

Abstract
Ductal plate malformations (DPM) are developmental anomalies considered to result from lack of ductal plate remodeling during bile duct morphogenesis. In mice, bile duct development is initiated by the formation of primitive ductal structures lined by two cell types, namely ductal plate cells and hepatoblasts. During ductal plate remodeling the primitive ductal structures mature to ducts as a result from differentiation of the ductal plate cells and hepatoblasts to cholangiocytes. We here report that this process is conserved in human fetal liver. These findings prompted us to evaluate how DPM develop in three mouse models, namely mice with livers deficient in Hepatocyte Nuclear Factor (HNF)6, HNF1β or cystin-1 (cpk mice). Human liver from a patient with a HNF1B/TCF2 mutation, and from fetuses affected with Autosomal Recessive Polycystic Kidney Disease (ARPKD) were also analysed. Despite the epistatic relationship between HNF6, HNF1β and cystin-1, the three mouse models displayed distinct morphogenic mechanisms of DPM. They all developed biliary cysts lined by cells with abnormal apico-basal polarity. However, the absence of HNF6 led to an early defect in ductal plate cell differentiation. In HNF1β-deficient liver, maturation of the primitive ductal structures was impaired. Cpk mouse livers and human fetal ARPKD showed normal differentiation and maturation but abnormal duct expansion.
Conclusion
DPM is the common end-point of distinct defects initiated at distinct stages of bile duct morphogenesis. Our observations provide a new pathogenic classification of DPM.
Keywords: liver, development, polycystic disease, polarity, cilium
Ductal plate malformations (DPM) are characterized by the persistence of embryonic biliary structures after birth (1). They consist of biliary cell clusters or duct-like structures with elongated lumina and variable shape, and are found in several congenital diseases (2). DPMs are considered to result from lack of remodeling of the ductal plate during the fetal period. However, recent insight into the mode of biliary tubulogenesis identified a new step in biliary tubulogenesis (3), prompting the need to re-assess how DPM develop.
Bile duct development in the mouse starts around embryonic day (E)12.5 with the differentiation of hepatoblasts into biliary precursor cells. The latter form the ductal plate, a single-layered sleeve of cells located around the portal mesenchyme. Around E15.5 tubulogenesis is initiated by the formation of primitive ductal structures (PDS), which are developing ducts asymmetrically lined on the portal side by ductal plate cells, and on the parenchymal side by hepatoblast-like cells. When the PDS mature to ducts (E17.5 to birth), the cells on the portal and parenchymal sides differentiate to mature cholangiocyte which then symmetrically line the duct lumina; simultaneously the ducts become integrated in the portal mesenchyme, and the ductal plate cells not involved in tubulogenesis involute (4, 5).
For this paper we took advantage of three mouse models with DPM to investigate the dysmorphogenesis leading to DPM and to study the epistatic relationship between the transcription factors Hepatocyte Nuclear Factor (HNF)6 and HNF1β and their downstream targets potentially involved in DPM. Mice knockout for HNF6 or with liver-specific inactivation of HNF1β show DPM associated with cholestasis (6, 7). HNF6 directly stimulates expression of HNF1β (6), but the effectors of HNF6 and HNF1β are not known. In pancreatic ducts HNF6 controls primary cilia formation and stimulates expression of Cystin1 (Cys1) and Polycystic kidney and hepatic disease-1 (Pkhd1) (8), two genes that control ciliogenesis and whose mutations are associated with cyst formation (9–14); in kidneys HNF1β stimulates expression of Pkhd1 (15, 16). Moreover, mice deficient in cystin-1 (congenital polycystic kidney (cpk)) display DPM (17, 18).
Here we thus analyse the dysmorphogenesis causing DPM in HNF6- and HNF1β-deficient mice, as well as in livers deficient in cystin-1, which is identified as a common target of HNF6 and HNF1β. We focused on differentiation, apico-basal polarity and ciliogenesis, and found that distinct defects initiated at distinct stages of bile duct morphogenesis may lead to DPM.
EXPERIMENTAL PROCEDURES
Animals
Wild-type, Hnf6, Hnf1bloxP/loxP-Alfp-Cre and cpk mice (6, 7, 19) were treated according to the principles of laboratory animal care of the NIH and with approval from Institutional animal welfare committees.
Human fetal liver
Tissue samples were obtained in compliance with the French and the Belgian legislations, the 1975 Declaration of Helsinki, and the European Guidelines for the use of human tissues. ARPKD fetuses had kidneys with radially oriented dilations of the medullary collecting ducts (20) and had mutations in the PKHD1 gene (21).
Immunofluorescence
Human samples (5 normal at 11 weeks of gestation (W), 2 ARPKD at 13W and 1 ARPKD at 22W, 1 patient with HNF1B mutation (4 days after birth) were formalin-fixed and paraffin-embedded. Mouse liver preparation and immunofluorescence analysis of 5 μm- (human) or 9 μm (mouse)-thick sections were as described (22) (Supplementary Table I).
