

ABSTRACT
Amebiasis is an enteric infection caused by Entamoeba histolytica, with symptoms ranging in severity from asymptomatic colonization to dysentery. Humans with the Q223R leptin receptor mutation have increased susceptibility to amebiasis, but the mechanism has been unclear. Using a mouse model expressing the mutation, we tested the impact of the Q223R mutation on the innate immune response to E. histolytica infection. The 223R mutation resulted in delayed clearance of amebae from the cecum, as had been previously observed. We found that neutrophil influx to the site of the infection was reduced 12 h after infection in 223R mice. Depletion of neutrophils with anti-Ly6G monoclonal antibody increased susceptibility of wild-type mice to infection, supporting the importance of neutrophils in innate defense. Leptin expression was increased in the cecum by E. histolytica infection, suggesting that leptin could serve as a homing signal for neutrophils to the gut. Interestingly, neutrophils from mice with the 223R mutation had diminished chemotaxis toward leptin. This impaired chemotaxis likely explained the reduced gut infiltration of neutrophils. The newly recognized effect of the leptin receptor Q223R mutation on neutrophil chemotaxis and the impact of this mutation on multiple infectious diseases suggest a broader impact of this mutation on susceptibility to disease.
IMPORTANCE
The Q223R leptin receptor mutation results in increased susceptibility of children and adults to E. histolytica, one of the leading causes of diarrhea morbidity and mortality in children of the developing world. Here we show that the mutation results in reduced neutrophil infiltration to the site of infection. This decreased infiltration is likely due to the mutation’s impact on neutrophil chemotaxis toward leptin, an inflammatory agent upregulated in the cecum after infection. The significance of this work thus extends beyond understanding E. histolytica susceptibility by also providing insight into the potential impact of leptin on neutrophil function in other states of altered leptin signaling, which include both malnutrition and obesity.
INTRODUCTION
Entamoeba histolytica is a protozoan parasite transmitted through the fecal-oral route. It causes a range of symptoms, from mild diarrhea to dysentery, and is an important pathogen in children (1, 2). A hallmark of infection is a strong innate response; neutrophilia is often seen at the site of infection (3). A large proportion of cases are asymptomatic, suggesting that host factors, including host genetics, may play an important role in determining susceptibility to infection and disease outcomes.
A study by Duggal et al. (4) identified a glutamine-to-arginine mutation (Q223R) in the leptin receptor that increased a child’s susceptibility to E. histolytica. Children homozygous for the R223 allele were 4 times more likely to have E. histolytica diarrhea than those homozygous for the wild-type Q223 allele. The Q223R mutation is located in the extracellular domain of the leptin receptor. Binding of leptin to the receptor is not affected by the mutation, as measured in vitro through the use of surface plasmon resonance (5). However, signal transduction through the activation of STAT3 by the R223 receptor is impaired in vitro (6), indicating that the mutation does have a functional effect on the receptor.
Leptin is a pleiotropic molecule, and its receptor is ubiquitously expressed (7). Leptin signaling appears to be important for a variety of tissues, affecting cellular functions such as apoptosis and cytokine expression as well as acting as an inflammatory cytokine (8). Malnutrition, which results in reduced leptin levels, increases susceptibility to many infections, including E. histolytica (9, 10). The Q223R mutation is the first leptin receptor mutation to be associated with an infectious disease and lends strength to the importance of leptin signaling in disease resistance. This study investigated the effect on host defense conferred by this mutation. We discovered that neutrophil chemotaxis toward leptin was impaired, likely contributing to reduced neutrophil influx to the site of infection and eventual susceptibility.
RESULTS
R223 mice exhibited reduced inflammation and infiltration after infection with E. histolytica.
