reactions, samples were separated by chromatography using anion exchange (10). In some cases, substrate and product were co-eluted as the case for UDP-GlcNAc and UDP-4-keto-6-deoxy-GlcNAc.
Pat and Pyl Enzymatic Assays and Product Analysis
The activity of recombinant UDP-4-keto-6-deoxy-HexNAc C-4″-aminotransferase (Pat) and UDP-4-amino-FucNAc 4–2OG transferase (Pyl) was examined by time-resolved 1H NMR spectroscopy and by HILIC-HPLC with UV or electrospray ionization mass spectrometry (ESI-MS). For HPLC-based assays, the total Pat aminotransferase reaction volume was 60 μl and included 50 mm Tris-HCl, pH 7.5, 0.7 mm UDP-4-keto-6-deoxy-HexNAc, 10 mm l-Glu, 0.1 mm pyridoxal phosphate (PLP), and 1.7 μg of purified Pat. Reactions continued for up to 3 h at 30 °C. The reaction was terminated by heating for 2 min at 95 °C. Chloroform was then added as described (12), and an aliquot (30 μl) of the upper layer phase was removed and mixed with acetonitrile (57 μl) and 0.5 m ammonium acetate, pH 4.35 (3 μl). A portion (10 μl) of this mixture was analyzed using HILIC chromatography coupled to a Shimadzu ESI-MS/MS. Assays were also analyzed by HPLC with UV diode array detection. HPLC-MS were carried out on the Shimadzu ESI-IT-TOF MS detector operated in the negative mode with a Nexera LC-30AD pump, autosampler (Sil30), and column heater (at 37 °C). HPLC with UV detection was carried out with an Agilent 1260 pump, autosampler, column heater (at 37 °C), and diode array detector. HPLC was performed using an Accucore 150-amide HILIC column (150 × 4.6 mm, 2.6 μm particle size, ThermoScientific) using a gradient system composed of 40 mm ammonium acetate, pH 4.35 (solvent A), and acetonitrile (solvent B). The column was equilibrated at 0.4 ml/min with 25% solvent A and 75% solvent B. After injection, chromatography conditions were as follows: from 0 to 2 min, flow rate 0.4 ml/min at 25% A, 75% B, followed by a 23-min gradient to 60% A, and then a 10-min gradient to 50% A. Subsequently, column flow rate was increased to 0.6 ml/min, and the column was washed for 10 min with 25% A, 75% B prior to the next injection. Peaks of enzymatic products detected by A261 (max for UDP-sugars) and A259 (max for ATP) were collected, lyophilized, and either suspended in 99.9% D2O for NMR analyses or in H2O for MS/MS analysis.
An HPLC-based assay monitoring the activity of recombinant Pyl ligase was carried out in a total volume of 67 μl. First, a Pat reaction (60 μl) was allowed to proceed for 3 h. Recombinant Pyl (1 μg), 2-oxoglutarate (1 mm, 2OG), ATP (1 mm), and MgCl2 (3 mm) were then added, and the reaction was allowed to proceed for 30 min at 37 °C. Pyl reactions were terminated and analyzed by HILIC chromatography.
For the NMR-based aminotransferase assay, a Pat reaction in a volume of 180 μl consisted of 120 μl of D2O and 60 μl of water-based reagents (50 mm Tris-HCl, pH 7.5, 0.7 mm UDP-4-keto-6-deoxy-HexNAc (UDP-4-keto-6-deoxy-GlcNAc), 10 mm l-Glu, 0.1 mm PLP, 0.1 mm 2,2-dimethyl-2-silapentane-5-sulfonate ((DSS) which served as internal NMR standard), and 5.1 μg of purified Pat). The reaction mixture was transferred to a 3-mm NMR tube, and the products were analyzed by 1H NMR spectroscopy for up to 3 h at 25 °C using a Varian DirectDrive 600-MHz spectrometer equipped with a cryogenic probe. For the NMR-based ligase assay, 3 μg of recombinant Pyl was added along with 1 mm 2OG, 1 mm ATP, and 3 mm MgCl2 to a 180-μl Pat reaction. The products were analyzed for up to 1 h at 25 °C in the 600-MHz spectrometer. Real time 1H NMR spectra were obtained using a 600-MHz spectrometer. Data were acquired before the addition of the enzyme as time 0 (t0). After adding the enzyme, acquisition was started after ∼3 min to allow the spectrometer operating parameters to be optimized. Sequential one-dimensional proton spectra with presaturation of the water resonance were acquired over the course of the reaction. All NMR spectra were referenced to the resonance of DSS set at 0.00 ppm. Processing of the data were performed with MestreNova (MestreLab Research).
NMR Analysis of Enzymatic Products
Individual enzyme reaction products were collected during HILIC HPLC. After lyophilization, the UDP-sugar products were dissolved in 100% D2O and characterized by NMR. Two-dimensional NMR spectra were obtained at room temperature on Varian INOVA 800 and 900 MHz spectrometers. Each purified UDP-sugar was identified using COSY (13), TOCSY (80-ms mixing time) (14), HSQC (15), HSQC-TOCSY (80-ms mixing time) (16), and HMBC (17) experiments.
Characterization and Kinetic Analyses of the Recombinant Enzymes Pat and Pyl
The activities of the recombinant enzymes were examined using buffers with different pH ranges, at different temperatures, and in the presence of potential inhibitors. To determine the optimal pH of Pat, a reaction volume of 50 μl was set and included 0.5 mm UDP-4-keto-6-deoxy-HexNAc (HPLC-purified; dissolved in 50 mm Tris-HCl between pH 7 and 9.5), 10 mm l-Glu, and 0.1 mm PLP. 1 μl Pat was then added, and the amount of product formed after 1 h at 30 °C was determined by HPLC. The optimal pH of Pyl was determined in a similar fashion using 0.5 mm UDP-amino-FucNAc (HPLC-purified) for 30 min at 37 °C. The activity of Pat and Pyl at pH 7.5 in Tris-HCl, phosphate, HEPES, and MOPS buffers was also compared. Assays (in triplicates) were performed for 1 h (Pat) or 30 min (Pyl) at 25, 30, 37, and 42 °C, and the amount of product formed was determined by HPLC. Inhibition assays were performed by first mixing the enzyme and buffer with various additives on ice for 10 min and then adding either UDP-4-keto-6-deoxy-HexNAc (for Pat) or UDP-4-amino-FucNAc (for Pyl). After each assay (1 h at 30 °C for Pat and 30 min at 37 °C for Pyl), the amounts of UDP-sugar formed and the amounts of substrate remaining were determined by HPLC.
To determine the kcat and Km values of Pat, the enzyme (7.43 nmol) in 50 mm Tris-HCl, pH 7.5, was reacted at 25 °C for 15 min with different concentrations of UDP-4-keto-6-deoxy-HexNAc (60, 120, 180, 240, 300, 360, 480, and 600 μm) in the presence of 10 mm l-glutamate and 0.1 mm PLP. Between 5 and 30 min, product formation is linear with respect to time. For Pyl kinetic studies, the recombinant enzyme (4.77 nmol) in 50 mm Tris-HCl, pH 8.0, was reacted at 25 °C for 20 min with different concentrations of UDP-4-amino-FucNAc (25, 50, 75, 100, 125, 150, 200, and 250 μm) in the presence of 1 mm ATP, 3 mm MgCl2, and 1 mm 2OG. The products formed were analyzed by HPLC. Initial velocities were fitted to the Michaelis-Menten equation using Prism (GraphPad software).