Quantitative reverse-transcriptase polymerase chain reaction
RNA from mouse liver was extracted with Tripure RNA Isolation reagent (Roche, Vilvoorde, Belgium), and quantitative reverse-transcriptase polymerase chain reaction (Supplementary Table II) was performed with SYBR Green Master Mix Reagent (Invitrogen, Merelbeke, Belgium). For quantification of Sec63, Pkhd1, and Cys1 copy number for each mRNA was normalized to β-actin mRNA copy number using standard calibration curves, and reported by reference to control values set at 100%.
RESULTS
Transient asymmetry during bile duct development is differentially controlled by HNF6 and HNF1β
To investigate how DPM develop in the absence of HNF6 or HNF1β, we first determined the differentiation status of the ductal cells lining DPM in livers of Hnf6−/− mice and of mice with liver-specific inactivation of Hnf1b (Hnf1bloxP/loxP-Alfp-Cre). Since differentiation progresses from the hilum to the periphery, all livers were analyzed near the hilum. At E17.5, the cells lining biliary structures in Hnf6−/− mice did not express the biliary marker SRY-related HMG box transcription factor 9 (SOX9), but most cells expressed the hepatocyte/hepatoblast marker HNF4. Higher E-cadherin expression in biliary cells as compared to parenchymal cells is typical for mouse fetal liver (Fig. 1A). The ectopic expression of HNF4 in biliary structures (arrowheads) was in line with our earlier observation that the lack of HNF6 generates hybrid hepato-biliary cells (23). Such cells were observed on the portal and parenchymal sides of the biliary structures, suggesting that HNF6 is required for differentiation of the two biliary layers. In control mice expression of the Transforming Growth Factor Receptor type II (TβRII) is absent from the portal side of PDS at E15.5 but is detected on their parenchymal side (3). At E17.5 the expression on the parenchymal side progressively waned, leading to ducts with a limited number TβRII-positive cells (open arrowheads, Supplementary Figure 1), and devoid of TβRII at E18.5 (3). In the absence of HNF6, expression of TβRII persisted on both sides of the biliary structures at E17.5 (arrowheads, Supplementary Figure 1), and this was again in line with our earlier data at E15.5 which showed elevated expression of TβRII in the liver (23).
Fig. 1.

Abnormal biliary differentiation in embryonic Hnf6−/− livers and perturbed duct maturation in livers deficient in HNF1β. (A) Wild-type biliary cells (E17.5) are all SOX9+/HNF4−. In Hnf6−/− livers (E17.5), biliary cells fail to express SOX9 while most cells express HNF4. In HNF1β-deficient mouse livers (E17.5), cells on the portal side are normal, whereas cells on the parenchymal side are still SOX9−/HNF4+. (B) Ductal plate malformation in a 4 day-old patient with a mutation in HNF1B. The portal mesenchyme contains short cords of SOX9+ cells (arrows) and HNF4− cells (arrowheads). (C) Primitive ductal structures (PDS) with asymmetrical expression of SOX9 are found in a normal human fetal liver (W11), indicating that duct morphogenesis proceeds in humans as in mice. PDS and mature ducts lined by SOX9+ cells are found within the same liver. Dotted lines, developing ducts; pv, portal vein; *, duct lumen; scale bar, 40 μm.
In the absence of HNF1β at E17.5, differentiation of cells that lined the portal side of the DPM was normal throughout the liver: these cells were SOX9+/HNF4−/Ecadhigh. In contrast, cells lining the parenchymal side were SOX9−/HNF4+/Ecadlow, indicating that they had not matured to biliary cells but that HNF1β-deficient biliary structures were still asymmetrical (Fig. 1A). This was supported by the observation at E17.5 that all cells on the parenchymal side of the biliary structures expressed TβRII (arrowheads), whereas cells on the portal side no longer expressed TβRII (open arrowheads, Supplementary Figure 1).
At postnatal day 7, biliary cells had differentiated in Hnf6−/− and in Hnf1bloxP/loxP-Alfp-Cre livers as they were SOX9+/HNF4− (arrows, Supplementary Figure 2A). Therefore, embryonic biliary differentiation defects seemed to resolve but this was not sufficient to allow normal tubulogenesis: Hnf6−/− livers showed DPM, and HNF1β-deficient livers showed heterogeneity, combining DPM and dysplastic ducts (Supplementary Figures 2A–B) within the same liver. Therefore, in HNF1β-deficient mice a homogenous embryonic phenotype gives rise to a heterogenous postnatal phenotype. We concluded that the absence of HNF6 induces an early defect in biliary cell differentiation while the lack of HNF1β leads to deficient maturation of PDS; both defects ultimately give rise to DPM.