The mouse model of an E. histolytica cecal infection was used to determine the differences between Q223 and R223 mice early during an infection. Previous work had shown that by 12 h after infection a difference could be observed in infection rates between Q223 and R223 mice, with complete eradication in Q223 but not R223 mice by 72 h (11). This 12-h time point was therefore chosen to investigate the very early host processes that could account for the differences in phenotypes. Q223 and R223 allele-expressing mice were infected with 2 × 106 E. histolytica trophozoites for 12 h, after which the cecum was harvested and evaluated for inflammation and infiltrating immune cells in the cecum. Histology scores revealed a reduction in inflammation in R223 mice, as graded by submucosal edema and epithelial blunting (Fig. 1A and B). Epithelial destruction, not included in the histology measurements but commonly seen in previous histologic analyses of late amebic infection, was not observed in either genotype at this early time point.
FIG 1 .

R223 mice exhibit reduced cecal inflammation. (A) Representative images of histology staining with H&E show reduced inflammation in the cecum of R223 mice. (Top) Q223 cecal section; (bottom) R223 cecal section. (B) Histological scoring of epithelial blunting and submucosal edema was performed blinded. *, P = 0.03 by two-tailed t test.
The 12- to 72-h time interval for eradication of amebae in Q223 mice suggested an innate mechanism could be responsible. At 12 h after infection, cecal lamina propria tissue samples from mice were reduced to a single-cell suspension, and flow cytometry was used to identify cell types. Neutrophils, identified as Ly6G and CD11b double-positive cells (Fig. 2A), were observed to make up a smaller percentage of the cells in the R223 mice (Fig. 2B). Other innate cell types, such as macrophages (CD11b+) and inflammatory monocytes (CD11b+ Ly6C+) were present at equivalent rates in both genotypes (data not shown). Naive Q223 and R223 mice had very low but similar amounts of neutrophils in the cecal lamina propria (Fig. 2C) and equivalent amounts of circulating blood neutrophils (Fig. 2D). The defect in neutrophil number therefore was not present at rest but only locally in the gut during an infection, suggesting a potential defect in infiltration or survival.
FIG 2 .

R223 mice had reduced neutrophil infiltration after infection. (A) Neutrophils were identified in the lamina propria by double expression of Ly6G and CD11b (top, Q223; bottom, R223). (B) Analysis of cell populations in the cecal lamina propria 12 h after infection revealed reduced neutrophil infiltration in R223 mice compared to Q223 mice. (C and D) Neutrophil populations are present at equivalent frequencies in the cecum (C) and blood (D) of naive mice. *, P = 0.01 by two-tailed test.
Neutrophil infiltration was important for amebic clearance.
The importance of neutrophils specifically for clearance of an E. histolytica infection was unknown. To test if diminished neutrophil presence in the cecum was responsible for the susceptible phenotype of the R223 mice, neutrophils were depleted with an anti-Ly6G monoclonal antibody (MAb) in C57BL/6 mice (Fig. 3A), an ameba-resistant strain of mouse (12). Administration of anti-Ly6G MAb achieved significant depletion of neutrophils in the lamina propria (Fig. 3B). After 12 h of infection, mice that had received the anti-Ly6G MAb had double the infection rate of mice that had received the isotype control. This suggested that a neutrophil response is critical for clearance of amebae early during infection and that a reduction in this response is an important mechanism for the susceptibility observed in mice with the R223 allele.
FIG 3 .
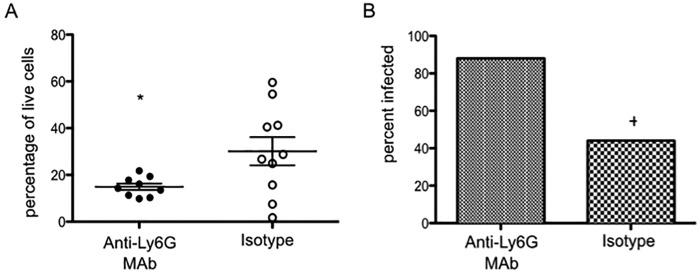
Depletion of neutrophils increases susceptibility to amebiasis. (A) C57BL/6 mice were treated with 50 µg of anti-Ly6G MAb or IgG isotype control 12 h before infection with E. histolytica. Depletion of neutrophils was confirmed in the cecal lamina propria 12 h after infection by using flow cytometry. (B) Infection was evaluated after 12 h of infection by culture positivity on the day of sacrifice. *, P = 0.03 by two-tailed t test; +, P = 0.04 by Mann-Whitney test.