For acceptor and donor specificity studies, Pat (50 μl) was reacted with the UDP-4-keto-sugar (∼0.5 mm), l-Glu (10 mm), and PLP (0.1 mm) as a control and with several donors (10 mm each l-alanine, l-glutamine, citrulline, GABA, and l-tryptophan) for 3 h at 30 °C. Pyl reactions (50 μl) were performed for 1 h at 37 °C with ∼0.5 mm UDP-4-amino-6-deoxy-HexNAc with 1 mm ATP, 3 mm MgCl2, and 1 mm 2OG as a control and with various additives (1 mm each isocitric acid, dl-malic acid, malonic acid, oxalate, oxaloacetic acid, pyruvic acid, and succinic acid). After each enzyme reaction, the amounts of product formed and the amounts of substrate remaining were determined by HPLC. We indicated as nondetected if a product was not shown (under these assay conditions) by the UV-HPLC or by LC-MS/MS.
RESULTS
Identification of Pat and Pyl from B. cereus ATCC 14579
We postulated two biosynthetic mechanisms that could be used to link FucNAc to galactose via 2-oxoglutarate in the polysaccharide produced by B. cereus ATCC 14579. The first requires an enzyme that catalyzes the transfer of 2-oxoglutarate to FucNAc during polysaccharide synthesis. The second route, which we provide evidence for here, involves the formation of an activated 2-oxoglutarate-4-amino-sugar-nucleotide. A specific glycosyltransferase likely then catalyzes the transfer of the 4-amido-2OG sugar to an oligosaccharide acceptor. We suspected that genes involved in the formation of such an activated nucleotide sugar would likely reside in an operon that contains at least one gene that encodes an enzyme with C4″-aminotransferase activity. This led us to identify B. cereus ATCC 14579 Bc5273 (which we named Pat) as a candidate even though this protein has relatively low amino acid sequence homology with other aminotransferases. For example, the B. cereus ATCC 14579 Pat protein has 33% amino acid sequence identity with the UDP-4-amino-6-deoxy-arabinose aminotransferase from Burkholderia cenocepacia and E. coli ArnB (18, 19) and 31% sequence identity with the GDP-perosamine synthase of Caulobacter crescentus (20). B. cereus ATCC 14579 Pat also has 33% amino acid identity with a 4″-aminotransferase from Campylobacter jejuni, but the end product sugar has an l-configuration (rather than d-configuration), and the 6-deoxy-HexNac sugar has a 5-epimer configuration (l-AltNAc). Despite the low protein sequence identity for aminotransferase, we noticed that the adjacent gene, Bc5272 (that we named Pyl), encodes a protein annotated as a carbamoyl-phosphate synthase small subunit. Pyl has low amino acid sequence identity with a few members of the ATP-Grasp protein family. Some of these proteins are involved in carboxylate-amine ligase reactions. For example, the biotin carboxylase from C. jejuni (21) shares 25% amino acid sequence identity with Pyl. Albeit the low amino acid sequence identity of Pat and Pyl for known aminotransferase and ATP-Grasp proteins, respectively, we decided to clone and express these genes in E. coli and examine the catalytic activities of the recombinant proteins.
Pat Encodes a l-Glu:UDP-4-keto-6-deoxy-HexNAc C″-4-Aminotransferase
E. coli cells expressing recombinant Pat (Bc5273-His6) were used to isolate and purify the recombinant protein using a nickel affinity column (Fig. 2a, E6 and E7). The purified Pat (calculated 45.8 kDa) was initially used to determine the specific activity by a UV-HPLC and mass spectrometry-based assay, and the enzymatic products were finally characterized by NMR. HILIC analysis of the products formed when purified recombinant Pat was reacted with UDP-4-keto-6-deoxy-HexNAc in the presence of l-glutamate and PLP showed the appearance of a new peak with a retention time of 17.5 min (Fig. 3a, panel 2). This peak was not detected in a reaction lacking l-glutamate (Fig. 3a, panel 3). When the Pat enzymatic reactions were chromatographed and analyzed in the negative mode by ESI-MS, the new peak gave a major ion at m/z 589.05 (Fig. 3b, panel 2), which likely corresponds to [M − H]− for a UDP-amino-6-deoxy-HexNAc. MS-MS analysis of this parent ion gave fragment ions at m/z 402.968 and 323.007 that are consistent with UDP and UMP, respectively. The neutral loss of 186.08 mass units implies a mass for a 6-deoxy-HexNac-amino-sugar. The results of these initial analyses led us to suspect that the newly formed product is a UDP-4-amino-6-deoxy-HexNac. However, the chirality of the 4-amino group (gluco- or galacto-configuration) could not be determined by MS analyses. Therefore, the peak was collected and analyzed by NMR spectroscopy. One-dimensional proton NMR analysis and chemical shift assignments (Table 1) indicated that the enzymatic product is UDP-4-amino-d-FucNAc (Fig. 4). The 4-amino-FucNAc H1″ anomeric region of the proton spectrum contains a quadruplet signal with chemical shifts of 5.52 ppm. The distinct chemical shift of the anomeric proton and the coupling constant values of 3.8 Hz for JH1″, H2″ and 7.2 Hz for JH1″, P are consistent with an α-linkage to the phosphate of UDP. The chemical shifts for each H6″ had a value of 1.31 ppm that is distinct from the C-6″ methyl protons (1.23 ppm) of the initial substrate. The methyl proton resonance of the N-acetylated group (C-2″-NAc) at 2.07 ppm is consistent for a C2 acetamido moiety of the product. The d-configuration of the linked FucNAc was established based on coupling constants. An l-sugar would have larger coupling between α-phosphate and the H1″ proton and also a larger coupling between H2″ and H3″ (22). Further support for the presence of UDP-4-amino-d-FucNAC product was established using two-dimensional NMR experiments. COSY and TOCSY experiments confirmed the assignments for H2″, H3″, H4″, and H5″ in this 4-amino-sugar. The JH2″, H3″ coupling constant of 10 Hz and the JH3″, H4″ coupling constant of 3.8 Hz is consistent with a galacto-configuration. 13C HSQC and HSQC-TOCSY experiments established carbon assignments and the presence of an N-acetylated carbon.
FIGURE 2.
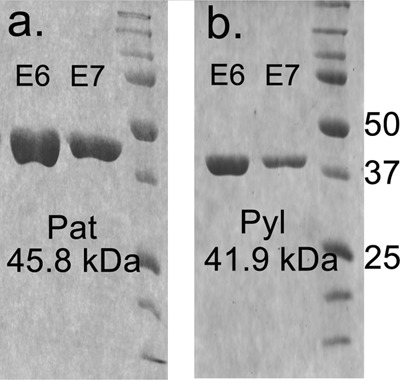
SDS-PAGE analysis of the B. cereus ATCC 14579 purified recombinant Pat and Pyl proteins involved in the biosynthesis of UDP-Yelosamine. a, protein standards are shown on the right in kDa. 1st and 2nd lanes, final elution (E6 and E7) of His6Bc5273 (Pat) are from the affinity column. b, 1st and 2nd lanes, final elution (E6 and E7) of His6Bc5272 (Pyl) are from the affinity column.
FIGURE 3.
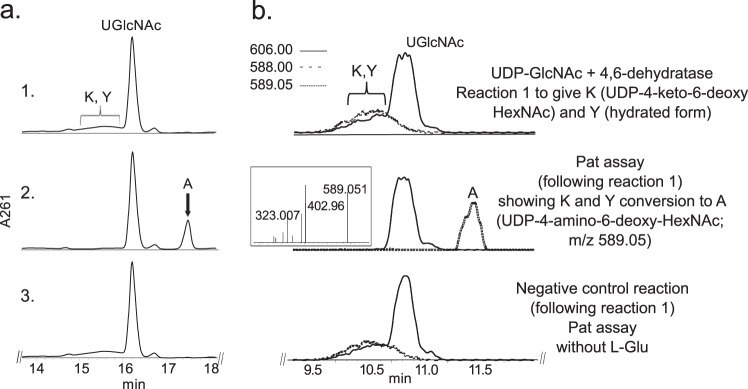
Analysis of Pat reaction by UV-HPLC and LC-ESI-MS-MS. UDP-GlcNAc 4,6-dehydratase reaction 1 (see panel 1 in a (UV) and b (ms)), is the conversion of UDP-GlcNAc to UDP-4-keto-6-deoxy-HexNAc. Note the broad peak (K and Y) denoting 4-keto and 4-hydrated-keto form of the UDP-4-keto-6-deoxy-sugar with m/z 588 and 606, respectively. Following reaction 1, purified Pat was added (panel 2 in a (UV) and b (ms)) and reacted with UDP-4-keto-6-deoxy-HexNAc in the presence of l-glutamate and PLP to give product A, UDP-amino-sugar, with m/z 589 and ms/ms of 402 and 323 (b, boxed inset in panel 2). Pat negative control reaction carried out without l-glutamate (panel 3 in a and b). The Pat enzymatic reactions in a and b were separated on different HILIC columns, and products were detected by UV (a) or by ESI-MS and MS/MS (b).