Transient asymmetry in human bile duct development and ductal plate malformation in a patient with a HNF1B mutation
There are no HNF6 mutations reported in humans. In contrast, patients with HNF1B (TCF2) mutations present with renal cysts and diabetes syndrome (MIM#137920). There is phenotypic variability and in rare cases this syndrome is associated with bile duct paucity (24, 25). We therefore looked for the presence of DPM in a patient with HNF1B mutation. This patient had multicystic kidneys and died at 4 days from pulmonary insufficiency; analysis of the HNF1B gene revealed heterozygous deletion of exon 6. Immunostaining of sections showed DPM constituted of clusters of SOX9+/Ecad+ cells (arrows) and short cords of HNF4−/Ecad+ cells (arrowheads) in the portal mesenchyme (Fig. 1B). Dysplastic ducts were also found (Supplementary Figure 3). Therefore, HNF1B mutation in humans can be associated not only with bile duct paucity but also with DPM and duct dysplasia.
The limited availability of samples from patients with HNF1B mutations precludes analysis of the morphogenesis of DPM. Therefore, we speculated that DPM develop similarly in patients and in Hnf1bloxP/loxP-Alfp-Cre mice. This was supported by our observations that bile duct development in humans proceeds by transient asymmetry, like in mice (Fig. 1C). Indeed, the liver of a normal fetus at the 11th week of gestation (W) showed PDS with asymmetrical expression of SOX9. Since maturation of ducts is not equal throughout the liver, ducts entirely lined by SOX9+ cells were also found within the same liver.
Ciliogenesis and polarization of cholangiocytes is impaired in the absence of HNF6 and HNF1β
HNF6 controls the formation of primary cilia in the pancreas and HNF-1β regulates genes involved in cilium function in mouse kidneys (8, 15). Therefore, we investigated how ciliogenesis proceeds in the biliary tract of Hnf6−/− and Hnf1bloxP/loxP-Alfp-Cre mice. Primary cilia were identified as acetylated tubulin-stained dots in wild-type livers at E17.5 (Fig. 2). In contrast, little or no cilia were detected on HNF6- and HNF1β-deficient biliary cells. In wild-type cholangiocytes, the basal body which is detectable by γ-tubulin staining and constituted by two centrioles, is assembled near the apical pole of the cells. In HNF6- and HNF1β-deficient biliary cells, the centrioles were randomly distributed (Fig. 2).
Fig. 2.
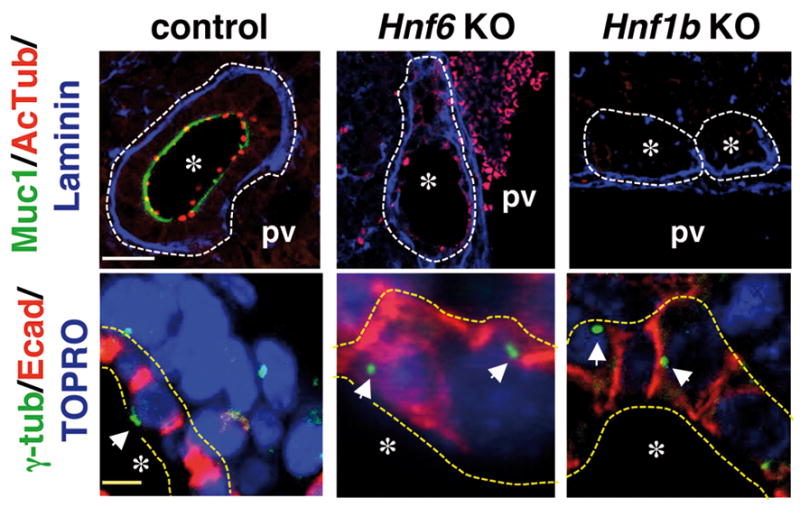
Deficient ciliogenesis and apico-basal polarity in biliary cells in the absence of HNF6 or HNF1β at E17.5. Biliary cells do not show primary cilia (Acetylated tubulin) in Hnf6−/− and Hnf1bloxP/loxP-Alfp-Cre livers. Mucin-1 is absent and centrioles (γ-tubulin, arrows; yellow dotted lines; biliary epithelium) are randomly located instead of being apical as in controls. White dotted lines; developing ducts or cysts; pv, portal vein; *, duct lumen; white and yellow scale bars, 50 and 10 μm.
The lack of cilia at the apical pole was associated with polarity defects. The apical markers mucin-1 and osteopontin (OPN) were not expressed in the absence of HNF6 or HNF1β (Fig. 2 and Supplementary Figure 4). The Golgi marker GM130 was randomly distributed instead of being located along the apico-basal axis between the nucleus and apical pole (Supplementary Figure 4). The basement membrane component laminin, normally connected to the basal pole, formed a continuous layer encircling the ducts in wild-type livers. In the absence of HNF6 or HNF1β the laminin layer was continuous along the basal pole of cells located at the portal side of biliary structures, but was irregular and discontinuous along the cells lining the parenchymal side (arrowheads in Supplementary Figure 4). Phenotypic variability was observed in Hnf6−/− livers, in which some cystic structures were entirely lined by a near continuous layer of laminin (Fig. 2); this variability was unrelated to the hilar-periphery axis. Tight junctions separate the apical pole from the basolateral pole of cells. In the absence of HNF6, ZO-1 was localized at the apical/lateral boundary on cells of the parenchymal and portal side (Supplementary Figure 4). When Hnf1b was deleted, ZO-1 was barely detected in cells of the parenchymal side but was clearly detectable at the portal side. On the portal side, ZO-1 staining did not show the expected punctuate pattern of tight junctions but showed coverage of the apical surface on serial confocal sections, suggesting lack of apical pole (Supplementary Figure 4).
