

Abstract
Endogenous retroviruses (ERVs) comprise 8% of the human genome and are common in all vertebrate genomes. The only retrovirus known to be currently transitioning from exogenous to endogenous form is the koala retrovirus (KoRV), making koalas (Phascolarctos cinereus) ideal for examining the early stages of retroviral endogenization. To distinguish endogenous from exogenous KoRV proviruses, we isolated koala genomic regions flanking KoRV integration sites. In three wild southern Australian koalas, there were fewer KoRV loci than in three captive Queensland koalas, consistent with reports that southern Australian koalas carry fewer KoRVs. Of 39 distinct KoRV proviral loci examined in a sire–dam–progeny triad, all proved to be vertically transmitted and endogenous; none was exogenous. Of the 39 endogenous KoRVs (enKoRVs), only one was present in the genomes of both the sire and the dam, suggesting that, at this early stage in the retroviral invasion of a host germ line, very large numbers of ERVs have proliferated at very low frequencies in the koala population. Sequence divergence between the 5′- and 3′-long terminal repeats (LTRs) of a provirus can be used as a molecular clock. Within each of ten enKoRVs, the 5′-LTR sequence was identical to the 3′-LTR sequence, suggesting a maximum age for enKoRV invasion of the koala germ line of approximately 22,200–49,900 years ago, although a much younger age is possible. Across the ten proviruses, seven LTR haplotypes were detected, indicating that at least seven different retroviral sequences had entered the koala germ line.
Keywords: insertional polymorphisms, koala, koala retrovirus, long terminal repeats, sire–dam–progeny triad
Introduction
Endogenous retroviruses (ERVs) are present in the genomes of all vertebrates, comprising 8% of the human genome (Lander et al. 2001; Bromham 2002). ERVs descend from exogenous retroviruses that integrated into the germ line of the ancestral host lineage, and that are now transmitted vertically from parent to offspring through Mendelian inheritance (Boeke and Stoye 1997; Bromham 2002; Coffin 2004; Stoye 2012). ERVs may follow various evolutionary trajectories: Although most decay through mutation, some ERVs may recombine with other endogenous or exogenous viruses, protect the host against similar exogenous viruses, retain the ability to produce viral proteins, or even become co-opted into a role functional for the host (Boeke and Stoye 1997; Bromham 2002; Coffin 2004; Stoye 2012). ERV-encoded Env proteins are reported to block infection by some exogenous murine and avian leukemia retroviruses (Stoye 2012; Weiss 2013). One ERV that has been co-opted to play a role in human health is syncytin, a gene derived from an ERV that is now vital for normal human placentation (Mi et al. 2000). ERVs may also play a role in human disease. Derepression of the LTR of an ERV was found to play a critical role in the pathogenesis of Hodgkin’s lymphoma (Lamprecht et al. 2010). Despite their important roles in human health and disease, and their implications for host genome origins and evolution, the process by which retroviruses endogenize has until recently been difficult to study. Almost all ERVs are quite ancient, often many millions of years old (Johnson and Coffin 1999; Coffin 2004; Stoye 2006) making it difficult to reconstruct the early steps of germ line invasions by ERVs.
Recently, the koala retrovirus (KoRV) was found to be in the midst of transitioning from exogenous to endogenous form (Stoye 2006; Tarlinton et al. 2006, 2008), enabling study of the process of retroviral endogenization. KoRV is a gammaretrovirus closely related to the gibbon ape leukemia virus (Hanger et al. 2000). The grassland melomys (Melomys burtoni), a rodent native to Australia, has also been found to carry a related retrovirus (Simmons et al. 2011). Analyses of the genomes of humans and other vertebrates suggest that retroviruses have, from a long-term evolutionary perspective, frequently jumped from one species to another and invaded the germ lines of new hosts (Fiebig et al. 2006; Denner 2007; Hayward et al. 2013) and KoRV is likely to be the result of a transspecies transmission.
The geographic distribution of koalas spans the east coast of Australia from northern Queensland to South Australia and historically three subspecies have sometimes been recognized: Phascolarctos cinereus adustus in Queensland, Phascolarctos cinereus cinereus in New South Wales, and Phascolarctos cinereus victor in Victoria and South Australia. The subspecies were designated based on differences in physical features such as body size and fur color, although these differences may be clinal (Lee and Martin 1988; Department of the Environment, Canberra 2014). Analyses of mitochondrial DNA control region sequences have detected different haplotypes between P. c. adustus and the other subspecies, but such differences were not evident between P. c. cinereus and P. c. victor (Houlden et al. 1999). Genetic diversity among southern Australian koalas has been shown to be very low compared with northern populations using both mitochondrial DNA and microsatellite markers (Houlden et al. 1996, 1999).
Koala populations in northern Australia exhibit 100% prevalence of KoRV, with a relatively high average of 165 copies of KoRV per cell, whereas in southern Australian populations many koalas are completely free of the virus (Tarlinton et al. 2006; Simmons et al. 2012). This suggests that KoRV initially affected koalas in northern Australia and is currently spreading to southern populations (Tarlinton et al. 2006, 2008). Using museum specimens of koalas, KoRV was found to be ubiquitous in northern Australian koalas by the late 19th century, with their sequences resembling that of modern KoRV (Avila-Arcos et al. 2013). KoRV has been associated with high rates of leukemia and lymphoma in koalas, and may play a role in susceptibility to Chlamydia infections (Tarlinton et al. 2005, 2008). The process of retroviral endogenization, at least in the case of koalas, may have involved centuries of reduced fitness and susceptibility to disease in the host species (Avila-Arcos et al. 2013). There also appear to be KoRV variants with more limited distributions that are believed to be of more recent origin and possibly exogenous (Shojima, Hoshino, et al. 2013; Shojima, Yoshikawa, et al. 2013; Xu et al. 2013; Shimode et al. 2014).
One issue in interpreting past studies of KoRV has been that the proviruses of KoRV that were detected could have been endogenous or exogenous. In this study, we isolate KoRV flanking sites in the koala genome using a modified genome-walking approach (Reddy et al. 2008). We then determine whether KoRV proviruses in the genome are endogenous, by establishing Mendelian inheritance using a sire–dam–progeny triad of northern Australian (Queensland) koalas kept in North American zoos. A provirus found at a particular locus in the progeny would be established as endogenous if it was also found in either parent at the same locus, as two ERVs independently integrating at the same locus in two individuals would be an extremely rare event (Johnson and Coffin 1999). We also sought to determine the time since integration of endogenous KoRV (enKoRV) using a molecular clock based on the sequence divergence within a provirus between the 5′- and 3′-long terminal repeats (LTRs), which are identical at the time of proviral integration into the germ line (Johnson and Coffin 1999; Bromham 2002; Coffin 2004). In addition to the zoo koala triad, we also examined KoRV flanks in three samples of koalas from different localities in southern Australia. Previous studies have shown that koalas from southern Australia carry fewer KoRV proviruses per cell than northern Australian koalas (Tarlinton et al. 2006; Simmons et al. 2012).
Results
Identification of KoRV Integration Sites
We sought to identify KoRV integration sites in six koalas known to be positive for KoRV, three from northern and three from southern Australia. Due to their morphological distinctions and historic classification into different subspecies, North American zoos have treated koalas from northern Australia (Queensland) and from southern Australia as distinct management units. Our study involved three northern Australian koalas from US zoos (table 1) that comprised a sire–dam–progeny triad (offspring: Pci-SN404, sire: Pci-SN248, and dam: Pci-SN345). The three southern Australian koalas were unrelated, wild-caught and chosen for the diversity of their geographic origins (table 1). One koala each was from the Stony Rises (Pci-157) and the Brisbane Ranges (Pci-106) of Victoria, and Kangaroo Island (Pci-187) of South Australia.
Table 1.