TABLE 1.
1H chemical shifts of UDP-4-amino-d-FucNAc obtained by NMR spectroscopic analysis of the purified reaction product in D2O
Chemical shifts were measured at 800 MHz at 25 °C and referenced to DSS. (Note: q means quadruplet, “m” as multiplet, “dd” as doublet of doublet, and “s” as singlet.)
| Proton |
|||||||
|---|---|---|---|---|---|---|---|
| H1 | H2 | H3 | H4 | H5 | H6 | NAc | |
| UDP-4-amino-FucNAc | |||||||
| Chemical shift (ppm), peak multiplicity | 5.52 (q) | 4.07(m) | 4.26(dd) | 3.66(m) | 4.56(q) | 1.31(d) | 2.07(s) |
| J coupling constants (Hz) | J1,p = 7.2 J1,2 = 3.8 | J2,3 = 10 | J3,4 = 3.8 | J4,5 = 1 | J5,6 = 6.6 | ||
| Ribose | |||||||
| Chemical shift (ppm), peak multiplicity | 5.98(d) | 4.36 | 4.35 | 4.27 | 4.24,4.18(d) | ||
| J coupling constant (Hz) | J1,2 = 3.9 | J5,5′ = 12 | |||||
| Uracil | |||||||
| Chemical shift (ppm) peak multiplicity | 5.97(d) | 7.96(d) | |||||
| J coupling constant (Hz) | J5,6 = 7.9 | ||||||
FIGURE 4.

Analysis of Pat C4″-aminotransferase reaction products by 1H NMR indicates formation of UDP-4-amino-d-FucNAc. The product of the Pat reaction (peak A, marked by an arrow in Fig. 3a, panel 2) was collected and analyzed at 800 MHz NMR. a, full proton spectrum of the Pat enzyme product UDP-4-amino-d-FucNAc, A. b, expanded proton spectra between 3.6 and 4.6 ppm that shows the FucNAc-4N sugar ring. The short line above NMR “peaks” denotes specific chemical shifts belonging to a UDP-4-amino-FucNAc. * denotes column contamination, and # denotes DSS.
We next used time-resolved 1H NMR to monitor the conversion of UDP-GlcNAc to UDP-4-keto-6-deoxy-GlcNAc by UDP-GlcNAc 4,6-dehydratase, followed by the C4″- aminotransferase (Pat) reaction (Fig. 5). The dehydratase reaction generates UDP-4-keto-6-deoxy-GlcNAc (K in Fig. 5) and its C4″-hydrated counterpart (Y in Fig. 5). The ratio of the 4-keto to the 4-hydrated form is 1:9 at steady state. When the recombinant Pat C4″-aminotransferase is added, additional signals (AH 1″) corresponding to the anomeric proton of UDP-4-amino-FucNAc appear and are accompanied by a decrease in the intensity of the signals corresponding to the anomeric protons of the keto (KH-1″) and the hydrate (YH-1″). The 6-deoxy proton signal (YH-6″) also decreases, whereas the signal (AH-6″) assigned to the newly formed C4″-amino-sugar increases. Time-resolved NMR (see Fig. 6) also detected the 2OG product that was generated when the amine of l-glutamate was transferred to the C4″-position of the sugar. The H3 and H4 protons of 2OG have distinct chemical shifts of 2.98 and 2.42 ppm, respectively. Together, these data confirm that Pat encodes a C-4″ aminotransferase that transfers the amino group of l-glutamate to UDP-4-keto-6-deoxy-HexNAc to form UDP-4″-amino-d-FucNAc (Fig. 1).
FIGURE 5.

Time-resolved 1H NMR analysis of Pat reaction products showing the conversion of both hydrated and 4-keto-UDP-sugar to UDP-4-amino-FucNAc sugar. Spectra were collected for the first 60 min of the reaction that was conducted at 25 °C and included Pat, UDP-4-keto-6-deoxy-HexNAc (keto and hydrated forms, K and Y), l-Glu, and PLP, after 4,6-dehydratase reaction (time 0). Two selected regions of the UDP-4-amino-FucNAc formed over time can be observed with a diagnostic H-6″ (a) and the anomeric proton (b). Proton signals of UDP-4-keto-6-deoxy-HexNAc and product, UDP-4-amino-FucNAc, are labeled as KH-6″ and KH-1″ for the keto form, as YH-6″ and YH-1″ for the hydrated form, and as AH-6″ and AH-1″, respectively. Note the chemical shifts of anomeric protons for G, UDP-GlcNAc, and A, UDP-amino-FucNAc, are very close to each other (slightly shifted), but the methyl protons AH-6″ of the product has a distinct chemical shift when compared with the substrate YH-6″.
FIGURE 6.
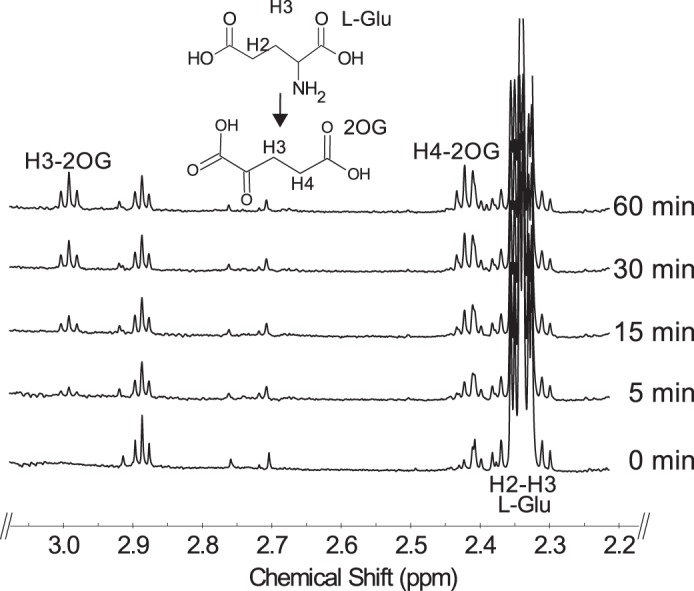
Time-resolved 1H NMR analysis of Pat reaction showing the conversion of l-glutamate to 2-oxoglutarate. Pat reaction at 25 °C included UDP-4-keto-6-deoxy-HexNAc (keto and hydrated forms, K and Y, respectively), l-Glu, and PLP. Selected spectral region for the diagnostic H3 and H4 of the product 2OG formed over the reaction time is shown.