At postnatal day 7, a few Hnf6−/− biliary cells showed normal location of cilia (acetylated tubulin, arrows), mucin-1 and laminin, indicating partial restoration of apico-basal polarity (Supplementary Figure 2A). A fraction of HNF1β-deficient cells expressed mucin-1; no cilia were detected on these cells, the basal bodies remained randomly distributed, and ZO-1 still showed apical coverage (Supplementary Figure 2A). In patients with HNF1B mutation, ZO-1 was irregularly expressed in dysplastic ducts and the DPM did not express ZO-1 (Supplementary Figure 3). We concluded that HNF6 and HNF1β are required for normal development of cilia and for polarization of the cells.
HNF6 and HNF1β stimulate the expression of cystin-1 in liver
The lack of cilia in HNF6- and HNF1β-deficient livers prompted us to investigate whether the expression of genes controlling ciliogenesis or cilium function was affected in these mice. These genes included a set that is regulated by HNF1β in kidneys (15, 16, 26), as well as those associated with hepato-renal fibrocystic diseases (27) (Supplementary Table II). In the absence of HNF6 at E17.5, Pkhd1 and Sec63 mRNA levels were upregulated, but were unaffected in Hnf1b knockout livers (Fig. 3). In contrast, expression of Cys1 was reduced in both mutants. The other genes tested had normal expression (data not shown); this suggests that HNF1β targets in kidney, such as Pkhd1, Pkd2, Nphp1, IFT88 and Kif12, are not regulated by HNF1β in liver, and that the transcriptional network operating in bile duct morphogenesis is distinct from that in kidneys.
Fig. 3.
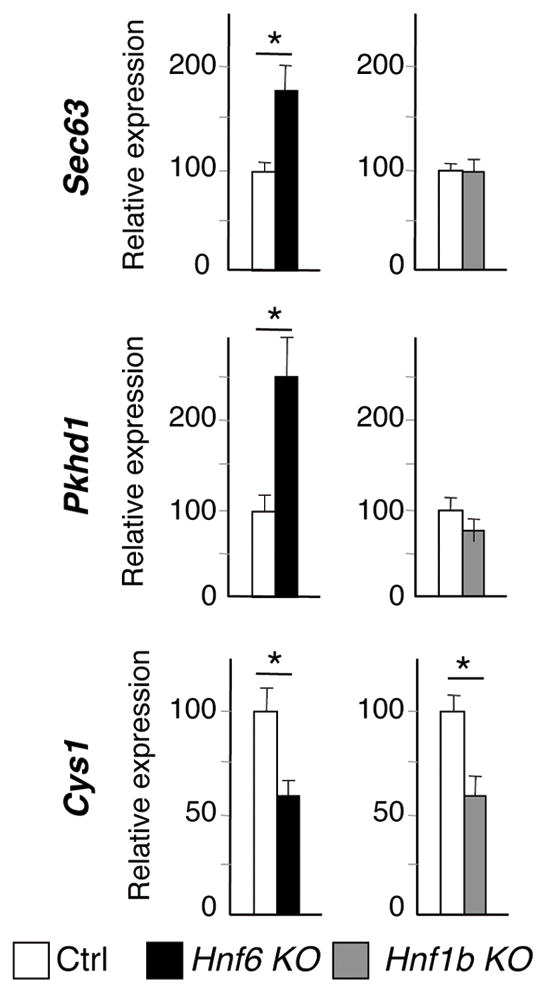
Expression of genes required for cilium function at E17.5. (A) The list of genes tested is shown in supplementary table II. Expression of Sec63 and Pkhd1 is upregulated in the absence of HNF6. HNF6 and HNF1β are required to stimulate expression of Cys1. Means +/− SEM, n = 5 (Hnf6−/−) or n=6 (Hnf1bloxP/loxP-Alfp-Cre), * P< 0.05.
Cystin-1 is required prenatally for normal polarization of biliary cells and for duct morphogenesis
Cys1 was the only common target of HNF6 and HNF1β. Cpk mice have a mutation in the Cys1 gene and display renal disease resembling ARPKD, in association with DPM (12, 17). Therefore, Cys1 is a candidate effector of HNF6 and HNF1β. We investigated how cyst morphogenesis is initiated in cpk mice and human ARPKD fetuses. Cysts were already prominent in cpk mice at E17.5, and were lined by SOX9+/HNF4− cells. Except for a few cells (arrowhead), most biliary cells no longer expressed TβRII (open arrowhead, Supplementary Figure 5). Cysts in human ARPKD fetuses at 13W and 22W were lined on the parenchymal and portal sides by cells expressing SOX9 (Fig. 4A). Therefore, biliary cells in cpk embryos and human fetal ARPKD showed normal differentiation. This was also the case after birth (Supplementary Figure 2A).