Northern Australian Koala Triad Description, Southern Koala Description, and KoRV Flank Statistics.
| Subspecies | Northern |
Southern |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | Pci-SN404 | Pci-SN248 | Pci-SN345 | Pci-157 | Pci-106 | Pci-187 | ||||||||
| Genealogy | Son of SN248/345 | Sire of SN404 | Dam of SN404 | Unknown | Unknown | Unknown | ||||||||
| Birth date | October 31, 2006 | September 29, 1998 | February 18, 2003 | Unknown | Unknown | Unknown | ||||||||
| Place sample collected | Columbus Zoo | San Diego Zoo | San Diego Zoo | Stony Rises | Brisbane Ranges | Kangaroo Island | ||||||||
| Year sample collected | 2010 | 2010 | 2010 | 1988 | 1988 | 1988 | ||||||||
| 5′- or 3′-LTR and flank | 5′ | 3′ | 5′ | 3′ | 5′ | 3′ | 5′ | 3′ | 5′ | 3′ | 5′ | 3′ | ||
| Total 454 sequencing reads | 21,556 | 19,554 | 7,869 | 15,633 | 18,640 | 21,223 | 14,608 | 9,804 | 1,275 | 3,987 | 1,041 | 1,240 | ||
| KoRV LTR plus flank reads | 8,391 | 7,212 | 2,465 | 7,263 | 7,392 | 10,167 | 6,134 | 3,907 | 9 | 1,198 | 1 | 1 | ||
| Distinct flanksa | 76 | 68 | 43 | 45 | 43 | 39 | 11 | 8 | 0 | 10 | 0 | 0 | ||
| Proviral loci identifieda | 108 | 73 | 69 | 16 | 10 | 0 | ||||||||
aDistinct flanks and proviral loci are likely to be underestimated due to technical factors and to stringent criteria used to screen sequences.
To identify host genomic DNA flanking the 5′- and 3′-KoRV LTRs in each koala, a genome-walking method (Reddy et al. 2008) was implemented, but modified to use next-generation sequencing, as illustrated in supplementary figure S1, Supplementary Material online. The flanks were sequenced using the Roche 454 GS FLX+ platform. A unique multiplex identifier (MID) was used for each flanking sequence of each koala, generating 12 sets of sequences. A total of 136,430 reads were generated across the koalas. The number of reads was high for all attempts on the triad (table 1), as was the average percentage of reads that contained the koala genomic flanks (31–48%). The total number of reads and the average proportion of reads that contained KoRV flanking regions were both much lower for three of the six attempts on southern Australian koalas—the 5′-attempt for Pci-106, and both flanks for Pci-187 (table 1). The reason for this reduced success was unclear, especially as we had initially verified that KoRV was present in each of the koalas through polymerase chain reaction (PCR). LTR primer mismatch or technical factors may be potential causes. As the focus of our study was the sire–dam–progeny triad of northern Australian koalas, for which all attempts were very successful, the genome-walking method was not repeated for less successful southern Australian koalas.
The koala genomic sequences flanking KoRV integration sites were queried against genomic scaffolds of the Meug_1.1 assembly of the tammar wallaby (Macropus eugenii) genome (Renfree et al. 2011), the closest relative of the koala with genome sequence available. In some cases, 5′- and 3′-flanking sequences were found to match adjacent regions of the wallaby genome, suggesting that the two flanks would correspond to koala genomic sequence on either side of the integration site of a single KoRV locus. Comparing the 5′- and 3′-host genomic flanks for a provirus at a single locus also permitted identification of the “target site duplication” on either side of the provirus. The target site duplication is a region of host DNA that is replicated during integration of a retrovirus, so that the same host sequence appears immediately upstream and downstream of the integrated provirus. We determined that the length of the target site duplication is 4 bp for KoRV.
The number of distinct flank sequences was estimated using a bioinformatics algorithm, taking for each individual the higher of the 5′- or 3′-count of distinct flank sequences having each of the 256 potential values for the 4-bp target site duplication (supplementary table S1, Supplementary Material online). The number of KoRV integration sites detected in the three northern Australian koalas was 73 for the sire, 69 for the dam, and 108 for the progeny (table 1 and supplementary table S1, Supplementary Material online). Among southern Australian koalas, Pci-157 had a count of 16, whereas Pci-106 had a count of 10 (with only one flank successful) (table 1 and supplementary table S1, Supplementary Material online), consistent with lower copy numbers for KoRV previously reported for southern Australian koalas (Simmons et al. 2012). Given the stringent criteria used in the bioinformatics approach and the poor quality of many reads, and given that two loci may share the same target site duplication, these numbers likely underestimate the number of distinct flanks. The mismatch in counts for each koala between the 5′- and 3′-flanks, possibly due to amplification biases, also indicated that the method did not identify all flanks comprehensively.
KoRV Insertional Polymorphisms
The target site duplication was used to designate individual proviral loci. For example, if the target site duplication on either side of the KoRV provirus had a sequence of ACGT, the provirus was designated “KoRV-ACGT” (in cases where the same target site duplication may be shared by proviruses at more than one locus, the two loci could be distinguished by appending a number to the designation). Subsequent PCR and sequencing of individual proviral loci (below) confirmed the 4 bp length of the target site duplication. There was one exceptional provirus that had a 5-bp target site duplication, KoRV-AAAAG. The integration site for this provirus included four adenine bases in tandem, suggesting that the longer target site duplication may have resulted from strand slippage of the host DNA during retroviral endogenization (Levinson and Gutman 1987; Craigie and Bushman 2012; Ballandras-Colas et al. 2013), although other target site duplications were 4 bp in length despite the presence of homopolymers (e.g., AAAG, AAAT, CCCC).
As there were potentially many hundreds of different loci present across the koalas, we selected 39 loci as representative, including KoRVs present in different frequencies among the reads, that is, some were based on flanks that had few reads in the data set whereas others were based on flanks that were common. All 39 KoRV loci were successfully amplified by locus-specific PCR. Among the 39, both of the flanks could be identified for ten of the loci (table 2; for the other 29 loci, we only identified one flank, see below). The wallaby and koala lineages diverged more than 50 Ma (Meredith et al. 2009), so that only eight loci could be identified by homology to the tammar wallaby genomic sequence. For locus KoRV-CCTT, one flank was identified in the flank sequence data set for Pci-SN345, and was used to query low-coverage GS FLX genomic sequence from Pci-SN404, identifying the other flank in a chromosome without the provirus. For locus KoRV-GCCT, matching 5′- and 3′-target site duplication sequences were detected after single-flank analyses were conducted (see below); PCR combining a primer from each of the two flanks established (after amplification and sequencing in a chromosome without the provirus) that the two flanks corresponded to the same locus.
Table 2.
Insertional Polymorphisms of KoRVs.
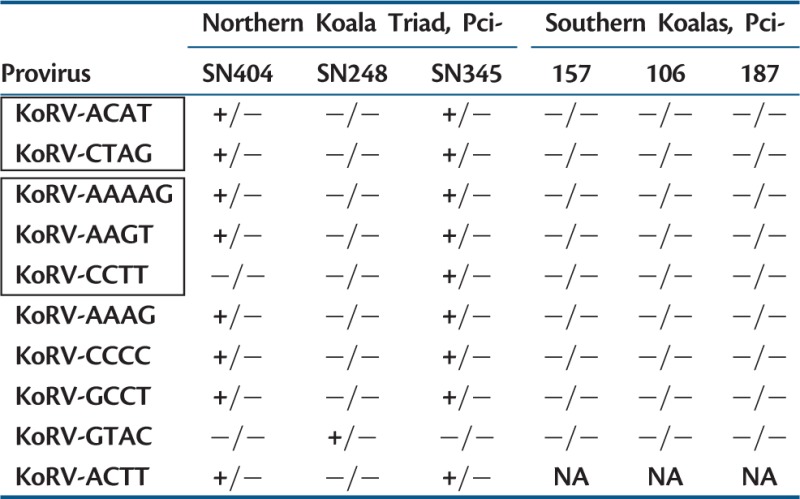 |
Note.—For the proviruses listed, flanks and LTRs were identified upstream and downstream of the integration site. +/+, provirus present on both chromosome homologues; +/−, provirus present on only one of the two homologs; −/−, neither chromosome had a provirus at the locus. KoRVs were first identified in Pci-SN404 except for CCTT (Pci-SN345) and GTAC (Pci-SN248). Boxes enclose proviral loci with identical LTR sequence. For KoRV-ACTT, there was no amplification (NA) in southern Australian koalas.
At each of the ten KoRV loci for which both flanks were identified, three different primer pairs were used, each pair in a separate PCR reaction, to evaluate insertion sites (fig. 1) (Roca et al. 2004). Two of the primer pairs established the presence of the 5′- or the 3′-flank and LTR, whereas the other primer pair would amplify only if KoRV was not present at the locus in at least one of the two chromosomes. Together, these would determine for a koala whether KoRV was present at a locus in both chromosomes, in one chromosome, or in neither of the two chromosomes (fig. 1). Using this strategy, the six koalas were screened for insertional polymorphisms across the ten proviruses (fig. 1 and table 2).