Immune cell death and amebic trogocytosis were not enhanced by the R223 polymorphism.
Previous work worked showed that R223 cecal epithelial cells have increased caspase-3 activation and subsequent apoptosis after an E. histolytica infection, suggesting that leptin signaling is responsible for cell survival in the face of amebic challenge. The reduction in signal engendered by the R223 polymorphism could result in increased death of immune cells. Amebic trogocytosis has been identified as a killing mechanism of E. histolytica for several cell types, including Jurkat cells (13). We tested if reduced leptin signaling resulted in an increase in amebic trogocytosis and cell death. This was done by comparing the death and ingestion rates of splenocytes isolated from ob/ob (leptin-deficient) mice and control C57BL/6 mice after incubation with E. histolytica. Cell death was increased when splenocytes were incubated with amebae, as expected, but there was no significant difference in cell death between the ob/ob and C57BL/6 cells (Fig. S1A). Amebic trogocytosis, measured by the percentage of amebae that had ingested fluorescently labeled fragments of splenoctye cell membrane, was also not significantly different between the two splenocyte populations (see Fig. S1B in the supplemental material). A similar experiment was conducted with Q223 and R223 neutrophils, which also revealed no significant differences (data not shown). Decreased leptin signaling with the 223R mutation therefore did not increase the susceptibility of immune cells to amebic killing.
Neutrophil attractant chemokines and leptin were produced at equivalent levels in both genotypes.
To determine if the host environment was responsible for the reduction in neutrophil infiltration, we measured production of an array of neutrophil chemoattractants produced by epithelial cells and inflammatory cells. CXCL1, CXCL2, CCL3, CCL4, and granulocyte colony-stimulating factor (G-CSF) were measured in cecal tissue lysates from mice challenged for 6 and 12 h; for this we used Luminex bead assays and normalized results to total protein content. We found that all chemokines tested were produced equally in Q223 and R223 mice at both 6 h (Fig. 4A) and 12 h (Fig. 4B) after infection.
FIG 4 .

Neutrophil chemoattractant expression in cecum after infection. Protein levels of key neutrophil attractant chemokines after 6 h (A) and 12 h (B) after infection with E. histolytica trophozoites were equivalent between Q223 and R223 mice. All levels were normalized to total protein and log transformed.
Leptin is also considered a neutrophil chemoattractant and is known to increase after many types of infection and to act as an inflammatory cytokine. To assess if there were any baseline differences between the genotypes that could result in a rapid imbalance of neutrophils, leptin was measured in the cecal tissue lysates and serum of naive Q223 and R223 mice in an enzyme-linked immunosorbent assay (ELISA). Leptin levels in uninfected mice were revealed to be equivalent in both the serum (Fig. 5A) and the cecum (Fig. 5B) of both genotypes. Leptin was then measured in cecal lysates after 6 and 12 h of E. histolytica infection to test if leptin production diminished or failed to increase in R223 mice (Fig. 5B). There was an increase in cecal production of leptin between 6 hours and 12 hours in both Q223 and R223 mice; this increase was significant in R223 mice (P = 0.02) and showed a strong trend in Q223 mice (P = 0.06). There were, however, no differences in cecal leptin levels between the genotypes at 6 h or 12 h. We concluded that leptin was a potential neutrophil chemoattractant to the gut and was capable of being produced as a response to infection in both genotypes.
FIG 5 .