Fig. 4.
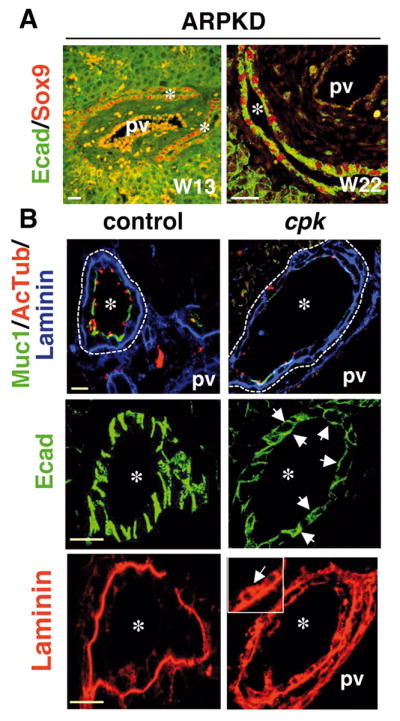
Abnormal biliary tubulogenesis in ARPKD fetuses and in cpk embryos. (A) In ARPKD fetuses at W13 and W22, developing ducts are expanded and lined by SOX9+ cells. (B) Biliary cell polarity is perturbed in cpk embryos at E17.5. The number of cells expressing mucin-1 (Muc1) and showing primary cilia (Acetylated Tubulin) is reduced; developing ducts give rise to cysts. In cpk livers, E-cadherin (Ecad) expression often extends towards the apical pole and more extensively covers the basal pole (arrows). Laminin expands along the lateral (arrows in inset) and apical poles of biliary cells. pv, portal vein; *, duct or cyst lumen, white and yellow scale bars, 50 and 20 μm.
The apical pole marker OPN was equally expressed in wild-type and cpk biliary cells at E17.5, but a lower number of cells showed cilia and mucin-1 in cpk mice (Fig. 4B). E-cadherin did not show the expected basolateral location in cpk biliary cells, as it extended towards the apical pole and covered more extensively the basal pole (arrows; Fig. 4B). The laminin layer was thickened and irregular, and was fragmented along the parenchymal side of the cysts. Laminin also expanded along the lateral and apical membranes (arrow; Fig. 4B), suggesting that the basal and lateral poles were not correctly set. This phenotype persists after birth: some cells did not express mucin-1 and showed apical location of laminin (arrow, Supplementary Figure 2A). The localization of ZO-1 was not restricted to the apical/lateral boundaries, but often extended to cover the apical surface. In wild-type mice at E17.5, serial sections (1.5 μm) by confocal microscopy disclosed the expected belt of ZO-1 expression, i.e. two dots when the focal plane intersects the belt, and linear staining when the focal plane sections an extended belt-like structure (Supplementary Figure 6). In cpk cysts, the intensity of ZO-1 staining was stronger and a higher number of successive confocal sections showed a linear staining of ZO-1, revealing that ZO-1 extensively covered the apical pole (scheme in Supplementary Figure 6). Interestingly, fetuses with ARPKD resulting from PKHD1 mutation had a phenotype similar to that of cpk mice (Supplementary Figure 6). In addition, the same extension of ZO-1 staining was detected on the portal side of DPM in HNF1β-deficient livers (Supplementary Figure 4).
We conclude that DPM and cyst formation in cpk mice and human ARPKD results from dysmorphogenesis affecting the parenchymal and portal sides of developing ducts, with normal differentiation but perturbed apico-basal polarity. Expression of cpk is controlled by HNF6 and HNF1β, but cystin-1 is not their main effector: except for the fragmented laminin layer surrounding the cysts and perturbed apico-basal polarity, there is limited phenotypic overlap between HNF6 and HNF1β deficient livers andcpk livers. Therefore, the analysis of the various mutants indicates that distinct mechanisms operate to generate DPM and cysts.
DISCUSSION
Recent insight into the mechanism of bile duct morphogenesis, and in particular the transient asymmetry (3) - shown here to occur in humans like in mice - prompted us to evaluate the morphogenesis of DPM, by investigating differentiation, polarity and ciliogenesis. We studied three mouse models with DPM and mainly focused on embryos at E17.5. This stage enabled us to investigate the ductal plate, the PDS and their maturation. The morphogenic mechanisms leading to DPM differed in the three models (Fig. 5): differentiation of biliary precursors from hepatoblasts was perturbed in HNF6-deficient fetuses, maturation of PDS failed in HNF1β-deficient livers, and abnormal expansion of ducts occured in cpk mice and human ARPKD. Considering that DPM is the common end-point in these animal models and human cases, but that the pathogenic mechanism leading to DPM differs in all those instances, we propose to classify the DPM according to distinct defects in (I) differentiation of biliary precursor cells, (II) maturation of PDS, or (III) duct expansion during development (Fig. 5).