Fig. 1.
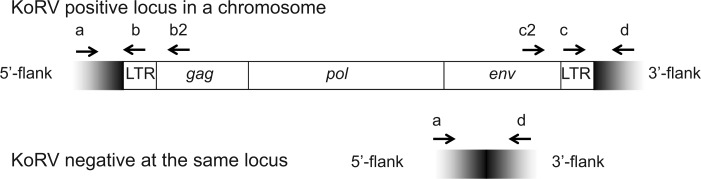
Strategy used to examine KoRV at a genomic locus. PCR was used to determine the presence or absence of KoRV in a particular koala at a particular genomic locus. Upper panel: Once flanking sequences in the koala genome (shaded) were identified for a proviral locus of KoRV, primers were designed to target the flanks (a, d) or to target KoRV (b, b2, c, c2). The presence or absence of a particular proviral locus of KoRV could be determined using three PCR reactions with limited elongation times. The primer pair a with b (or b2) would amplify when KoRV is present at a locus; as would primer pair d with c (or c2), although a with d will not amplify if KoRV is present at the locus. Lower panel: However, if KoRV is not integrated at a locus, primer pair a with d will amplify (the two contiguous flanks), but the other primer combinations will not. If no primer pairs amplify, the DNA may be of poor quality; whereas if all primer pairs amplify, it is an indication that KoRV is present at the locus in one of the two homologous chromosomes but not in the other. Note that primers b2 and c2 target KoRV genes, so that primers a with b2, or c2 with d, would amplify the complete 5′-LTR and 3′-LTR, respectively. However, b and c are within the LTR so that primers a with b, or c with d, would amplify only a portion of the LTR.
The screening involved six koalas, ten proviral loci, and two chromosomes for each locus, a total of 120 potential integration sites. Across the 120 sites, a provirus was detected only in 16 cases. In every one of these cases, the provirus was present at a locus in only one of the two chromosomes in an individual. If the progeny koala Pci-SN404 is excluded, since he could have received enKoRVs vertically from either parent, and only the five unrelated koalas are considered, then each of these ten KoRV proviruses was detected in only one koala individual. The lack of shared KoRV proviral loci among unrelated individuals, and the presence of each KoRV provirus in only one of the two chromosomes in a single individual, suggested that the KoRV proviruses were present at low frequencies across the koala population.
Identifying KoRVs as Endogenous Using Koala Kin
KoRV proviruses at each of the ten loci (table 2) were evaluated to determine whether they were endogenous. If a provirus detected in progeny koala Pci-SN404 could be detected in either parent, this would establish vertical transmission of an endogenous retrovirus due to Mendelian inheritance, as the probability that two proviruses would independently integrate at the same site in two individuals is minuscule (Johnson and Coffin 1999). The eight KoRVs present in progeny koala Pci-SN404 were also present in at least one of the parents, and therefore vertically transmitted and endogenous. The other two proviruses had been originally detected in one of the parents, but were not present in progeny Pci-SN404. Therefore, PCR was attempted using DNA that had been extracted from other koalas known to be kin (see description of these additional kin under Materials and Methods). The provirus KoRV-GTAC of Pci-SN248 was also detected in his daughter, Pci-SN374. The provirus KoRV-CCTT of Pci-SN345 was also detected in two of her siblings, Pci-SN339 and Pci-SN356. Thus all ten of these proviruses were shared among family members, and therefore were vertically transmitted and endogenous.
To determine at an even larger number of loci whether KoRV proviruses were endogenous, we examined the 29 additional proviral loci for which only one host genomic flank had been identified. These 29 loci had been detected in the progeny koala Pci-SN404 (table 3). Pci-SN404 is the son of Pci-SN248 (sire) and Pci-SN345 (dam). PCR was conducted using primers based on the 5′-flank and 5′-LTR (a and b in fig. 1) for 18 distinct proviruses for which the 5′-flank had been identified in Pci-SN404; or primers based on the 3′-LTR and 3′-flank (c and d in fig. 1) for 11 other proviruses for which the 3′-flank had been identified in Pci-SN404. Thus, 29 additional proviruses were successfully screened by PCR in the sire–dam–progeny triad of koalas. In every case, the provirus detected at a locus in Pci-SN404 was also present at the same locus in at least one of the parents (table 3). The provirus was detected at the locus in both of the parents in only one case. Thus, across a total of 39 loci (10 for which both flanks had been identified, and 29 for which a single flank could be screened), all 39 proved to be vertically transmitted and endogenous, although only one of these was present in the two unrelated parents. We did not find a locus in Pci-SN404 that was absent from both parents, which would have been indicative of an exogenous KoRV (or possibly a newly integrated enKoRV) in Pci-SN404. The 95% confidence interval for the proportion of enKoRVs among the distinct KoRV proviruses in the data set was calculated as 0.92–1.00 (using a “modified Wald method”) (Agresti and Coull 1998). Given our results, exogenous KoRVs would comprise (with 95% confidence) less than 0.08 of distinct KoRVs in the data set, if they were present at all.
Table 3.
KoRV Proviruses Screened in a Parent–Progeny Triad.
| Provirus | Progeny | Sire | Dam |
|---|---|---|---|
| Pci-SN404 | Pci-SN248 | Pci-SN345 | |
| 5′-LTR and flank | |||
| KoRV-5-AAGG | + | + | + |
| KoRV-5-AGTC | + | − | + |
| KoRV-5-ATAGa | + | − | + |
| KoRV-5-ATGG | + | − | + |
| KoRV-5-CAAC | + | + | − |
| KoRV-5-CCCCa | + | + | − |
| KoRV-5-CTAGa | + | + | − |
| KoRV-5-CTAT | + | + | − |
| KoRV-5-GTTG | + | + | − |
| KoRV-5-GAAG | + | − | + |
| KoRV-5-GAGC | + | − | + |
| KoRV-5-GCTT | + | − | + |
| KoRV-5-GTGC | + | + | − |
| KoRV-5-TCAT | + | − | + |
| KoRV-5-TGCA | + | + | − |
| KoRV-5-TTAC | + | − | + |
| KoRV-5-TTATa | + | + | − |
| KoRV-5-TTCC | + | − | + |
| 3′-LTR and flank | |||
| KoRV-3-AAAT | + | + | − |
| KoRV-3-AGAT | + | − | + |
| KoRV-3-AGGC | + | + | − |
| KoRV-3-ATAGa | + | − | + |
| KoRV-3-ATCA | + | − | + |
| KoRV-3-CTCC | + | − | + |
| KoRV-3-GATC | + | + | − |
| KoRV-3-GGAT | + | − | + |
| KoRV-3-GGCC | + | − | + |
| KoRV-3-GGTA | + | + | − |
| KoRV-3-TTATa | + | + | − |
Note.—For the KoRVs listed, the host flanking sequence was identified for only one side (5′ or 3′) of the integration site. + indicates presence of the provirus-flank region. − indicates that the provirus-flank region failed to amplify. All proviruses were initially detected in Pci-SN404. Proviruses included in table 2 are not listed here.
aThe 4-bp target site duplication of these KoRVs is also found in another KoRV (listed here or in table 2), but the other KoRV was at a different host locus.
Molecular Dating of enKoRVs in the Koala Germ Line
KoRV is common in koala museum specimens of northern Australian provenance collected in the late 1800s (Avila-Arcos et al. 2013). This establishes a minimum date for widespread infection of the northern Australian koala population by KoRV. To estimate a maximum date for the entry of KoRV into the koala germ line, we used a molecular clock relying on the divergence between the 5′- and 3′-LTR sequences within the same provirus. Retroviral 5′- and 3′-LTRs result from a duplication event at the time of integration, so that the two LTRs are initially identical in sequence. Once a provirus invades the host germ line, mutations will occur at the host nuclear mutation rate. Random mutations would thereafter cause proviral 5′- and 3′-LTR sequences to gradually accumulate differences, so that divergence between the two LTRs within the same provirus can be used to estimate the time since integration into the host germ line (Johnson and Coffin 1999; Bromham 2002; Coffin 2004; Roca et al. 2004). For the ten enKoRV proviruses for which flanking sequences on both sides of the integration site had been identified, we amplified each LTR separately. PCR used one primer based on a flank and another primer based on the proviral region beyond the LTR (gag for the 5′-LTR, env for the 3′-LTR) (fig. 1 and table 2). Both LTRs were then sequenced. As each provirus was present at the locus in only one of the two chromosomes, 5′- and 3′-LTR sequences of a provirus from the same individual would be in phase.