Pyl Encodes 2OG:UDP-4-Amino-FucNAc 4″N-2OG Transferase and Forms UDP-Yelosamine
To ascertain the function of Bc5272 (Pyl), E. coli cells expressing the gene were used to isolate and purify the recombinant protein (Fig. 2b, E6 and E7; 41.9 kDa). During the initial characterization of the Pyl protein, we determined that its activity required magnesium, ATP, 2OG, and UDP-4-amino-sugar, as no activity was observed without ATP or with UDP-4-keto-sugars or UDP-6-deoxy-sugars. As shown in Fig. 7a (panel 2) purified recombinant Pyl in the presence of ATP, MgCl2, and 2OG readily converted UDP-4-amino-FucNAc to a new UV peak migrating on a HILIC column with a retention time of 19 min (Fig. 7a, panel 2). This peak was not formed in the absence of added ATP (Fig. 7a, panel 3). Negative ion LC-ESI-MS analyses of the new product gave a major ion at m/z 717 (Fig. 7b, panel 2), suggesting the 2OG moiety is linked to the UDP-4-amino-sugar. MS-MS analysis of this ion gave ion fragments at m/z 402.968 and 304.996 consistent with UDP and UMP [UMP − H2O − H]−, respectively. The neutral loss of 314.03 mass units suggested that the 2OG is linked to the 4-amino group of the sugar nucleotide. One- (Fig. 8) and two-dimensional NMR spectroscopic analyses of the isolated product provided compelling evidence that the enzyme formed UDP-d-FucNAc-4N-(2)-oxoglutarate (herein referred as UDP-Yelosamine). The chemical shift assignments for UDP-Yelosamine are summarized in Table 2. NMR signals characteristic of 2OG 4-N-linked UDP-FucNAc are the terminal COOH (13C at 176.01ppm), the CONH (13C at 178.9 ppm), and the hydrogens (1H at 2.69 and 3.09 ppm) linked to C3 and to C4 (13C of 31.48 and 36.81 ppm) of 2OG, respectively. An HMBC experiment identified heteronuclear coupling between the H4 (4.32 ppm) of the sugar and the carbonyl (178.9 ppm) of the 2OG residue and provides unambiguous evidence for the 4″-N-acyl linkage that was formed by the carboxylate-amine ligation reaction (Fig. 9). NMR signals at 52.47 and 55.98 ppm for C2 and C4 of the sugar moiety of UDP-Yelosamine established that they were substituted by nitrogen atoms. A 1H signal at 2.06 ppm indicated that the C2 position of the sugar was N-acetylated. The JH2″, H3″ and JH3″, H4″ coupling constants of 10 and 3.8 Hz, respectively, are consistent with a galacto sugar. The chemical shift of 5.51 ppm of the anomeric proton of the Yelosamine residue and JH1″, P and JH1″, H2″ coupling constants of 7.2 and 3.8 Hz, respectively, are consistent with an α-linkage. These data established that the product is UDP-2,4,6-trideoxy-d-GalNac-4-amido-(2)-oxoglutarate (abbreviated UDP-Yelosamine). Collectively, we concluded that Pyl encodes an enzyme that ligates a carboxylate of 2OG to the 4-amino group of a UDP-sugar to form UDP-Yelosamine.
FIGURE 7.

Analysis of Pyl reaction by UV-HPLC and LC-ESI-MS-MS. Pat reaction 2 *see panel 1 in a (UV) and b (ms)), gives A, UDP-4-amino-FucNAc, with m/z 589 (b, panel 1). Following reaction 2, purified Pyl was added (2 in a (UV) and, (ms)) and reacted in the presence of ATP, MgCl2, and 2-oxoglutarate to convert A to F-OG, UDP-FucNAc-4N-2OG (UDP-Yelosamine), with parent ion m/z 717 and ms/ms ion fragments of 402.9 and 304.9 (b, boxed inset in panel 2). Pyl negative control reaction was carried out without ATP (panel 3 in a and b). The Pyl enzymatic reactions in a and b were separated on different HILIC columns, and products were detected by UV (a) or by ESI-MS and MS/MS (b). ATP was added to a, panel 1, as standard to view elution time by UV-HPLC.
FIGURE 8.
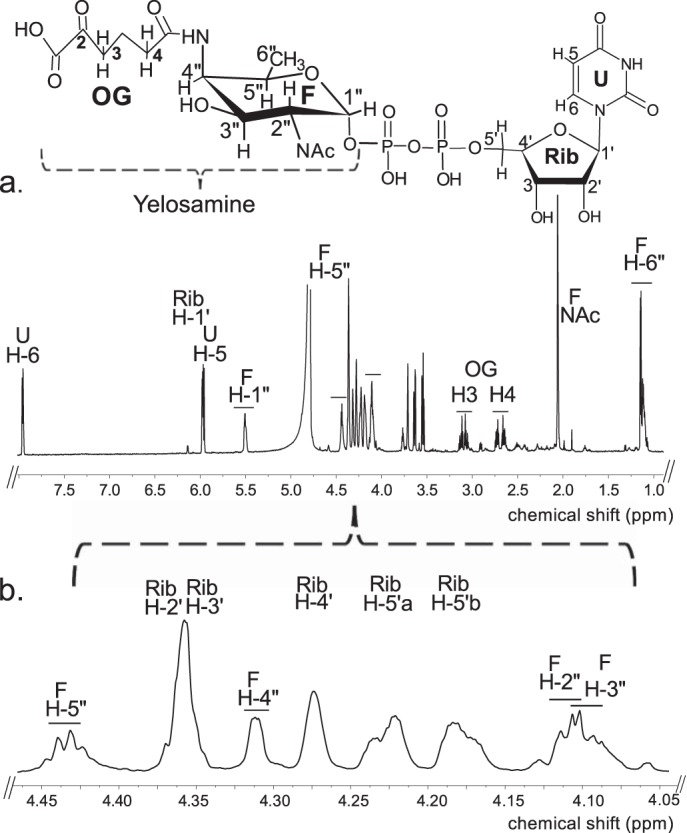
Analysis of Pyl reaction products by 1H NMR indicates formation of UDP-Yelosamine. The product of the Pyl reaction (peak F-OG, marked by an arrow in Fig. 7a, panel 2) was collected and analyzed at 800 MHz NMR. a, full proton spectrum of HPLC-collected product. b, expanded proton spectra between 4.05 and 4.46 ppm that shows the Yelosamine sugar ring. The short line above NMR peaks denotes specific chemical shifts belonging to a UDP-Yelosamine.
TABLE 2.
1H chemical shifts of UDP-Yelosamine obtained by NMR spectroscopic analysis of the purified reaction product in D2O
Chemical shifts were measured at 800 MHz at 25 °C and referenced to DSS. (Note: q means quadruplet, “m” as multiplet, “dd” as doublet of doublet, and “s” as singlet.)
| Proton |
|||||||
|---|---|---|---|---|---|---|---|
| H1 | H2 | H3 | H4 | H5 | H6 | NAc | |
| UDP-Yelosamine | |||||||
| Chemical shift (ppm), peak multiplicity | 5.51 (q) | 4.11(m) | 4.10(dd) | 4.32(m) | 4.44(q) | 1.13(d) | 2.06(s) |
| J coupling constants (Hz) | J1,p = 7.2 J1,2 = 3.8 | J2,3 = 10 | J3,4 = 3.8 | J4,5 = 1 | J5,6 = 6.6 | ||
| 2OG | |||||||
| Chemical shift (ppm), peak multiplicity | 3.09(m) | 2.69(m) | |||||
| Ribose | |||||||
| Chemical shift (ppm), peak multiplicity | 5.98(d) | 4.36 | 4.35 | 4.27 | 4.24,4.18(d) | ||
| J coupling constant (Hz) | J1,2 = 3.9 | J5,5′ = 12 | |||||
| Uracil | |||||||
| Chemical shift (ppm) peak multiplicity | 5.97(d) | 7.96(d) | |||||
| J coupling constant (Hz) | J5,6 = 7.9 | ||||||
FIGURE 9.