Fig. 5.

Classification of DPM based on distinct pathogenic mechanisms. Normal bile duct morphogenesis is illustrated at the top. DPM can arise by three mechanisms: (I) differentiation of hepatoblasts to ductal plate cells is abnormal and associated with perturbed polarization and cyst formation; (II) PDS are formed but fail to mature to bile ducts; this is associated with formation of cysts and abnormal polarity; (III) differentiation of ductal plate cells and maturation of PDS proceeds but duct expansion is perturbed; this is associated with defects in cell polarity.
Cilia were mostly absent in HNF6- and in HNF1β-deficient livers. The presence of cilia in cpk mice dismissed the possibility that reduced expression of Cys1 in HNF6- and HNF1β-deficient livers causes the near absence of cilia in the latter two mouse models. However, the lack of cilia in the absence of HNF6 or HNF1β at E17.5 may result from mispositioning of the basal body, and from the global perturbation of biliary cell polarity. Apico-basal polarity was strongly affected, as shown by the absence of OPN, abnormal location of centrioles and Golgi apparatus, and fragmented appearance of laminin. Whereas these polarity criteria were equally affected in the absence of HNF6 or HNF1β, the location of tight junctions differed in the two mouse models. Tight junctions were detected normally by ZO-1 immunostaining and electron microscopy (data not shown) in HNF6 knockout livers. In contrast, in the absence of HNF1β, ZO-1 was barely detected in cells of the parenchymal side of the biliary structures and was mislocated on the cells of the portal side. Cpk mice and human fetuses with ARPKD also showed mislocation of ZO-1. They displayed the same apical coverage of ZO-1 as on the portal side of developing ducts in HNF1β-deficient livers. Therefore, an HNF1β - cystin-1 cascade may control ZO-1 location. Earlier work has shown that HNF1β controls bile duct polarity in vitro and that this process requires laminin (28, 29). Our data now show that laminin expression in vivo is reduced in HNF1β-deficient livers, thereby uncovering a cross-talk between the biliary cells and extracellular matrix in the control of tubulogenesis.
In HNF6 knockout livers, biliary cell differentiation is abnormal. Perturbed TGFβ signaling generates hybrid hepato-biliary cells (23), and this hybrid character persists at later stage of gestation as shown here at E17.5 by the coexpression of HNF4 and SOX9, and by the presence of microvilli, glycogen, well-developed endoplasmic reticulum and large nuclei with large nucleoli (data not shown). Our work also reveals an unexpected regulation of SOX9. Indeed, SOX9 mRNA levels are reduced in Hnf6−/− livers at E15.5, while normal levels are restored at E17.5 (3). SOX9 protein is undetectable at E15.5 (not shown) and here we found that it remains absent at E17.5, indicating that SOX9 expression is controlled by HNF6 at the transcriptional and post-transcriptional levels.
In HNF1β-deficient livers, biliary cells on the portal side appeared well-differentiated since they were SOX9+/HNF4−/TβRII−. They also expressed the Notch effector Hes1 (data not shown). In contrast, biliary cells on the parenchymal side were SOX9−/HNF4+/TβRII+ and expressed low levels of Hes1 (data not shown). Therefore, at E17.5, the biliary structures still displayed a PDS-like configuration. It cannot be determined if perturbed Notch or TGFβ signaling, which is suggested by the PDS-like expression of Hes1 and TβRII, causes or results from the lack of PDS maturation. Still, our data identify HNF1β as a critical regulator of PDS maturation and show for the first time that deficient maturation is a cause of DPM.
During normal biliary tubulogenesis, differentiation and polarity progress concomitantly (3). When HNF6 or HNF1β is inactivated, differentiation, polarity and tubulogenesis are all affected. In contrast, in cpk mice and ARPKD patients, differentiation does not seem affected while polarity and tubulogenesis are perturbed. Therefore, polarity and differentiation are associated or separated, depending on the DPM model studied. Interestingly, in all models lumina still formed. We also measured the expression of the key planar polarity genes in the three mouse models, but found no strong evidence for a HNF6-HNF1β-cystin1 cascade regulating planar polarity (Supplementary Figure 7).
Cyst expansion in cpk mouse kidneys depends on excess proliferation, but at E17.5 in the liver, no excess proliferation was seen: the percentage of proliferating biliary cells measured by phospho-histone H3 staining was 2.06% (58 PHH3+ biliary cells/2811 biliary cells; quantification on 3 livers) as compared to 1.8% (41 PHH3+ biliary cells/2254 biliary cells). Therefore, the mechanism of cyst formation in liver may differ from that in kidneys. This is in line with our observations that the transcriptional network regulating bile duct development is distinct from that in kidneys.
Previous work suggests that Hnf6 and Hnf1b are linked in a common gene network (6, 7). Importantly, the present work reveals that HNF1β cannot be considered as the sole effector of HNF6, and that cystin-1 is not the main effector of HNF6 or HNF1β in differentiation and morphogenesis. We speculate that the three genes may coordinately regulate biliary functions not uncovered in the present study.