For each of the ten enKoRV loci, the 5′-LTR sequence was identical to the 3′-LTR sequence of the same provirus, although LTR sequences differed across proviruses. The complete lack of divergence between 5′- and 3′-LTR sequences within the same provirus could be used to estimate a maximum time since integration. For nine of the KoRV proviruses, each LTR had a length of 502 bp, whereas in the tenth provirus (KoRV-GTAG) the LTRs were each 483 bp. A mutation in either LTR would cause the two sequences to diverge; thus, no mutation had occurred in the 10,002 bp across the enKoRV LTRs since integration into the germ line. Although a nuclear mutation rate estimate for koalas is not available, we reasoned that it would fall between the human (slow evolving lineage) mutation rate of approximately 2 × 10−9 and the mouse (fast evolving) mutation rate of approximately 4.5 × 10−9 mutations per site per year (Kumar and Subramanian 2002; Waterston et al. 2002; Xue et al. 2009). Using these rates, we calculated that the first mutation anywhere along the LTRs of the ten enKoRVs would be expected to occur within 22,200–49,990 years of integration. As no mutations at all were detected in the LTRs, this would represent a maximum estimate, and the time of integration may be much more recent.
Although 5′- and 3′-LTR sequences were identical for each provirus, there were differences across the ten proviruses sequenced. The two proviruses KoRV-ACAT and KoRV-GTAC had identical LTR sequences, as did the three proviruses KoRV-AAAAG, KoRV-AAGT, and KoRV-CCTT (fig. 2 and table 2). The other five KoRV proviruses each had unique LTR sequences. The number of mutations separating the LTR sequences of different KoRVs was low, with at most seven mutations separating LTR sequences between proviruses (fig. 2), counting as a single mutation a 19-bp deletion in KoRV-GTAG spanning positions 129–147.
Fig. 2.
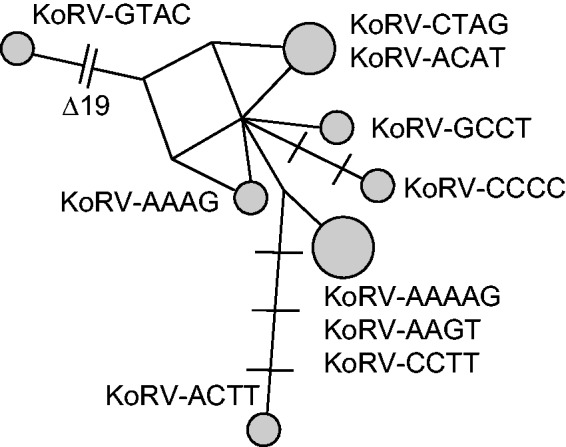
Relationships among KoRV LTR sequences. The MJ network (Bandelt et al. 1999) was generated using an alignment of complete enKoRV LTR sequences from ten proviruses. There were no differences between 5′- and 3′-LTR sequences within a provirus, for any of the enKoRVs. Across proviral sequences, the number of nucleotide differences is indicated by hatch marks (a branch without hatch marks represents a single difference). Node sizes are proportional to the number of proviruses in which the LTR sequence was detected; each provirus was designated by the sequence of its target site duplication (4-bp host sequence duplicated upstream and downstream of the provirus; except for KoRV-AAAAG, which had a 5-bp target site duplication). KoRV-GTAC had a 19-bp deletion (indicated by “Δ19”) in addition to other nucleotide differences when compared with the other proviruses.
Discussion
LTR sequences within ten proviruses allowed us to estimate a maximum date for the invasion of the germ line by endogenizing KoRVs of 22,200–49,990 years ago, although a much more recent date is possible. The estimated time of integration of less than 50,000 years is quite recent compared with those of endogenous retroviruses in other species (Johnson and Coffin 1999; Coffin 2004; Roca et al. 2004; Niebert and Tonjes 2005; Martins and Villesen 2011). Individual proviral copies in other species may be insertionally polymorphic, such as endogenous feline leukemia viruses (enFeLVs) in cats (Roca et al. 2004) and some HERV-K loci in humans (Turner et al. 2001; Moyes et al. 2007). One important difference distinguishes KoRV from these other ERVs. All individuals in the domestic cat lineage carry multiple copies of enFeLVs (Roca et al. 2005; Polani et al. 2010) and all humans carry multiple copies of HERV-K (Turner et al. 2001; Moyes et al. 2007). In contrast, KoRV is still in the process of spreading across koala populations, and many southern Australian koalas carry no copies of KoRV at all (Tarlinton et al. 2006; Simmons et al. 2012).
Although there were no LTR mutations detected within each enKoRV provirus, the LTR sequences did vary across the proviruses (fig. 2). Among the ten proviruses, seven LTR haplotypes were identified, indicating that at least seven different KoRVs have entered the koala germ line. No new mutations have occurred in the LTRs of the ten enKoRVs since the time of integration (otherwise there would be differences between 5′- and 3′-LTRs within a provirus). Thus, the LTR differences across proviruses existed before they invaded the germ line. Even across the KoRV proviruses, only a small number of differences separated LTR sequences (fig. 2), indicating that KoRV strains invading the koala germ line differed little from each other before their endogenization. A high rate of mutation often characterizes exogenous retroviruses (Katz and Skalka 1990), and the differences reported across KoRVs believed to be exogenous (Shimode et al. 2014) are greater than those separating the enKoRVs in figure 2. The low sequence diversity across KoRV LTRs before they endogenized may be an indication that retroviral invasion of the koala germ line occurred across a limited period of time, and it may be plausible that all of the enKoRVs formed as part of a single outbreak.
Of 39 distinct KoRV proviruses examined, all proved to be endogenous and none was potentially exogenous. If the 39 proviruses examined represent an unbiased sampling of KoRVs present in the genome, then our best estimate is that all of the KoRVs in the progeny were endogenous, with a 95% confidence interval of 0.92–1.00. As exogenous KoRVs may be present at lower copy number than enKoRVs (the latter would be integrated in every cell), bias was minimized by examining KoRVs with flanks that were present at both high and low frequencies among the reads (supplementary fig. S2 and table S2, Supplementary Material online). Although we tried to take steps to minimize bias, we cannot completely rule out the possibility that exogenous KoRVs might be present in the koalas, for example, if sequence differences in exogenous KoRVs would have prevented their being targeted by primers, or if their copy number was very low relative to that of enKoRVs. The genome-walking method (Reddy et al. 2008) involved nested PCR and this also might have produced some bias. Alternatively, it is also possible that exogenous KoRVs were not present among the koala triad. Simmons et al. (2012) has previously noted that the high proviral copy number of Queensland koalas may be due to endogenous transmission.
Each of the 39 enKoRVs examined was shown to be vertically transmitted; each locus was present in the progeny and at least one parent, or in a parent and one or more other koalas known to be kin. Although no novel enKoRVs were detected between the generations, the degree to which additional proliferation of new enKoRVs may continue among koalas is not yet clear. In studies of endogenous murine leukemia viruses (MuLVs), the proliferation of novel ERVs has been examined in highly inbred strains of mice. Novel endogenous proviral integrations into the germ line are very rare in low viremic strains of mice, although they occur more commonly in highly viremic strains (Rowe and Kozak 1980; Herr and Gilbert 1982; Jenkins et al. 1982; Jenkins and Copeland 1985). Yet even in mouse congenic strains in which highly expressed endogenous MuLV loci are bred into a background strain permissive for endogenous MuLV expression (SWR/J), the number of newly acquired proviruses was low (Jenkins and Copeland 1985). In the permissive conditions, only 18.6% of progeny acquired new germ line proviruses with only an average of 0.5 proviruses per individual (Jenkins and Copeland 1985). Although no novel enKoRVs were detected in the koala progeny, the mouse studies suggest that new ERVs may be generated only gradually through transposition or through exogenous KoRV integration.