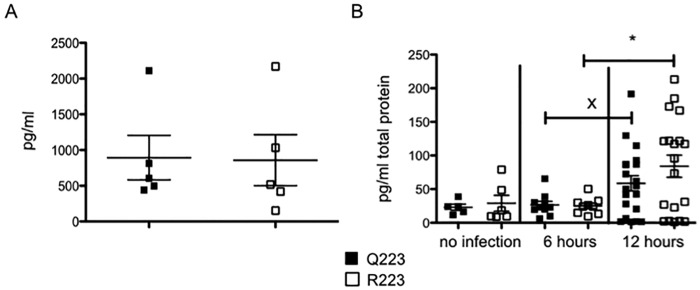
Leptin is increased in the cecum during amebic colitis. (A) Leptin levels were measured in naive Q223 and R223 mice in serum. (B) Leptin was quantified in the cecal tissue lysate of mice with no infection and after 6 and 12 h of infection. *, P = 0.02; x, P = 0.06 by two-tailed t test.
R223 neutrophils had an inhibited chemotactic response to leptin.
The preceding experiments demonstrated the absence of an impact of the 223R mutation on neutrophil survival from amebic killing or on neutrophil chemoattractants in the gut. This suggested that reduced neutrophil infiltration in the R223 mouse was a result of an intrinsic defect of the neutrophils to respond to gut homing signals. As we had demonstrated that leptin markedly increased at 12 h after infection compared to earlier time points, we hypothesized that leptin was a signal for gut recruitment and, additionally, that R223 neutrophils would exhibit reduced chemotaxis toward leptin. To test this, bone marrow neutrophils were isolated using magnetic bead separation and applied to a transwell filter plate that contained leptin or a combination of CXCL1 and CXCL2 in the lower well. Chemotaxis was allowed to proceed for 1.5 h before the filter was fixed and stained. Leptin was able to induce a strong chemotactic response in both populations of neutrophils. R223 neutrophils demonstrated reduced chemotaxis compared to Q223 neutrophils toward 10 ng and 50 ng of leptin (Fig. 6A and B). Chemotaxis toward CXCL1 and CXCL2 was, however, equal in both neutrophil genotypes (Fig. 6C). We concluded that there was a leptin-specific defect in chemotaxis that was conferred by the 223R mutation.
FIG 6 .

R223 neutrophils exhibit reduced chemotaxis toward leptin but not CXCL1 and -2. Chemotaxis of neutrophils from Q223 and R223 mice toward 10 ng leptin (A), 50 ng leptin (B), or a mixture of 10 ng of CXCL1+CXCL2 (C) was compared. Neutrophils isolated from the bone marrow were added to the top of a 3-µm-pore-size filter and allowed to migrate for 1.5 h. Migration counts are presented as the percentage of the Q223 average neutrophil migration toward the chemoattractant. *, P = 0.02; **, P = 0.008 (by one-tailed t test).
DISCUSSION
In this study, we investigated the mechanism behind the increased susceptibility to amebiasis of children who have the R223 leptin receptor mutation. We explored the differences in the innate immune response to amebic colitis in mice expressing the R223 mutation. The R223 mutation resulted in decreased neutrophil recruitment to the site of infection at 12 h after infection, the time at which the increased clearance of infection by wild-type Q223 mice manifests. Neutrophils with the R223 mutation had decreased chemotaxis to leptin in vitro. Finally, neutrophil depletion resulted in increased infection susceptibility, supporting the role of neutrophil recruitment to clear infection.
The Q223R leptin receptor mutation is common, expressed in up to 50% of Caucasian and Bangladeshi populations (4). In addition to impacting E. histolytica susceptibility, the mutation has also been shown to affect host resistance to peritonitis and chronic bronchitis (14, 15). The results of studies of the impact of the mutation on host response to amebiasis may therefore suggest a common mechanism of susceptibility.