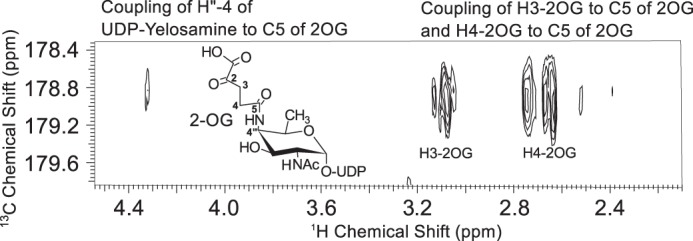
HMBC NMR spectrum of UDP-Yelosamine confirms the linkage between 2OG and the UDP-4-amino-FucNAc. H-4″ of UDP-Yelosamine is coupled to carboxylic carbon C5 of 2OG. Couplings between H3 and C5 of 2OG and between H4 and C5 of 2OG are also shown in the spectrum.
Time-resolved 1H NMR used to monitor the reaction progress of Pyl is shown in Fig. 10. As time progresses, the peak corresponding to the anomeric proton (AH-1″) of the UDP-4-amino-sugar substrate is decreased, whereas peaks of the enzymatic product, UDP-FucNAc-4N-2OG (FH-1″), are increased. The distinct chemical shifts for the proton signals belonging to the methyl group of the substrate and product are also an indication of the enzymatic reaction's progress with the 6-deoxy (AH-6″) substrate peak decreasing and the product methyl peak (FH-6″) of the amino sugar increasing (Fig. 10b). As the 2OG carboxylate moiety is ligated via the 4-amino group of UDP-4-amino-FucNAc, a distinct and diagnostic proton signal is observed in the time-resolved NMR experiment. The proton signals of H3 and H4 of FucNAc-2OG appear around 3.1 and 2.7 ppm, respectively (Fig. 11). Finally, members of the ATP-Grasp proteins are believed to carry out a reaction in the presence of ATP. Pyl requires ATP for 2OG ligase reaction, and it can be observed in the real time NMR-based assay as well. The proton signal of ATP around 8.52 ppm decreases, whereas the signal of ADP around 8.54 ppm increases (Fig. 12), suggesting the transfer of the γ-phosphate group of ATP to the 2OG to yield a 2OG-phosphate intermediate, as proposed in Fig. 13.
FIGURE 10.
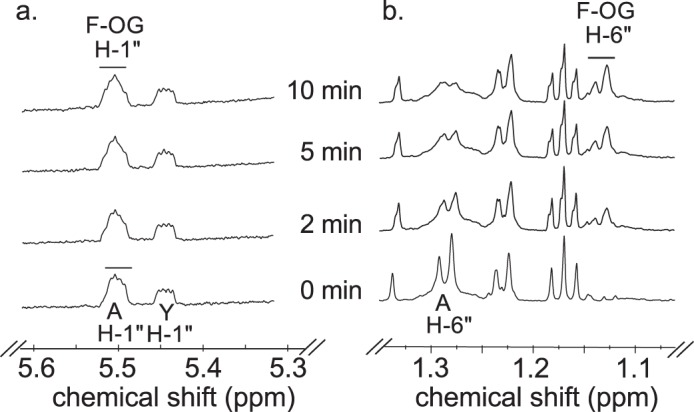
Time-resolved 1H NMR analysis of Pyl reaction products showing the conversion of UDP-4-amino-FucNAc to UDP-Yelosamine (F-OG). Spectra were collected for the first 10 min of the reaction that was carried out at 25 °C and included Pyl, A (UDP-4-amino-FucNAc), ATP, MgCl2, and 2-oxoglutarate after Pat reaction. Two selected regions of the UDP-Yelosamine formed over time can be observed with a diagnostic anomeric proton product (F-OG H-1″, a) and the diagnostic H-6″ (b). Proton signals of UDP-4-amino-FucNAc are labeled as AH-6″ and AH-1″. Note that the chemical shift of the anomeric proton of UDP-4-amino-FucNAc (AH-1″) is very close to the proton of UDP-Yelosamine (F-OG H-1″) (slightly shifted). YH-1″ is the hydrated form of UDP-4-keto-6-deoxy-GlcNAC.
FIGURE 11.
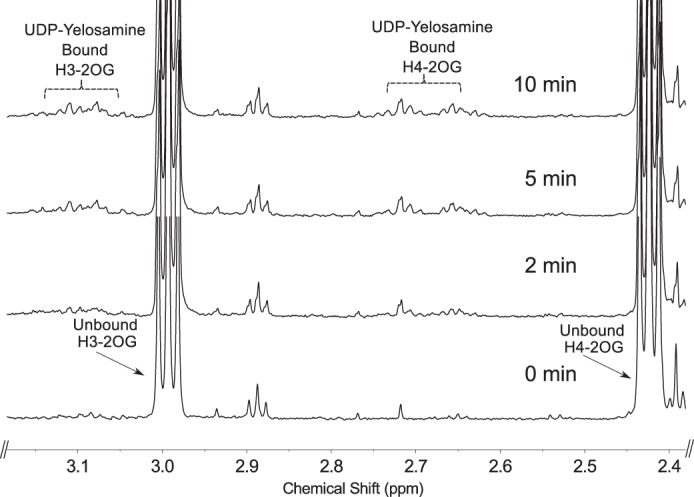
Time-resolved 1H NMR analysis of Pyl reaction showing the conversion of unbound 2OG (i.e. unreacted substrate) to UDP-Yelosamine-bound 2OG (i.e. 2OG-ligated to the 4-amino-UDP-sugar). Following Pat reaction (time 0), Pyl was added with ATP, MgCl2, and 2-oxoglutarate, and reaction was carried out for 10 min at 25 °C. A chemical shift change of H3 and H4 of 2OG is shown due to attachment to the 4-amino group of the FucNAc-4N of UDP-Yelosamine.
FIGURE 12.
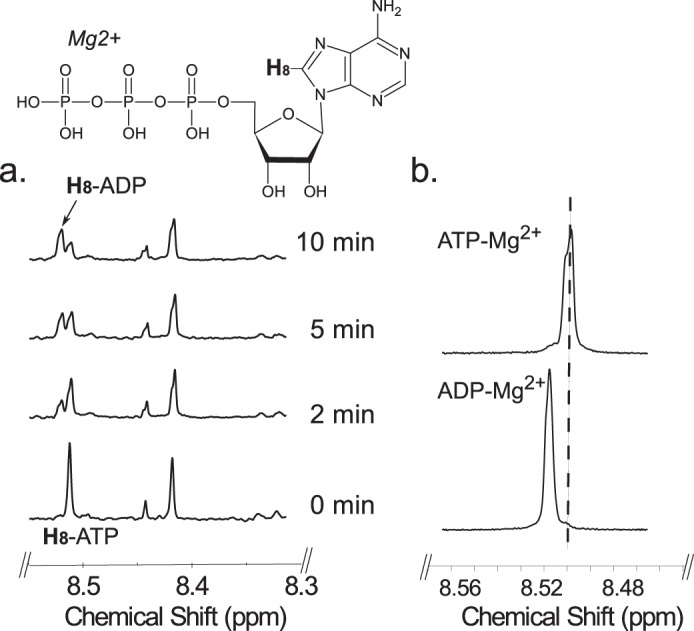
Time-resolved 1H NMR analysis of Pyl reaction showing the conversion of ATP to ADP. Following Pat reaction (time 0), Pyl was added with ATP, MgCl2, and 2-oxoglutarate, and the reaction was carried out for 10 min at 25 °C. a, the H-8 of adenosine ring of ATP change its chemical shift from 8.52 to 8.54 ppm due to formation of ADP over time during Pyl reaction. b, 1H NMR spectra of standard ADP or ATP mixed in reaction buffer containing Mg(II) shows a chemical shift for H-8.
FIGURE 13.