Although all 39 KoRV proviruses examined proved to be endogenous, only one was shared between the unrelated sire and dam koalas (tables 2 and 3). This suggests that a proliferation (table 1) (Simmons et al. 2012) of low frequency enKoRVs (tables 2 and 3) has occurred among the germ lines of northern Australian koalas. The low frequency for each enKoRV would also explain why enKoRV loci were not detected in both chromosomes within individual koalas: Only enKoRVs present at high frequencies in their natal population would tend to be present in both chromosomes of an individual. Previous estimates suggest that northern Australian koalas carry an average of 165 copies of KoRV per cell (Simmons et al. 2012). Although the population size of northern Australian koalas is uncertain, one estimate placed it at approximately 167,000 (Department of the Environment, Canberra 2014). Given that only one of 39 enKoRVs was shared between the two parent koalas (zoo animals descended from Queensland koalas), one can extrapolate that the wild koalas of Queensland may have carried many thousands of distinct enKoRV loci, each present in only a small proportion of the individual koalas.
The low frequencies at which distinct enKoRVs are present across koala individuals and chromosomes (tables 2 and 3) would almost certainly be due to their very recent integration, as each enKoRV would have originated in a single germ line chromosome. The lower the frequency of an enKoRV, the greater the probability of its removal by genetic drift. Over time, genetic drift would remove most enKoRV loci from the koala population. However, a small proportion of loci would increase in frequency and become fixed through random drift (a process that would take many thousands of generations).
The low diversity of KoRV may reflect a low mutation rate for enKoRVs in the koala nuclear genome, which should be much slower than the mutation rate of exogenous KoRV retroviruses (Katz and Skalka 1990). The limited polymorphism reported for KoRV among modern and museum archive koalas has suggested that selection among KoRV sequence variants may not have played a strong role in KoRV evolution (Avila-Arcos et al. 2013; Tsangaras et al. 2014). This would not rule out the possibility that enKoRVs may greatly vary in their effects on koala fitness depending on where they integrated in the host genome. It seems likely that selection would favor koalas with fewer enKoRVs, or with enKoRVs that had fewer combined deleterious effects. The overall reduction in fitness of an individual due to enKoRVs would likely depend on the total number of enKoRVs present in the genome (Simmons et al. 2012), on the chromosomal locations of the enKoRVs (Buzdin et al. 2006; Lamprecht et al. 2010), and on whether they had one or two chromosomal copies at enKoRV loci (Bellone et al. 2013).
Every northern Australian koala carries many copies of KoRV (Simmons et al. 2012). Although the KoRV copy number estimated for northern Australian koalas is 165 copies/cell (Simmons et al. 2012), the variance across these koalas is limited (range 139–199 copies/cell) (Simmons et al. 2012). The limited range in copy number may reflect a tendency of random mating to equilibrate the number of enKoRVs per individual within a population. In contrast, across populations, studies of genetic diversity in koalas suggest that gene flow may be limited (Houlden et al. 1999). This may be particularly true between koala populations in northern and southern Australia, as the average copy number for KoRV is very low in the south relative to the north, whereas KoRV has been ubiquitous in the north for more than a century (Tarlinton et al. 2006; Simmons et al. 2012; Avila-Arcos et al. 2013). To the degree that gene flow can occur between north and south, this would be expected to eventually equilibrate the copy number of enKoRVs at a level intermediate between those currently found in northern and southern Australian koalas.
In summary, the northern Australian koala population is now marked by a very large number of enKoRV loci, but with each distinct enKoRV at low frequency in the population. Thus only a small proportion of enKoRVs would be shared between individuals, or present in both chromosomes of an individual. Our results suggest that the initial emergence of ERVs involves a massive proliferation of proviruses in the germ lines of one or more populations of the host species. After stabilization, the number of copies of the ERV would be reduced by selection against deleterious integrants; the number of ERV loci would be reduced by drift (with most disappearing but a small proportion becoming fixed), whereas admixture with populations that carry few or no copies of the ERV would lead to dilution and equilibration of ERV copy number.
Materials and Methods
Koala Samples
Ethical approval for this study was granted by the University of Illinois Institutional Animal Care and Use Committee, approved protocol number 12040. Blood samples from northern Australian koalas were obtained during regular physical examinations by trained staff at the Columbus Zoo and the San Diego Zoo, USA. The American Zoo Association’s Species Survival Plan manages northern (Queensland) and southern koalas separately. Three northern Australian koalas comprised a parent–progeny triad (progeny: Pci-SN404, sire: Pci-SN248, and dam: Pci-SN345). The pedigree of these individuals was available in the North American Studbook for koalas. Inbreeding was known to be limited in their pedigree. The parents shared only a single distant ancestor (great grandparent to the sire and great–great grandparent to the dam) and thus had a low estimated relatedness (r ≅ 0.008). In addition to the triad, other zoo samples that were kin to the triad included Pci-SN374, the daughter of Pci-SN248, and Pci-SN345; and two patrilineal siblings of Pci-SN345: Pci-SN339 and Pci-SN356. For zoo koalas, genomic DNA was extracted from buffy coat using the QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA). The southern Australian koala DNA samples were provided by the National Cancer Institute (NCI), USA, used the NCI sample numbers, and had been collected from free-ranging wild koalas in Australia. Pci-157 was from the Stony Rises of Victoria, Pci-106 was from the Brisbane Ranges of Victoria, and Pci-187 was from Kangaroo Island of South Australia (table 1). The blood samples had been collected under permit no. 87-150 issued by the Department of Conservation, Forests and Land, Victoria (Taylor et al. 1991). The DNA had been extracted using a phenol–chloroform method at NCI.
Screening Koala Samples for the Presence of KoRV
As some koala individuals and populations are largely free of KoRV, the DNA samples used in this study were screened to determine that they were KoRV positive by PCR using primers that were previously published (Tarlinton et al. 2006) or newly designed based on conserved regions of the LTRs (3′-LTR-F2: AGTTGTGTTCGCGTTGATCC, KoRV3LTR_F2R: TACCTCCCGTCGGTGGTT). The primer 3′-LTR-F2 was also used to isolate KoRV flanking regions (the next section has details). The PCR setup is described below, whereas the algorithm used was as previously described (Ishida et al. 2011).
Isolation and Sequencing of Koala Genomic Regions Flanking KoRV Proviruses
To identify host genomic regions flanking KoRV proviral integration sites, the genome-walking method established by Reddy et al. (2008) was implemented, although modified to use next-generation sequencing as illustrated in supplementary figure S1, Supplementary Material online. The REPLI-g Mini Kit (Qiagen) was used. Approximately 100 ng of each koala genomic DNA was denatured, following the REPLI-g kit protocols. Four different walker-adapter primers were then attached to each denatured DNA (Reddy et al. 2008) (supplementary table S3, Supplementary Material online), using a mix that consisted of 10 units of Phi29 DNA polymerase, 1× Phi29 DNA polymerase reaction buffer, 200 µM dNTPs, and 20 µM of each walker-adapter primer. The mixture was incubated at 30 °C for 90 min to initiate multiple primer extension events, then incubated at 65 °C for 10 min to inactivate the polymerase. The QIAquick PCR Purification Kit (Qiagen) was used to remove unincorporated walker-adapter primers, following the manufacturer’s protocol. The purified DNA fragments with walker-adapter primers were eluted using 40 µl of TLE buffer.