The rapid divergence of infection phenotypes observed, with full eradication of infection by 72 h in mice with the ancestral Q223 leptin receptor allele (11), suggested that the innate immune system was responsible for the differences in infection clearance. R223 mice were observed to have a reduced neutrophil population in the lamina propria compared to Q223 mice 12 h after infection. Depletion of neutrophils resulted in a doubled infection rate at 12 h, confirming their importance for the early clearance of amebae. This echoes previous work indicating that depleting all GR1+ cells resulted in increased amebic pathology (16). Activated neutrophils have also been shown to kill E. histolytica (17). Together, these data suggest that a vigorous neutrophil response is needed to achieve early elimination of E. histolytica and could represent a critical crossroad in the host response that determines whether the pathogen is eradicated.
Previous work had found that leptin receptor presence on the epithelial barrier was critical for E. histolytica infection defense, as demonstrated by increased infection and disease severity with an intestinal epithelial knockout cells (18). Bone marrow chimeras of wild-type and db/db mice revealed that the presence of leptin receptor on epithelial cells was critical for defense against E. histolytica (18). Neutrophil infiltration to a site of infection is controlled by chemotactic signals; intestinal epithelial cells have the capacity to make a variety of these chemoattractants in response to a pathogen challenge, including leptin (19–22), suggesting a possible mechanism for reduced neutrophilic infiltration through decreased chemoattractant production. We found that all neutrophil-attractant chemokines tested for were detected in cecal lysate after infection, but there was no difference between Q223 and R223 mice in the concentrations of these chemokines. Leptin was also produced equally between Q223 and R223 mice, both at baseline in serum, replicating the initial human cohort findings, and in the cecum before and after infection. We concluded there were no discernible differences between the abilities of Q223 and R223 mice to make neutrophil-attractant chemokines.
Immune cell survival after amebic challenge was also not affected by the R223 mutation. Leptin has known antiapoptotic effects (23–25) and likely protects intestinal epithelial cells against ameba-mediated cell death. Previously, transfection of HEK cells with the Q223 receptor had been shown to provide leptin-dependent protection from cell death that was greater than that from the R223 leptin receptor (6), and the intestinal epithelial cells in R223 mice showed greater caspase-3 activation after infection (4). In contrast to the importance of leptin signaling in epithelial cell defense, in immune cells we found no impact of the R223 mutation on increasing the susceptibility to amebic killing. In the context of protection from amebic trogocytosis leptin, signaling appears to have a different role in neutrophils than in epithelial cells.
Increased chemotaxis toward leptin was the most likely explanation for the increased neutrophil presence in the gut of Q223 mice. We observed between 6 hours and 12 hours after infection that there was a significant increase in leptin in the cecum. Other investigators have also observed an increase in leptin after infection. A human study of H. pylori infection also reported increased leptin expression in gastric epithelial cells after infection, which was subsequently reduced after eradication of infection (26); lipopolysaccharide inhalation also resulted in increased broncheoalveolar lavage (BAL) fluid leptin levels (27). In mice this elevation has been observed in peritonitis and pneumonia models; leptin was observed to rise significantly at 12 and 24 h after cecal ligation and puncture, and both bacterial and viral pneumonia resulted in greater lepitn concentrations in the BAL fluid (27–29). Furthermore, these studies observed that increased leptin resulted in increased neutrophila at the sites of injury. Leptin thus appears to be part of the inflammatory response by epithelial cells under attack. The rise of leptin at 12 h coincided with the observed neutrophil infiltration, suggesting a likely signal for the neutrophil influx apart from the traditionally described chemoattractants.