Proposed Pyl enzymatic reaction to form UDP-Yelosamine. Conversion of ATP, 2OG, and UDP-4-amino-FucNAc to UDP-Yelosamine by Pyl. The carboxylate group of 2OG attacks (step 1) the γ-phosphate of ATP, forming an activated intermediate (2OG-phosphate) and ADP. The 4″-amino group of UDP-amino-FucNAc attacks the “activated” C-5 carbonyl moiety attached to a 2OG-phosphate intermediate (step 4). This yields presumably an unstable tetrahedral intermediate that is collapsed, releasing UDP-Yelosamine, a phosphate, and ADP.
Selected Enzymatic Properties of Pat and Pyl
The recombinant Pat C4″-aminotransferase had its highest activity between 25 and 30 °C and between pH 7 and 8.5 irrespective of the buffer used. Similar pH and temperature profiles were observed for Pyl, the ATP-dependent 2OG 4″-amino-sugar ligase. Kinetics parameters for the recombinant Pat and Pyl activities are summarized in Table 3. Recombinant Pat eluted from a Superdex 75 size-exclusion column in the region for a protein with a mass of 90,000 Da, suggesting that the enzyme is active as a dimer. By contrast, recombinant ATP-Grasp Pyl eluted from the same column in the region for a protein with mass of 157,500 Da, implying this enzyme is active predominantly as a trimer-tetramer. Further analyses have shown that Pat requires PLP to convert UDP-4-keto-6-deoxy-HexNAc to UDP-4-amino-FucNAc, and Pyl requires ATP and Mg2+ or Mn2+ to convert UDP-4-amino-FucNAc to UDP-Yelosamine. To test the specificity of Pat, we enzymatically generated several different UDP-4-keto-sugar substrates, including UDP-4-keto-d-xylose, UDP-4-keto-6-deoxy-d-glucose, and UDP-4-keto-6-deoxy-l-AltNAc. LC-MS/MS analysis provided clear evidence that all of these UDP-4-keto-sugars are substrates (see scheme in Fig. 14). In addition, several compounds that are structural analogs of l-glutamate were tested as a possible substrate for Pat activity. Pat appears specific to l-Glu, and marginal activity (less than 15%) was observed with l-alanine and l-glutamine, but no Pal activity was detected with γ-aminobutyric acid (GABA) or l-tryptophan. We were unable to determine whether co-factors other than PLP can be used in the 4-aminotransferase reaction because PLP had to be added to the recombinant protein to maintain its activity during purification and storage.
TABLE 3.
Enzymatic properties of recombinant l-Glu:UDP-4-keto-6-deoxy-HexNAc C″-4-aminotransferase (Pat, Bc5273-His6) and 2OG:UDP-4-amino-FucNAc 4N″-transferase (Pyl, Bc5272-His6)
| Pat | Pyl | |
|---|---|---|
| Optimal pHa | 7.0–8.5 | 7.5–8.5 |
| Optimal temperaturea | 25–30 | 25–37 |
| Km (μm)b | 0.353 ± 0.07 | 0.243 ± 0.05 |
| Vmax (μm min−1) | 22.96 ± 2.3 | 7.03 ± 0.8 |
| kcat (min−1) | 3.09 ± 0.3 | 1.47 ± 0.1 |
| kcat/Km (μm−1 min−1) | 8.77 ± 0.92 | 6.07 ± 0.87 |
| Protein mass (kDa) | 45.8 | 41.9 |
a Optimal pH and temperature assays were determined using Tris-HCl. Virtually no differences in activity (± 5%) were observed using phosphate buffer, MOPS, HEPES at pH 7.5.
b The reaction was determined by HPLC-UV after 15 min at 25 °C incubation for Bc5273 (Pat) and 20 min at 25 °C incubation for Bc5272 (Pyl). Km values for both reactions are for the UDP-sugars. For Pat assays, the reaction consisted of various concentrations of UDP-4-keto-6-deoxy-HexNAc (60, 120, 180, 240, 300, 360, 480, and 600 μm) with fixed amounts of co-factor (10 mm l-glutamate, 0.1 mm PLP, and 0.34 μg of recombinant Pat. For Pyl assays, the reaction included various concentrations of UDP-4-amino-FucNAc (25, 50, 75, 100, 125, 150, 200, and 250 μm) with fixed amounts of co-factor (1 mm ATP, 3 mm MgCl2, 1 mm 2OG, and 0.2 μg of recombinant Pyl).
FIGURE 14.

Various UDP-4-keto-sugars are substrates for Pat and subsequently for Pyl. Pat has a C4″-aminotransferase activity and converts UDP-4-keto-6-deoxy-d-glucose to UDP-4-amino-d-fucose with m/z 676.04 (a); UDP-4-keto-d-xylose to UDP-4-amino-arabinose with m/z 662.03 (b); and UDP-4-keto-6-deoxy-l-AltNAc to UDP-4-amino-6-deoxy-l-AltNAc with m/z 717.01 (c). Subsequently, Pyl ligates the 2OG to these UDP-4-amino sugars. We named the various 2OG derivatives amido-linked to UDP-4-amino-sugars the following: UDP-Yelose, UDP-Aravonose, and UDP-Solosamine. MS value for final 2OG-products is shown (box).
Our data also suggest that Pyl, the 2OG-4-amino-ligase encoded by Bc5272, can ligate 2OG to different 4-amino-sugars linked to UDP (Fig. 14). MS/MS analyses of these UDP-4-amino-sugars indicate that Pyl can form UDP-Yelose (UDP-d-fucose-4N-2OG), UDP-Aravonose (UDP-arabinose-4N-2OG), and UDP-Solosamine (UDP-l-AltNAc-4N-2OG). Further substrate specificity studies showed no discernible Pyl activity when the enzyme was reacting with structural analogs of 2OG, including isocitric acid, malic acid, malonic acid, ammonium oxalate, oxaloacetic acid, pyruvic acid, or succinic acid. Therefore, we concluded that recombinant Pyl is specific for 2OG but promiscuous with UDP-4-amino-sugars. Inhibition studies showed that l-Glu analogs, including l-alanine, citrulline, GABA, l-glutamine, and l-tryptophan, reduced recombinant Pat activity by 29, 26, 24, 23, and 13%, respectively. Co-factor analogs, including pyridoxal, pyridoxal amine, and pyridoxine, reduced Pat activity by 31, 18, and 0%, respectively. Similar inhibition studies were carried out with Pyl. Oxalate, isocitric acid, oxaloacetic acid, succinic acid, malonic acid, dl-malic acid, and pyruvic acid, which were tested as 2OG analogs, inhibited Pyl activity by 46, 39, 32, 27, 21, 11, and 2%, respectively. Kinetic analyses of the recombinant Pat and Pyl activities are summarized in Table 3. The apparent Km values were 0.353 and 0.243 μm for UDP-sugar substrates; the Vmax values were 22.96 and 7.03 μm min−1, and the kcat/Km values were 8.77 and 6.07 μm−1min−1 with Pat and Pyl, respectively.