The eluted DNA was used as template for PCR procedures involved in the genome-walking method (supplementary fig. S1, Supplementary Material online) (Reddy et al. 2008). Each PCR relied on one walker primer and one KoRV-specific primer (supplementary table S3, Supplementary Material online). The KoRV-specific primers were designed using Primer3 (http://fokker.wi.mit.edu/primer3/input.htm, last accessed September 29, 2014) (Rozen and Skaletsky 2000), and designed to target the 5′-end or 3′-end of the LTR based on regions conserved among published KoRV sequences available at the time: GenBank accession numbers AF151794 (Hanger et al. 2000), DQ683164, DQ683166, DQ683167, and DQ683168 (Tarlinton 2006). A primary PCR was conducted as previously described (Reddy et al. 2008). In the subsequent nested PCR, the primary PCR product was used as template, and amplified using a pair of HPLC-purified primers (Integrated DNA Technologies, Coralville, IA). Primers were prepared by following the manufacturer’s protocol for the Roche Genome Sequencer System (Roche Applied Science, Penzberg, Germany). One primer consisted of three concatenated segments: A GS FLX Titanium adapter “Primer A” segment (CCATCTCATCCCTGCGTGTCTCCGACTCAG), a MID, and a KoRV-specific primer (supplementary table S3, Supplementary Material online). The MID used was the same across the four different amplicons of walker-adapter but distinctive for each koala individual, and for each run (5′ or 3′). The second primer consisted of two concatenated segments: The GS FLX Titanium adapter “Primer B” (CCTATCCCCTGTGTGCCTTGGCAGTCTCAG) and a second walker primer previously described (Reddy et al. 2008) (supplementary table S3, Supplementary Material online). PCR was conducted using the FastStart High Fidelity PCR System (Roche Applied Science) and the PCR components and algorithm conformed to the manufacturer’s protocol. The resulting PCR amplicons were purified using AMPure XP beads (Beckman Coulter, CA) with a magnetic particle concentrator. The concentrations of the purified nested PCR amplicons were estimated using a Qubit 2.0 Fluorometer (Life Technologies Corp.) and amplicon sizes, quality, and quantity were measured using an Agilent 2100 Bioanalyzer at the Functional Genomics Unit, Biotechnology Center (Biotech Center) at the University of Illinois at Urbana-Champaign (UIUC). Amplicon concentrations were adjusted so that equal amounts would be pooled. The pooled sample was eluted on an agarose gel and separated into two size classes, one approximately 200–400 bp and the other approximately 400–1,000 bp at the High-Throughput Sequencing and Genotyping Unit, Biotech Center at UIUC. Each size class was run separately on 1/16th of a PicoTiterPlate (PTP) (1/8 PTP total) on the Roche 454 GS FLX+ platform at the UIUC High-Throughput Sequencing and Genotyping Unit.
Bioinformatics Processing of Next-Generation Sequences
Reads generated by the Roche 454 GS FLX platform were converted into FASTQ format using the Galaxy platform (Giardine et al. 2005; Blankenberg et al. 2010; Goecks et al. 2010). The experimentally ligated MID formed part of the sequence read and indicated which koala the sequence originated in, and whether the 5′-end of the LTRs or the 3′-end of the LTRs was the target. As 5′- and 3′-LTRs have nearly identical sequences, about half of the PCR amplicons and subsequent sequencing reads would be expected to identify sequence within the KoRV provirus rather than sequences in the host flanks. To remove reads matching the KoRV provirus, we used Bowtie2 (Langmead and Salzberg 2012), using the “very sensitive local alignment” preset, to attempt to map all reads to published KoRV sequence AF151794 (Hanger et al. 2000). Only reads that did not map to KoRV genes were further considered.
To identify the boundary between the KoRV LTR and the koala flanking genomic sequence, the flanks were mapped onto the published KoRV LTR sequence using Bowtie2 (Langmead and Salzberg 2012), using the “very sensitive local alignment” preset. The LTR sequences proved to be 2 bp shorter at the 5′-end and 1 bp shorter at the 3′-end of the LTRs (total 3 bp shorter) than the published reference sequence (Hanger et al. 2000).
A number of steps were taken to find the matching 5′- and 3′-flanks at a single proviral locus. First, the flank sequences were trimmed to include approximately 10 bp of the end of the LTR and 10 bp of the koala genomic regions. Each flank sequence was aligned to the Meug_1.1 assembly of the genome of the tammar wallaby (Renfree et al. 2011) using BLASTN (Altschul et al. 1990) using parameters for short local alignment. Flanks that aligned to more than three scaffolds were removed to reduce the possibility that multiple unique flanks of KoRV might be misidentified as one insertion. We wrote a routine using BioPython (Cock et al. 2009) to filter the BLAST results for pairs of 5′- and 3′-koala genomic flanks. The matched 5′- and 3′-flanks should be aligned within 10 bp due to target site duplication but not overlapping by more than 10 bp, on the same wallaby scaffold, in the proper orientation, to identify koala genomic sequences that corresponded to the 5′- and 3′-host genomic flanks of the same KoRV locus. The trio of sequences (5′ of the proviral integration site, 3′ of the proviral integration site, plus the matching wallaby segment) was then realigned and visually inspected in the software Sequencher 5.1 (Gene Codes Corp., MI).
To identify additional matched flanking sequences on either side of a single proviral locus, all flank sequences were queried against low-coverage koala genomic sequences. For this search, Bowtie2 (version 2.1.0) (Langmead and Salzberg 2012) was run on the Galaxy platform (Giardine et al. 2005; Blankenberg et al. 2010; Goecks et al. 2010). The koala genomic reads had been generated using DNA from Pci-SN404, sequenced on 1/16th of a PTP of the Roche 454 GS FLX+ platform (Roche Applied Science) run at the High-Throughput Sequencing and Genotyping Unit, UIUC, as has been previously described (Ruiz-Rodriguez et al. 2014).
To estimate the number of distinct retroviral integrations from the host flanks sequenced by the Roche 454 GS FLX platform (supplementary table S1, Supplementary Material online), the reads were trimmed to only include approximately 50 bp of host genomic flank adjacent to the proviral LTR. We retained only those reads that contained at least 50 bp of host genomic flank and had a base call quality of 99% for every position in the 50 bp. This minimized the possibility of an inflated count due to sequencing errors. For each MID data set iteration, we used the Megablast algorithm in BLASTN (Altschul et al. 1990) to cross-align all filtered reads from the same iteration, and grouped together those reads that were at least 80% similar, with each group of reads counted as a “distinct” flank sequence. These criteria for grouping the number of distinct flank sequences may have somewhat underestimated the total. For each distinct grouping of flank sequences, the consensus 4 bp at the LTR boundary was taken as the target site duplication for the integration site. The number of proviruses for each koala was estimated as the number of distinct flank sequences detected for 5′- and 3′-flanks separately, and present in at least two of the sequence reads (singletons were removed to minimize potential error). Then for each target site duplication, the number of 5′- and 3′-distinct sequences was compared, and the larger of the two for each target site duplication was used in estimating the total number of reads for each individual koala (supplementary table S1, Supplementary Material online). The number of sequencing reads per distinct flank is shown in supplementary figure S2, Supplementary Material online.
PCR and Sequencing of Flanks and LTRs, and Network Analysis of LTRs
PCR primers were designed using the software Primer3 (http://fokker.wi.mit.edu/primer3/input.htm, last accessed September 29, 2014) (Rozen and Skaletsky 2000), targeting koala genomic sequences flanking proviral integration sites, or targeting KoRV LTR sequence (supplementary tables S4 and S5, Supplementary Material online). Primers for identification of enKoRVs in the dam–sire–progeny triad were designed based on flank reads from the Roche 454 GS FLX+ platform for Pci-SN404 (progeny). To minimize potential bias in detecting endogenous over exogenous KoRVs, half of the primer sets were designed based on distinct flanks that were detected in high frequencies among the sequence reads, whereas the rest were designed based on distinct flanks that were detected in low frequencies among the sequence reads (supplementary table S2 and fig. S2, Supplementary Material online). Only the successful primers are shown in supplementary table S5, Supplementary Material online. To minimize the targeting of repetitive regions within the koala genome, flank primer sequences were queried against low coverage whole-genome sequence of Pci-SN404 from a 1/16th PTP run on the Roche 454 GS FLX+ platform (Ruiz-Rodriguez et al. 2014), although none of them was found to be in repetitive regions by using this low coverage sequence. When the same 4-bp target site duplication was identified upstream of a 5′-LTR and downstream of a 3′-LTR, PCR was conducted using a primer that targeted the 5′-flank with one that targeted the 3′-flank (table 3), to determine whether the two primers flanked the same locus, using DNA from a koala known not to carry the relevant KoRV(s) (fig. 1).