The leptin receptor mutation resulted in an intrinsic defect in neutrophil chemotaxis to leptin. Though only expressing the short form of the leptin receptor (30), neutrophils have been shown to migrate toward leptin (27, 31, 32). JAK2 signaling from this short form receptor is known to activate phosphhatidylinositol 3-kinase and potentially p38; inhibitors of both signaling molecules can inhibit neutrophil chemotaxis toward leptin (22, 26, 28). Little research has been performed regarding the significance of this chemotaxis, and until now there has been no clear link between disease susceptibility and neutrophil migration toward leptin. We found that both Q223 and R223 neutrophils were able to respond chemotactically to leptin in medium, but R223 exhibited reduced rates of chemotaxis. The lack of a difference in chemotaxis between the two genotypes toward a combination of CXCL1 and CXCL2 suggests that this chemotactic decline is restricted to leptin and does not interfere with other chemotaxis pathways. These data, combined with our observation of increased leptin production at the site of infection, suggest that infiltration into the cecum is inhibited by the R223 mutation. However, it is also possible that bone marrow mobilization of neutrophils is affected. A variety of chemokines and cytokines have been shown to cause the neutrophil efflux from the bone marrow seen after infection (33); leptin has not been identified as having a direct role in this migration as of yet, but as our experiments were performed with bone marrow-isolated neutrophils, it is possible this could be happening. In addition, leptin has been shown to upregulate the oxidative burst (34); upregulated leptin production could therefore be acting as a booster signal to more than just chemotaxis. Should the R223 receptor result in additional functional inhibitions, R223 neutrophils could be unable to efficiently kill the invading ameba and thus would become more susceptible to the ameba themselves. The role of leptin has started to be appreciated in immune mediators such as T cells (8, 10, 35), and now leptin signaling appears to have a powerful role in neutrophil influx and thus the response to infection by E. histolytica.
This study has identified a novel leptin-mediated mechanism of innate defense, adding to the epithelial defense we had previously observed. While we are open to the possibility that this mutation has other yet-to-be-discovered roles in inflammation and infection, the discovery that neutrophil chemotaxis is altered due to the R223 mutation is of significance, as the mutation is common in human populations. This work may have relevance for obesity and malnutrition, as both conditions result in an altered leptin environment. In conclusion, this study provided insights into the host defense against E. histolytica infection and to the role of leptin in disease and immunity through the demonstration of leptin-mediated chemotaxis.
MATERIALS AND METHODS
Mice.
Male 129/J mice homozygous for the Q223 or R223 allele (8 to 12 weeks old) were maintained and bred at the University of Virginia under pathogen-free conditions. C57BL/J6 and BV.6-Lepob/J mice were obtained from Jackson Laboratories.
Cecal lamina propria isolation.
Single-cell suspensions of cecal lamina propria tissue were prepared as described previously. In brief, cecal tissue was washed in Hanks balanced salt solution (HBSS) containing dithiothreitol and EDTA for 15 min at 37°C to remove the epithelial layer. Tissue was then diced and incubated in RPMI containing collagenase D and DNase for 30 min at 37°C. Digested tissue pieces were passed through 100-µm and 30-µm cell filters to obtain single-cell suspensions.
Histology.
Cecal tissue sections were stained with hematoxylin and eosin (H&E). Scoring for submucosal edema and epithelial blunting was based on a scale of 0 to 3 for increasing severity. Samples were read blinded by two investigators, and scores were averaged for all sections; scores were then added, for a final overall histology score between 0 and 6.
Murine infection with Entamoeba histolytica.
Trophozoites for infections were originally derived from laboratory strain HM1: IMSS that was sequentially passaged in vivo through mouse cecum. Virulence has been maintained with periodic passaging. For all intracecal inoculations, 2 × 106 trophozoites in 150 µl were injected intracecally after laparotomy. Mice were sacrificed 12 h after infection, and the contents were collected and cultured in complete trypsin-yeast-iron (TYI-33) medium supplemented with Diamond Vitamin (JRH Biosciences), 100 U/ml of penicillin and streptomycin, and bovine serum (Sigma-Aldrich). The presence of visible amebae 12 to 24 h after sacrifice determined the infection status. Murine infection results are representative of two independent experiments.
Neutrophil depletion.
Fifty micrograms of anti-Ly6G monoclonal antibody or 50 µg of IgG isotype (BioXcell) control was injected into the peritoneal cavity 12 h before infection. Depletion was confirmed with flow cytometric analysis of the lamina propria after infection.
Amebic trogocytosis assay.