DISCUSSION
In this study, we have identified two genes (Pat and Pyl, Bc5273 and Bc5272) in B. cereus ATCC 14579 that encode the enzymes capable of converting UDP-4-keto-d-GlcNAc to UDP-4-amino-FucNAc and then to UDP-Yelosamine (UDP-FucNAc-4N-2OG, Fig. 1). This pathway is initiated by a UDP-GlcNAc 4,6-dehydratase that converts UDP-GlcNAc to UDP-4-keto-6-deoxy-GlcNAc. Pat, a C4″-aminotransferase, in the presence of PLP, then transfers an amino group from l-glutamate to form UDP-4-amino-FucNAc and 2OG. Subsequently, the ATP and Mg(II)-dependent Pyl ligase (encoded by Bc5272) mediates the attachment of 2OG to the 4-amino moiety of UDP-sugar-4N to yield UDP-Yelosamine. This ATP dependence is specific, and no other nucleotide (TTP, CTP, and GTP) can be substituted for the 2OG transfer reaction. Time-resolved NMR analysis of this enzymatic reaction (Fig. 12) clearly shows the conversion of ATP to ADP and together with additional NMR and MS/MS analyses show that 2OG and UDP-FucNAc-4N must also be present to form ADP. Mn(II) can substitute for Mg(II), but its paramagnetic properties preclude analyses of the products by NMR. The ability of recombinant Pyl to form an amide bond between the carboxylate of 2OG and the 4-amino moiety of UDP-FucNAc-4N in an ATP-dependent fashion together with the release of ADP resembles the activities reported for several enzymes of the ATP-Grasp superfamily proteins. Hence, we proposed the following Pyl reaction mechanism (see Fig. 13). In step 1, the carboxylate moiety of 2OG acts as a nucleophile and attacks the γ-phosphate of ATP, yielding an activated intermediate (2OG-phosphate) and ADP, which are both bound to the enzyme. ADP is not yet released nor is the 2OG-phosphate intermediate. In the following steps, the 4-amino moiety (H3N+) of the UDP-sugar-4N molecule is deprotonated and acts as a nucleophile (H2N:) to attack the activated C-5 carbonyl moiety attached to a 2OG-phosphate intermediate. This presumably yields an unstable tetrahedral intermediate that is collapsed, releasing UDP-Yelosamine, a phosphate, and ADP. Although the Pat and Pyl enzyme pair was shown to utilize UDP-GlcNAc, the substrate specificity study shows that these recombinant enzymes can similarly modify other NDP-4-keto-sugars (Fig. 14), including UDP-sugars where the sugar ring lacks the 2-NAc moiety or where the sugar ring is in an l-configuration. This promiscuity may allow B. cereus to link other sugars together by organic acid and may allow the microbe to survive in different environmental niches. Such promiscuity may also facilitate the bacterial transition from one developmental stage to another.
Based on Pyl's ATP-dependent enzymatic function and despite the low amino acid sequence identity to other known enzymes, we suggest that this Bacillus protein belongs to the ATP-Grasp protein family. ATP-Grasp enzymes have been proposed to have a conserved ATP-binding fold domain based on studies of the crystal structure of an E. coli d-alanine:d-alanine ligase (23–25). Specific amino acid sequences likely mediate binding to the adenosine and ribose moieties of ATP, whereas cations, including Mg(II), are likely involved in binding to the ATP phosphates. The amino acids involved in these binding sites are somewhat conserved across members of the ATP-Grasp family proteins, although the proteins themselves typically have low amino acid sequence identities (9, 26, 27). Pyl has ∼20% amino acid sequence similarity with the dipeptide l-amino acid ligase from Bacillus licheniformis (8), the biotin carboxylase from Pseudomonas aeruginosa (28), the ATPase from Paracoccus denitrificans (29), and the d-alanyl-d-lactate ligase from Enterococcus faecium BM4147 (21). Nevertheless, these ATP-Grasp proteins and Pyl appear to have conserved amino acid sequences involved in ATP binding. The predicted amino acid of Pyl involved in ATP binding, based on sequence similarity, is shown (Fig. 15). The ATP-Grasp superfamily currently includes 17 groups of enzymes (30). Pyl likely belongs to ATP-Grasp family 4. This family includes diverse ligation activities with a wide array of substrates. A search of currently available bacterial genomic sequence data identified only a few species that carry a gene encoding a protein with high amino acid sequence similarity to Pyl (Bc5272). Homologs of Pyl were found in B. cereus Rock4-2 and AH676 (see Fig. 16). Additional homologs exist in Vagococcus lutrae, Helcococcus kunzii ATCC 51366, Parabacteroides distasonis CL09T03C24, Geobacillus caldoxylosilyticus NBRC 107762, and Bacteroides cellulosilyticus. Pyl-like proteins were also found in Staphylococcus carnosus, Clostridium difficile, Enterococcus sp., Erysipelotrichaceae sp., and Pseudomonas sp. M1. Further studies with Pyl homologs are needed to confirm the repertoire of sugar-nucleotides used by these types of enzymes. Interestingly, the pilin protein is known to be glycosylated with di-N-acetyl bacillosamine modified with glyceramido acetamido trideoxyhexose (31, 32), but the biochemical pathway and the enzyme(s) involved in this process remain elusive.
FIGURE 15.

Blast analysis of Pyl with closely related proteins of the ATP-Grasp family has identified conserved amino acids and domains likely involved in ATP binding and catalysis. Conserved amino acid sequences in Pyl are underlined.
FIGURE 16.

Phylogenetic relationships of selected ATP-Grasp proteins from diverse species and Pyl. Protein sequences from different species and Pyl were first aligned with MUSCLE 3.7 software. Alignments were subsequently analyzed using PhyML 3.0 to generate the phylogenetic tree.
There are only a limited number of published studies showing that an organic acid links two sugars together in an oligosaccharide. For example, the O-polysaccharide of the LPS from the pathogenic Gram-negative bacterium F. maritimus (7) is composed of the disaccharide repeating unit 2-acetamido-3-O-acetyl-4-[(S)-2-hydroxyglutar-5-ylamido]-2,4,6-trideoxy-β-glucose and 5-acetamido-7-[(S)-3-hydroxybutyramido]-8-amino-3,5,7,8,9-pentadeoxynonulopyranosonic acid (7). A similar organic residue linked to an amino-sugar in the oligosaccharide structure was described in Neisseria meningitidis species (31), but it remains unclear whether other glycosyl residues attach to the glyceramido-acetamido moiety of this sugar.
The formation of the SCWP containing FucNAc-2HG in B. cereus ATCC 14579 has been reported to occur only when the bacterium is grown in a defined HCT medium (6). This structure was also present in the planktonic stage when the bacterium was cultured with shaking. However, in static culture during the transition from the planktonic to the biofilm stage, it took 72 h to form the FucNAc-4N-2HG oligosaccharide in the biofilm (6). Thus, specific nutrients in the HCT media as well as elements contributing to the transition to a biofilm community may lead to the induction of genes involved in the formation of these oligosaccharides. Current effort is underway to determine factors involved in this regulation.
The characterization of the Pat and Pyl enzyme pair involved in linking an organic acid to an activated sugar has led to the identification of a new metabolic pathway involved in bacterial polysaccharide biosynthesis. Identifying similar pathways and the genes in other microbial species will provide insight into the ability of these organisms to alter or modify their cell surfaces in response to different growth and environmental conditions.
Acknowledgments
We thank Dr. Malcolm O'Neill of the Complex Carbohydrate Research Center for constructive comments on the manuscript. We also thank Dr. John Glushka of the Complex Carbohydrate Research Center for NMR technical assistance. Our research benefitted from instrumentation provided by an NIH grant, S10 RR027097.
This work was supported in part by BioEnergy Science Center Grant DE-PS02-06ER64304, which is supported by the United States Office of Biological and Environmental Research in the Department of Energy Office of Science.
- SCWP
- secondary cell wall polysaccharide
- COSY
- correlation spectroscopy
- TOCSY
- total correlation spectroscopy
- HSQC
- heteronuclear single quantum Coherence
- HMBC
- heteronuclear multiple bond correlation
- PLP
- pyridoxal phosphate
- DSS
- 2,2-dimethyl-2-silapentane-5-sulfonate
- 2OG
- 2-oxoglutarate
- HILIC
- hydrophilic interaction liquid chromatography.