PCR mixes used a final concentration of 0.4 µM of each primer, 1.5 mM MgCl2, 200 µM of each dNTP (Life Technologies Corp., CA), and 0.04 units/µl of AmpliTaq Gold DNA Polymerase (Life Technologies Corp.). The PCR algorithm consisted of an initial denaturation and activation of AmpliTaq Gold at 95 °C for 9:45 min; with cycles of 20-s denaturation at 94 °C, followed by 30-s annealing at 60 °C (first three cycles), decreasing the annealing temperature in 2 °C steps to 58, 56, 54 and 52 °C (five cycles each), or 50 °C (last 22 cycles), followed by 1-min extension at 72 °C; with a final extension of 7 min at 72 °C. An aliquot of each PCR amplicon was examined on an agarose gel with ethidium bromide under UV light. Amplicons were treated with Exonuclease I (USB Corporation, OH) and shrimp alkaline phosphatase (USB Corporation) to remove excess primers and unincorporated dNTPs (Hanke and Wink 1994). Sanger sequencing was performed in both directions using the BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies Corp.) with 2.5 µl of purified PCR product and 0.12 µM primer (M13 forward or reverse), as previously described (Ishida et al. 2011), and purified and resolved on an ABI 3730XL capillary sequencer at the High-Throughput Sequencing and Genotyping Unit, Biotech Center at UIUC. The software Sequencher 4.5 (Gene Codes Corp., MI) was used to examine and edit chromatograms. For the LTR sequence across ten KoRV proviruses, a median-joining (MJ) network was constructed using the software Network 4.6.1.1 (Bandelt et al. 1999). Flank and LTR sequences of ten proviruses were deposited in GenBank (accession numbers: KJ152809–KJ152818). For 30 proviruses, only the 5′- or the 3′-flank is known; these are included as supplementary sequences, Supplementary Material online.
For each distinct proviral flank verified by PCR and Sanger sequencing, the 50 bp of host genomic sequence flanking the provirus identified by Sanger sequencing was used as a query against the Roche 454 flank sequencing data set, and the number of matching reads was recorded (supplementary table S2, Supplementary Material online), in order to showthat proviruses were evenly distributed among flanks with low numbers of reads and flanks with high numbers of reads.
Statistical Analyses
Confidence intervals were calculated using the “modified Wald method” (Agresti and Coull 1998) implemented in GraphPad (http://graphpad.com/quickcalcs/confInterval1/, last accessed September 29, 2014) for the confidence interval of a proportion. As all 39 KoRVs examined were determined to be endogenous, with no exogenous KoRVs detected, the confidence intervals were adjusted to account for data in which the observed proportion is zero, and the true proportion cannot be lower than zero (i.e., uncertainty is unidirectional and not bidirectional).
Supplementary Material
Supplementary sequences, figures S1 and S2, and tables S1–S5 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank S.J. Golubovich for technical assistance. They also thank C. Wright, A.G. Hernandez, and M.F. Majewski of the Roy J. Carver Biotechnology Center of UIUC. They also thank the Cleveland Zoo, Columbus Zoo, Dallas Zoo, Riverbanks Zoo, San Diego Zoo, and San Francisco Zoo, as well as M. Malasky, R. Hanson, S. O’Brien, M. Bush, J. Graves, W. Sherwin and D. Wildt for koala samples. The project described was supported by Grant Number R01GM092706 from the National Institute of General Medical Sciences (NIGMS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS or the National Institutes of Health.
References
- Agresti A, Coull BA. Approximate is better than “exact” for interval estimation of binomial proportions. Am Stat. 1998;52:119–126. [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Avila-Arcos MC, Ho SY, Ishida Y, Nikolaidis N, Tsangaras K, Honig K, Medina R, Rasmussen M, Fordyce SL, Calvignac-Spencer S, et al. One hundred twenty years of koala retrovirus evolution determined from museum skins. Mol Biol Evol. 2013;30:299–304. doi: 10.1093/molbev/mss223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballandras-Colas A, Naraharisetty H, Li X, Serrao E, Engelman A. Biochemical characterization of novel retroviral integrase proteins. PLoS One. 2013;8:e76638. doi: 10.1371/journal.pone.0076638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Bellone RR, Holl H, Setaluri V, Devi S, Maddodi N, Archer S, Sandmeyer L, Ludwig A, Foerster D, Pruvost M, et al. Evidence for a retroviral insertion in TRPM1 as the cause of congenital stationary night blindness and leopard complex spotting in the horse. PLoS One. 2013;8:e78280. doi: 10.1371/journal.pone.0078280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: A web-based genome analysis tool for experimentalists. In: Current Protocols in Molecular Biology: John Wiley & Sons, Inc. 2010;89 doi: 10.1002/0471142727.mb1910s89. 19.10.1-19.10.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD, Stoye JP. Retrotransposons, endogenous retroviruses, and the evolution of retroelements. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997. pp. 343–436. [PubMed] [Google Scholar]
- Bromham L. The human zoo: endogenous retroviruses in the human genome. Trends Ecol Evol. 2002;17:91–97. [Google Scholar]
- Buzdin A, Kovalskaya-Alexandrova E, Gogvadze E, Sverdlov E. At least 50% of human-specific HERV-K (HML-2) long terminal repeats serve in vivo as active promoters for host nonrepetitive DNA transcription. J Virol. 2006;80:10752–10762. doi: 10.1128/JVI.00871-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin JM. Evolution of retroviruses: fossils in our DNA. Proc Am Philos Soc. 2004;148:264–280. [PubMed] [Google Scholar]
- Craigie R, Bushman FD. HIV DNA integration. Cold Spring Harb Perspect Med. 2012;2:a006890. doi: 10.1101/cshperspect.a006890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denner J. Transspecies transmissions of retroviruses: new cases. Virology. 2007;369:229–233. doi: 10.1016/j.virol.2007.07.026. [DOI] [PubMed] [Google Scholar]
- Department of the Environment, Canberra. 2014. Phascolarctus cinereus (combined populations of QLD, NSW and the ACT) in Species Profile and Threats Database [Internet] [cited 2014 Jun 26]. Available from: http://www.environment.gov.au/cgi-bin/sprat/public/publicspecies.pl?taxon_id=85104. [Google Scholar]
- Fiebig U, Hartmann MG, Bannert N, Kurth R, Denner J. Transspecies transmission of the endogenous koala retrovirus. J Virol. 2006;80:5651–5654. doi: 10.1128/JVI.02597-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanger JJ, Bromham LD, McKee JJ, O’Brien TM, Robinson WF. The nucleotide sequence of koala (Phascolarctos cinereus) retrovirus: a novel type C endogenous virus related to gibbon ape leukemia virus. J Virol. 2000;74:4264–4272. doi: 10.1128/jvi.74.9.4264-4272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke M, Wink M. Direct DNA sequencing of PCR-amplified vector inserts following enzymatic degradation of primer and dNTPs. Biotechniques. 1994;17:858–860. [PubMed] [Google Scholar]
- Hayward JA, Tachedjian M, Cui J, Field H, Holmes EC, Wang LF, Tachedjian G. Identification of diverse full-length endogenous betaretroviruses in megabats and microbats. Retrovirology. 2013;10:35. doi: 10.1186/1742-4690-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr W, Gilbert W. Germ-line MuLV reintegrations in AKR/J mice. Nature. 1982;296:865–868. doi: 10.1038/296865a0. [DOI] [PubMed] [Google Scholar]
- Houlden BA, Costello BH, Sharkey D, Fowler EV, Melzer A, Ellis W, Carrick F, Baverstock PR, Elphinstone MS. Phylogeographic differentiation in the mitochondrial control region in the koala, Phascolarctos cinereus (Goldfuss 1817) Mol Ecol. 1999;8:999–1011. doi: 10.1046/j.1365-294x.1999.00656.x. [DOI] [PubMed] [Google Scholar]
- Houlden BA, England PR, Taylor AC, Greville WD, Sherwin WB. Low genetic variability of the koala Phascolarctos cinereus in south-eastern Australia following a severe population bottleneck. Mol Ecol. 1996;5:269–281. [PubMed] [Google Scholar]
- Ishida Y, Demeke Y, van Coeverden de Groot PJ, Georgiadis NJ, Leggett KE, Fox VE, Roca AL. Distinguishing forest and savanna African elephants using short nuclear DNA sequences. J Hered. 2011;102:610–616. doi: 10.1093/jhered/esr073. [DOI] [PubMed] [Google Scholar]