Splenocytes were isolated by filtration through 100-µm cell filters followed by red blood cell lysis. Amebic trogocytosis and splenocyte death were quantified using imaging flow cytometry as described previously (13).
Determination of cellular populations in the lamina propria.
Isolated lamina propria cells were surface stained for markers Ly6G, CD11b, CD45, Ly6C, and CXCR2 according to a general flow cytometry protocol. All samples were run on a Beckman Coulter CyAn ADP LX apparatus and analyzed with FlowJo software (Treestar, Ashland, OH).
Quantification of cecal proteins.
Cecal tissue was homogenized in HBSS with 50 mM HEPES, Triton X-100, and HALT protease inhibitor cocktail (Pierce). Lysates were aliquoted and stored at −70°C. Aliquots were not freeze-thawed more than twice. CXCL1, CXCL2, CCL3, CCL4, and G-CSF Luminex assays (Bio-rad) were run on a Bio-Rad Bioplex 200 apparatus. Leptin quantification was performed in an ELISA (Peprotech). Results were normalized to total protein concentration, as measured in a bicinchoninic acid assay (Pierce) per aliquot used.
Neutrophil isolation and chemotaxis assay.
Neutrophils were isolated from bone marrow of hind legs of R223 and Q223 mice. Bones were cleaned and ground in sterile HBSS, then cells were filtered through 100-µm and 40-µm cell filters. Anti-Ly6G magnetically activated cell sorting bead separation was performed according to the manufacturer’s protocol (Miltenyi Biotec) on an AutoMACS Pro system. A total of 4 × 104 neutrophils were immediately added to the top of the transwell filter. A 600-µl volume of HBSS (plus Ca and Mg) containing 10 ng leptin, 50 ng leptin, or 10 ng CXCL1 and CXCL2 (5 ng each; Peprotech) was added to the bottom well. Chemotaxis was allowed to proceed for 1.5 h at 37°C. Filters were then removed from the plates, and excess liquid was removed from the top of the filter with a cotton-tipped applicator. Filters were cut out, fixed with methanol, and then stained with crystal violet. Neutrophils were counted on one field of view with 20× magnification by using an Olympus DP71.
Statistical analysis.
Statistical analyses were performed using Prism 5.0 software. Student’s t test was performed for comparisons between two groups, and the Mann-Whitney test was performed for infection rate comparisons.
SUPPLEMENTAL MATERIAL
Amebic trogocytosis of splenocytes is not enhanced by reduced leptin signaling.
Splenocytes isolated from WT and ob/ob mice (4 mice per group) were incubated with E. histolytica trophozoites and compared for their ability to resist amebic trogocytosis and cell killing using imaging flow cytometry. Cell death was assessed with fluorescent live/dead staining after incubation with amebae for 40 min or, as a control, incubation without amebae for 40 min (A). Amebic trogocytosis was assessed based on the percentage of amebae that had ingested fluorescently labeled splenocyte cell parts in 40 min (B). Four mice were analyzed per group, and technical replicates from each mouse were collected; the mean values of the technical replicates are plotted. The mean values and standard deviations for each group are shown. Download
ACKNOWLEDGMENTS
This work was supported by NIH grant 5R01 AI026649-25.
We thank Zannatan Noor and Carrie Cowardin for assistance in mouse surgery.
Footnotes
Citation Naylor C, Burgess S, Madan R, Buonomo E, Razzaq K, Ralston K, Petri, WA, Jr. 2014. Leptin receptor mutation results in defective neutrophil recruitment to the colon during Entamoeba histolytica infection. mBio 5(6):e02046-14. doi:10.1128/mBio.02046-14.