REFERENCES
- 1. Vilain S., Brözel V. S. (2006) Multivariate approach to comparing whole-cell proteomes of Bacillus cereus indicates a biofilm-specific proteome. J. Proteome. Res. 5, 1924–1930 [DOI] [PubMed] [Google Scholar]
- 2. Berg G., Eberl L., Hartmann A. (2005) The rhizosphere as a reservoir for opportunistic human pathogenic bacteria. Environ. Microbiol. 7, 1673–1685 [DOI] [PubMed] [Google Scholar]
- 3. Stenfors Arnesen L. P., Fagerlund A., Granum P. E. (2008) From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Rev. 32, 579–606 [DOI] [PubMed] [Google Scholar]
- 4. Bottone E. J. (2010) Bacillus cereus, a volatile human pathogen. Clin. Microbiol. Rev. 23, 382–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leoff C., Choudhury B., Saile E., Quinn C. P., Carlson R. W., Kannenberg E. L. (2008) Structural elucidation of the nonclassical secondary cell wall polysaccharide from Bacillus cereus ATCC 10987. Comparison with the polysaccharides from Bacillus anthracis and Bacillus cereus type strain ATCC 14579 reveals both unique and common structural features. J. Biol. Chem. 283, 29812–29821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Candela T., Maes E., Garénaux E., Rombouts Y., Krzewinski F., Gohar M., Guérardel Y. (2011) Environmental and biofilm-dependent changes in a Bacillus cereus secondary cell Wall Polysaccharide. J. Biol. Chem. 286, 31250–31262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vinogradov E., MacLean L. L., Crump E. M., Perry M. B., Kay W. W. (2003) Structure of the polysaccharide chain of the lipopolysaccharide from Flexibacter maritimus. Eur. J. Biochem. 270, 1810–1815 [DOI] [PubMed] [Google Scholar]
- 8. Suzuki M., Takahashi Y., Noguchi A., Arai T., Yagasaki M., Kino K., Saito J. (2012) The structure of l-amino-acid ligase from Bacillus licheniformis. Acta Crystallogr. D Biol. Crystallogr. 68, 1535–1540 [DOI] [PubMed] [Google Scholar]
- 9. Jitrapakdee S., Wallace J. C. (2003) The biotin enzyme family: Conserved structural motifs and domain rearrangements. Curr. Protein Pept. Sci. 4, 217–229 [DOI] [PubMed] [Google Scholar]
- 10. Gu X., Glushka J., Yin Y., Xu Y., Denny T., Smith J., Jiang Y., Bar-Peled M. (2010) Identification of a bifunctional UDP-4-keto-pentose/UDP-xylose synthase in the plant pathogenic bacterium Ralstonia solanacearum strain GMI1000, a distinct member of the 4,6-dehydratase and decarboxylase family. J. Biol. Chem. 285, 9030–9040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Martinez V., Ingwers M., Smith J., Glushka J., Yang T., Bar-Peled M. (2012) Biosynthesis of UDP-4-keto-6-deoxyglucose and UDP-rhamnose in pathogenic fungi magnaporthe grisea and Botryotinia fuckeliana. J. Biol. Chem. 287, 879–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gu X., Glushka J., Lee S. G., Bar-Peled M. (2010) Biosynthesis of a new UDP-sugar, UDP-2-acetamido-2-deoxyxylose, in the human pathogen Bacillus cereus subspecies cytotoxis NVH 391-98. J. Biol. Chem. 285, 24825–24833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rance M., Sorensen O. W., Bodenhausen G., Wagner G., Ernst R. R., Wuthrich K. (1983) Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 117, 479–485 [DOI] [PubMed] [Google Scholar]
- 14. Braunschweiler L. E. (1983) Coherence transfer by isotropic mixing-application to proton correlation spectroscopy. J. Magn. Reson. 53, 521–528 [Google Scholar]
- 15. Bodenhausen G., Ruben D. J. (1980) Natural abundance N-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 69, 185–189 [Google Scholar]
- 16. Marion D., Driscoll P. C., Kay L. E., Wingfield P. T., Bax A., Gronenborn A. M., Clore G. M. (1989) Overcoming the overlap problem in the assignment of 1H NMR spectra of larger proteins by use of 3-dimensional heteronuclear 1H-15N Hartmann-Hahn multiple quantum coherence and nuclear overhauser multiple quantum coherence spectroscopy–Application to interleukin-1-β. Biochemistry 28, 6150–6156 [DOI] [PubMed] [Google Scholar]
- 17. Bernassau J. M., Nuzillard J. M. (1994) Selective Hmbc experiments using soft inversion pulses. J. Magn. Reson. Ser. B 103, 77–81 [Google Scholar]
- 18. Breazeale S. D., Ribeiro A. A., Raetz C. R. (2003) Origin of lipid A species modified with 4-amino-4-deoxy-l-arabinose in polymyxin-resistant mutants of Escherichia coli–An aminotransferase (ArnB) that generates UDP-4-amino-4-deoxy-l-arabinose. J. Biol. Chem. 278, 24731–24739 [DOI] [PubMed] [Google Scholar]
- 19. Raetz C. R. (2002) Origin of lipid A species modified with 4-amino-4-deoxy-l-arabinose in Escherichia coli. Biochemistry 41, 8962 [Google Scholar]
- 20. Cook P. D., Holden H. M. (2008) GDP-perosamine synthase: structural analysis and production of a novel trideoxysugar. Biochemistry 47, 2833–2840 [DOI] [PubMed] [Google Scholar]
- 21. Madej T., Addess K. J., Fong J. H., Geer L. Y., Geer R. C., Lanczycki C. J., Liu C., Lu S., Marchler-Bauer A., Panchenko A. R., Chen J., Thiessen P. A., Wang Y., Zhang D., Bryant S. H. (2012) MMDB: 3D structures and macromolecular interactions. Nucleic Acids Res. 40, D461–D464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bubb W. A. (2003) NMR spectroscopy in the study of carbohydrates: characterizing the structural complexity. Concept. Magn. Reson. A 19A, 1–19 [Google Scholar]
- 23. Fan C., Moews P. C., Walsh C. T., Knox J. R. (1994) Vancomycin resistance–structure of d-alanine-d-alanine ligase at 2.3-angstrom resolution. Science 266, 439–443 [DOI] [PubMed] [Google Scholar]
- 24. Fan C., Moews P. C., Shi Y., Walsh C. T., Knox J. R. (1995) Common fold for peptide synthetases cleaving Atp to Adp–glutathione synthetase and d-alanine-d-alanine ligase of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 92, 1172–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fawaz M. V., Topper M. E., Firestine S. M. (2011) The ATP-Grasp enzymes. Bioorg. Chem. 39, 185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Esser L., Wang C. R., Hosaka M., Smagula C. S., Südhof T. C., Deisenhofer J. (1998) Synapsin I is structurally similar to ATP-utilizing enzymes. EMBO J. 17, 977–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Polekhina G., Board P. G., Gali R. R., Rossjohn J., Parker M. W. (1999) Molecular basis of glutathione synthetase deficiency and a rare gene permutation event. EMBO J. 18, 3204–3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mochalkin I., Miller J. R., Evdokimov A., Lightle S., Yan C., Stover C. K., Waldrop G. L. (2008) Structural evidence for substrate-induced synergism and half-sites reactivity in biotin carboxylase. Protein Sci. 17, 1706–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ludlam A., Brunzelle J., Pribyl T., Xu X., Gatti D. L., Ackerman S. H. (2009) Chaperones of F1-ATPase. J. Biol. Chem. 284, 17138–17146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Galperin M. Y., Koonin E. V. (1997) A diverse superfamily of enzymes with ATP-dependent carboxylate-amine/thiol ligase activity. Protein Sci. 6, 2639–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chamot-Rooke J., Rousseau B., Lanternier F., Mikaty G., Mairey E., Malosse C., Bouchoux G., Pelicic V., Camoin L., Nassif X., Duménil G. (2007) Alternative Neisseria spp. type IV pilin glycosylation with a glyceramido acetamido trideoxyhexose residue. Proc. Natl. Acad. Sci. U.S.A. 104, 14783–14788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nothaft H., Szymanski C. M. (2013) Bacterial protein N-glycosylation: new perspectives and applications. J. Biol. Chem. 288, 6912–6920 [DOI] [PMC free article] [PubMed] [Google Scholar]