- Jenkins NA, Copeland NG. High frequency germline acquisition of ecotropic MuLV proviruses in SWR/J-RF/J hybrid mice. Cell. 1985;43:811–819. doi: 10.1016/0092-8674(85)90254-5. [DOI] [PubMed] [Google Scholar]
- Jenkins NA, Copeland NG, Taylor BA, Lee BK. Organization, distribution, and stability of endogenous ecotropic murine leukemia virus DNA sequences in chromosomes of Mus musculus. J Virol. 1982;43:26–36. doi: 10.1128/jvi.43.1.26-36.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Coffin JM. Constructing primate phylogenies from ancient retrovirus sequences. Proc Natl Acad Sci U S A. 1999;96:10254–10260. doi: 10.1073/pnas.96.18.10254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz RA, Skalka AM. Generation of diversity in retroviruses. Annu Rev Genet. 1990;24:409–445. doi: 10.1146/annurev.ge.24.120190.002205. [DOI] [PubMed] [Google Scholar]
- Kumar S, Subramanian S. Mutation rates in mammalian genomes. Proc Natl Acad Sci U S A. 2002;99:803–808. doi: 10.1073/pnas.022629899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht B, Walter K, Kreher S, Kumar R, Hummel M, Lenze D, Kochert K, Bouhlel MA, Richter J, Soler E, et al. Derepression of an endogenous long terminal repeat activates the CSF1R proto-oncogene in human lymphoma. Nat Med. 2010;16:571–579. doi: 10.1038/nm.2129. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AK, Martin R. 1988. The koala: a natural history. Kensington (NSW): NSW University Press. [Google Scholar]
- Levinson G, Gutman GA. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol. 1987;4:203–221. doi: 10.1093/oxfordjournals.molbev.a040442. [DOI] [PubMed] [Google Scholar]
- Martins H, Villesen P. Improved integration time estimation of endogenous retroviruses with phylogenetic data. PLoS One. 2011;6:e14745. doi: 10.1371/journal.pone.0014745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith RW, Westerman M, Springer MS. A phylogeny and timescale for the living genera of kangaroos and kin (Macropodiformes: Marsupialia) based on nuclear DNA sequences. Aust J Zool. 2009;56:395–410. [Google Scholar]
- Mi S, Lee X, Li X, Veldman GM, Finnerty H, Racie L, LaVallie E, Tang XY, Edouard P, Howes S, et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature. 2000;403:785–789. doi: 10.1038/35001608. [DOI] [PubMed] [Google Scholar]
- Moyes D, Griffiths DJ, Venables PJ. Insertional polymorphisms: a new lease of life for endogenous retroviruses in human disease. Trends Genet. 2007;23:326–333. doi: 10.1016/j.tig.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Niebert M, Tonjes RR. Evolutionary spread and recombination of porcine endogenous retroviruses in the suiformes. J Virol. 2005;79:649–654. doi: 10.1128/JVI.79.1.649-654.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polani S, Roca AL, Rosensteel BB, Kolokotronis SO, Bar-Gal GK. Evolutionary dynamics of endogenous feline leukemia virus proliferation among species of the domestic cat lineage. Virology. 2010;405:397–407. doi: 10.1016/j.virol.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Reddy PS, Mahanty S, Kaul T, Nair S, Sopory SK, Reddy MK. A high-throughput genome-walking method and its use for cloning unknown flanking sequences. Anal Biochem. 2008;381:248–253. doi: 10.1016/j.ab.2008.07.012. [DOI] [PubMed] [Google Scholar]
- Renfree MB, Papenfuss AT, Deakin JE, Lindsay J, Heider T, Belov K, Rens W, Waters PD, Pharo EA, Shaw G, et al. Genome sequence of an Australian kangaroo, Macropus eugenii, provides insight into the evolution of mammalian reproduction and development. Genome Biol. 2011;12:R81. doi: 10.1186/gb-2011-12-8-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca AL, Nash WG, Menninger JC, Murphy WJ, O’Brien SJ. Insertional polymorphisms of endogenous feline leukemia viruses. J Virol. 2005;79:3979–3986. doi: 10.1128/JVI.79.7.3979-3986.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca AL, Pecon-Slattery J, O’Brien SJ. Genomically intact endogenous feline leukemia viruses of recent origin. J Virol. 2004;78:4370–4375. doi: 10.1128/JVI.78.8.4370-4375.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe WP, Kozak CA. Germ-line reinsertions of AKR murine leukemia virus genomes in Akv-1 congenic mice. Proc Natl Acad Sci U S A. 1980;77:4871–4874. doi: 10.1073/pnas.77.8.4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Ruiz-Rodriguez C, Ishida Y, Greenwood A, Roca A. Development of 14 microsatellite markers in the Queensland koala (Phascolarctos cinereus adustus) using next generation sequencing technology. Conserv Genet Resour. 2014;6:429–431. doi: 10.1007/s12686-013-0115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimode S, Nakagawa S, Yoshikawa R, Shojima T, Miyazawa T. Heterogeneity of koala retrovirus isolates. FEBS Lett. 2014;588:41–46. doi: 10.1016/j.febslet.2013.10.046. [DOI] [PubMed] [Google Scholar]
- Shojima T, Hoshino S, Abe M, Yasuda J, Shogen H, Kobayashi T, Miyazawa T. Construction and characterization of an infectious molecular clone of koala retrovirus. J Virol. 2013;87:5081–5088. doi: 10.1128/JVI.01584-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shojima T, Yoshikawa R, Hoshino S, Shimode S, Nakagawa S, Ohata T, Nakaoka R, Miyazawa T. Identification of a novel subgroup of koala retrovirus from koalas in Japanese zoos. J Virol. 2013;87:9943–9948. doi: 10.1128/JVI.01385-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G, Young P, McKee JJ, Meers J. The epidemiology of koala retrovirus. Jpn Soc Vet Epidemiol. 2011;15:1–9. [Google Scholar]
- Simmons GS, Young PR, Hanger JJ, Jones K, Clarke D, McKee JJ, Meers J. Prevalence of koala retrovirus in geographically diverse populations in Australia. Aust Vet J. 2012;90:404–409. doi: 10.1111/j.1751-0813.2012.00964.x. [DOI] [PubMed] [Google Scholar]
- Stoye JP. Koala retrovirus: a genome invasion in real time. Genome Biol. 2006;7:241. doi: 10.1186/gb-2006-7-11-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoye JP. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol. 2012;10:395–406. doi: 10.1038/nrmicro2783. [DOI] [PubMed] [Google Scholar]
- Tarlinton R, Meers J, Hanger J, Young P. Real-time reverse transcriptase PCR for the endogenous koala retrovirus reveals an association between plasma viral load and neoplastic disease in koalas. J Gen Virol. 2005;86:783–787. doi: 10.1099/vir.0.80547-0. [DOI] [PubMed] [Google Scholar]
- Tarlinton R, Meers J, Young P. Biology and evolution of the endogenous koala retrovirus. Cell Mol Life Sci. 2008;65:3413–3421. doi: 10.1007/s00018-008-8499-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlinton RE. Brisbane: University of Queensland; 2006. Characterisation of the epidemiology and molecular biology of koala retrovirus. [Google Scholar]
- Tarlinton RE, Meers J, Young PR. Retroviral invasion of the koala genome. Nature. 2006;442:79–81. doi: 10.1038/nature04841. [DOI] [PubMed] [Google Scholar]
- Taylor AC, Graves JAM, Murray ND, Sherwin WB. Conservation genetics of the koala (Phascolarctos cinereus) II. Limited variability in minisatellite DNA-sequences. Biochem Genet. 1991;29:355–363. doi: 10.1007/BF00554143. [DOI] [PubMed] [Google Scholar]
- Tsangaras K, Siracusa MC, Nikolaidis N, Ishida Y, Cui P, Vielgrader H, Helgen KM, Roca AL, Greenwood AD. Hybridization capture reveals evolution and conservation across the entire koala retrovirus genome. PLoS One. 2014;9:e95633. doi: 10.1371/journal.pone.0095633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner G, Barbulescu M, Su M, Jensen-Seaman MI, Kidd KK, Lenz J. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr Biol. 2001;11:1531–1535. doi: 10.1016/s0960-9822(01)00455-9. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Weiss RA. On the concept and elucidation of endogenous retroviruses. Philos Trans R Soc Lond B Biol Sci. 2013;368:20120494. doi: 10.1098/rstb.2012.0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Stadler CK, Gorman K, Jensen N, Kim D, Zheng H, Tang S, Switzer WM, Pye GW, Eiden MV. An exogenous retrovirus isolated from koalas with malignant neoplasias in a US zoo. Proc Natl Acad Sci U S A. 2013;110:11547–11552. doi: 10.1073/pnas.1304704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Wang Q, Long Q, Ng BL, Swerdlow H, Burton J, Skuce C, Taylor R, Abdellah Z, Zhao Y, et al. Human Y chromosome base-substitution mutation rate measured by direct sequencing in a deep-rooting pedigree. Curr Biol. 2009;19:1453–1457. doi: 10.1016/j.cub.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.