Supplementary Material
Acknowledgments
Grant Support. This work was supported by the IAP Program (Belgian Science Policy, to OD, PC and FL), the D.G. Higher Education and Scientific Research of the French Community of Belgium (to FL), the Alphonse and Jean Forton Fund (to OD and FL), the FRSM (Belgium, to FL), the European Community (EUNEFRON, GA#201590, to OD), and the NIH (DK55534; to LGW). MP was a recipient of “Bettencourt Schueller prix d’élan pour la recherche” and “Equipe Fondation pour la Recherche Médicale”. CEP is Senior Research Associate of the FRS-FNRS.
The authors thank Dr. Laure Collard for providing information on the patient with HNF1B mutation, and Chaozhe Yang for help.
Abbreviations
- DPM
ductal plate malformation
- ARPKD
autosomal recessive polycystic kidney disease
- E
embryonic day
- PDS
primitive ductal structures
- HNF
hepatocyte nuclear factor
- PKHD1
polycystic kidney and hepatic disease-1
- cpk
congenital polycystic kidney
- W
week of gestation
- SOX9
SRY-related HMG box transcription factor 9
- TβRII
Transforming Growth Factor Receptor type II
- ZO-1
Zonula Occludens-1
- OPN
osteopontin
References
- 1.Desmet VJ. Congenital diseases of intrahepatic bile ducts: variations on the theme “ductal plate malformation”. Hepatology. 1992;16:1069–1083. doi: 10.1002/hep.1840160434. [DOI] [PubMed] [Google Scholar]
- 2.Awasthi A, Das A, Srinivasan R, Joshi K. Morphological and immunohistochemical analysis of ductal plate malformation: correlation with fetal liver. Histopathology. 2004;45:260–267. doi: 10.1111/j.1365-2559.2004.01945.x. [DOI] [PubMed] [Google Scholar]
- 3.Antoniou A, Raynaud P, Cordi S, Zong Y, Tronche F, Stanger B, Jacquemin P, et al. Intrahepatic Bile Ducts Develop According to a New Mode of Tubulogenesis Regulated by the Transcription Factor SOX9. Gastroenterology. 2009;136:2325–2333. doi: 10.1053/j.gastro.2009.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raynaud P, Carpentier R, Antoniou A, Lemaigre FP. Biliary differentiation and bile duct morphogenesis in development and disease. Int J Biochem Cell Biol. 2011;43:245–256. doi: 10.1016/j.biocel.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 5.Roskams T, Desmet V. Embryology of extra- and intrahepatic bile ducts, the ductal plate. Anat Rec. 2008;291:628–635. doi: 10.1002/ar.20710. [DOI] [PubMed] [Google Scholar]
- 6.Clotman F, Lannoy VJ, Reber M, Cereghini S, Cassiman D, Jacquemin P, Roskams T, et al. The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development. 2002;129:1819–1828. doi: 10.1242/dev.129.8.1819. [DOI] [PubMed] [Google Scholar]
- 7.Coffinier C, Gresh L, Fiette L, Tronche F, Schutz G, Babinet C, Pontoglio M, et al. Bile system morphogenesis defects and liver dysfunction upon targeted deletion of HNF1beta. Development. 2002;129:1829–1838. doi: 10.1242/dev.129.8.1829. [DOI] [PubMed] [Google Scholar]
- 8.Pierreux CE, Poll AV, Kemp CR, Clotman F, Maestro MA, Cordi S, Ferrer J, et al. The transcription factor hepatocyte nuclear factor-6 controls the development of pancreatic ducts in the mouse. Gastroenterology. 2006;130:532–541. doi: 10.1053/j.gastro.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 9.Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 10.Nagasawa Y, Matthiesen S, Onuchic LF, Hou X, Bergmann C, Esquivel E, Senderek J, et al. Identification and characterization of Pkhd1, the mouse orthologue of the human ARPKD gene. J Am Soc Nephrol. 2002;13:2246–2258. doi: 10.1097/01.asn.0000030392.19694.9d. [DOI] [PubMed] [Google Scholar]
- 11.Ward CJ, Yuan D, Masyuk TV, Wang X, Punyashthiti R, Whelan S, Bacallao R, et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet. 2003;12:2703–2710. doi: 10.1093/hmg/ddg274. [DOI] [PubMed] [Google Scholar]
- 12.Gattone VH, 2nd, MacNaughton KA, Kraybill AL. Murine autosomal recessive polycystic kidney disease with multiorgan involvement induced by the cpk gene. Anat Rec. 1996;245:488–499. doi: 10.1002/(SICI)1097-0185(199607)245:3<488::AID-AR5>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 13.Tao B, Bu S, Yang Z, Siroky B, Kappes JC, Kispert A, Guay-Woodford LM. Cystin localizes to primary cilia via membrane microdomains and a targeting motif. J Am Soc Nephrol. 2009;20:2570–2580. doi: 10.1681/ASN.2009020188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muchatuta MN, Gattone VH, 2nd, Witzmann FA, Blazer-Yost BL. Structural and functional analyses of liver cysts from the BALB/c-cpk mouse model of polycystic kidney disease. Exp Biol Med. 2009;234:17–27. doi: 10.3181/0807-RM-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gresh L, Fischer E, Reimann A, Tanguy M, Garbay S, Shao X, Hiesberger T, et al. A transcriptional network in polycystic kidney disease. EMBO J. 2004;23:1657–1668. doi: 10.1038/sj.emboj.7600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hiesberger T, Bai Y, Shao X, McNally BT, Sinclair AM, Tian X, Somlo S, et al. Mutation of hepatocyte nuclear factor-1beta inhibits Pkhd1 gene expression and produces renal cysts in mice. J Clin Invest. 2004;113:814–825. doi: 10.1172/JCI20083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guay-Woodford LM, Green WJ, Lindsey JR, Beier DR. Germline and somatic loss of function of the mouse cpk gene causes biliary ductal pathology that is genetically modulated. Hum Mol Genet. 2000;9:769–778. doi: 10.1093/hmg/9.5.769. [DOI] [PubMed] [Google Scholar]
- 18.Hou X, Mrug M, Yoder BK, Lefkowitz EJ, Kremmidiotis G, D’Eustachio P, Beier DR, et al. Cystin, a novel cilia-associated protein, is disrupted in the cpk mouse model of polycystic kidney disease. J Clin Invest. 2002;109:533–540. doi: 10.1172/JCI14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Preminger GM, Koch WE, Fried FA, McFarland E, Murphy ED, Mandell J. Murine congenital polycystic kidney disease: a model for studying development of cystic disease. J Urol. 1982;127:556–560. doi: 10.1016/s0022-5347(17)53911-7. [DOI] [PubMed] [Google Scholar]
- 20.Villeneuve J, Pelluard-Nehme F, Combe C, Carles D, Chaponnier C, Ripoche J, Balabaud C, et al. Immunohistochemical study of the phenotypic change of the mesenchymal cells during portal tract maturation in normal and fibrous (ductal plate malformation) fetal liver. Comp Hepatol. 2009;8:5. doi: 10.1186/1476-5926-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denamur E, Delezoide AL, Alberti C, Bourillon A, Gubler MC, Bouvier R, Pascaud O, et al. Genotype-phenotype correlations in fetuses and neonates with autosomal recessive polycystic kidney disease. Kidney Int. 2010;77:350–358. doi: 10.1038/ki.2009.440. [DOI] [PubMed] [Google Scholar]
- 22.van Eyll JM, Pierreux CE, Lemaigre FP, Rousseau GG. Shh-dependent differentiation of intestinal tissue from embryonic pancreas by activin A. J Cell Sci. 2004;117:2077–2086. doi: 10.1242/jcs.01067. [DOI] [PubMed] [Google Scholar]
- 23.Clotman F, Jacquemin P, Plumb-Rudewiez N, Pierreux CE, Van der Smissen P, Dietz HC, Courtoy PJ, et al. Control of liver cell fate decision by a gradient of TGF beta signaling modulated by Onecut transcription factors. Genes Dev. 2005;19:1849–1854. doi: 10.1101/gad.340305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beckers D, Bellanne-Chantelot C, Maes M. Neonatal cholestatic jaundice as the first symptom of a mutation in the hepatocyte nuclear factor-1beta gene (HNF-1beta) J Pediatr. 2007;150:313–314. doi: 10.1016/j.jpeds.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 25.Devriendt K, Dooms L, Proesmans W, de Zegher F, Desmet V, Eggermont E. Paucity of intrahepatic bile ducts, solitary kidney and atrophic pancreas with diabetes mellitus: atypical Alagille syndrome? Eur J Pediatr. 1996;155:87–90. doi: 10.1007/BF02075756. [DOI] [PubMed] [Google Scholar]
- 26.Gong Y, Ma Z, Patel V, Fischer E, Hiesberger T, Pontoglio M, Igarashi P. HNF-1beta regulates transcription of the PKD modifier gene Kif12. J Am Soc Nephrol. 2009;20:41–47. doi: 10.1681/ASN.2008020238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams M, Smith UM, Logan CV, Johnson CA. Recent advances in the molecular pathology, cell biology and genetics of ciliopathies. J Med Genet. 2008;45:257–267. doi: 10.1136/jmg.2007.054999. [DOI] [PubMed] [Google Scholar]
- 28.Tanimizu N, Miyajima A, Mostov KE. Liver progenitor cells develop cholangiocyte-type epithelial polarity in three-dimensional culture. Mol Biol Cell. 2007;18:1472–1479. doi: 10.1091/mbc.E06-09-0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanimizu N, Miyajima A, Mostov KE. Liver progenitor cells fold up a cell monolayer into a double-layered structure during tubular morphogenesis. Mol Biol Cell. 2009;20:2486–2494. doi: 10.1091/mbc.E08-02-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.