REFERENCES
- 1. Haque R, Huston CD, Hughes M, Houpt E, Petri WA. 2003. Amebiasis. N. Engl. J. Med. 348:1565–1573. 10.1056/NEJMra022710. [DOI] [PubMed] [Google Scholar]
- 2. Taniuchi M, Sobuz SU, Begum S, Platts-Mills JA, Liu J, Yang Z, Wang XQ, Petri WA, Haque R, Houpt ER. 2013. Etiology of diarrhea in Bangladeshi infants in the first year of life analyzed using molecular methods. J. Infect. Dis. 208:1794–1802. 10.1093/infdis/jit507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mortimer L, Chadee K. 2010. The immunopathogenesis of Entamoeba histolytica. Exp. Parasitol. 126:366–380. 10.1016/j.exppara.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 4. Duggal P, Guo X, Haque R, Peterson KM, Ricklefs S, Mondal D, Alam F, Noor Z, Verkerke HP, Marie C, Leduc CA, Chua SC, Myers MG, Leibel RL, Houpt E, Gilchrist CA, Sher A, Porcella SF, Petri WA. 2011. A mutation in the leptin receptor is associated with Entamoeba histolytica infection in children. J. Clin. Invest. 121:1191–1198. 10.1172/JCI45294.of. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Verkerke H, Naylor C, Zabeau L, Tavernier J, Petri Wa, Marie C. 2014. Kinetics of leptin binding to the Q223R leptin receptor. PLoS One 9:e94843. 10.1371/journal.pone.0094843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marie CS, Verkerke HP, Paul SN, Mackey AJ, Petri WA. 2012. Leptin protects host cells from Entamoeba histolytica cytotoxicity by a STAT3-dependent mechanism. Infect. Immun. 80:1934–1943. 10.1128/IAI.06140-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Margetic S, Gazzola C, Pegg GG, Hill RA. 2002. Leptin: a review of its peripheral actions and interactions. Int. J. Obes. Relat. Metab. Disord. 26:1407–1433. 10.1038/sj.ijo.0802142. [DOI] [PubMed] [Google Scholar]
- 8. Faggioni R, Feingold KR, Grunfeld C. 2001. Leptin regulation of the immune response and the immunodeficiency of malnutrition. FASEB J. 15:2565–2571. 10.1096/fj.01-0431rev. [DOI] [PubMed] [Google Scholar]
- 9. Petri WA, Mondal D, Peterson KM, Duggal P, Haque R. 2009. Association of malnutrition with amebiasis. Nutr. Rev. 67(Suppl 2):S207–S215. 10.1111/j.1753-4887.2009.00242.x. [DOI] [PubMed] [Google Scholar]
- 10. Fernández-Riejos P, Najib S, Santos-Alvarez J, Martín-Romero C, Pérez-Pérez A, González-Yanes C, Sánchez-Margalet V. 2010, 2010 Role of leptin in the activation of immune cells. Mediators Inflamm. 2010:568343. 10.1155/2010/568343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mackey-Lawrence NM, Guo X, Sturdevant DE, Virtaneva K, Hernandez MM, Houpt E, Sher A, Porcella SF, Petri WA. 2013. Effect of the leptin receptor Q223R polymorphism on the host transcriptome following infection with Entamoeba histolytica. Infect. Immun. 81:1460–1470. 10.1128/IAI.01383-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Houpt ER, Glembocki DJ, Obrig TG, Moskaluk CA, Lockhart LA, Wright RL, Seaner RM, Keepers TR, Wilkins TD, Petri WA. 2002. The mouse model of amebic colitis reveals mouse strain susceptibility to infection and exacerbation of disease by CD4+ T cells. J. Immunol. 169:4496–4503. 10.4049/jimmunol.169.8.4496. [DOI] [PubMed] [Google Scholar]
- 13. Ralston KS, Solga MD, Mackey-lawrence NM, Somlata A, Bhattacharya A, Petri WA. 2014. Trogocytosis by Entamoeba histolytica contributes to cell killing and tissue invasion. Nature 508:526–530. 10.1038/nature13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bracho-Riquelme RL, Loera-Castañeda V, Torres-Valenzuela A, Loera-Castañeda GA, Sánchez-Ramírez JP. 2011. Leptin and leptin receptor polymorphisms are associated with poor outcome (death) in patients with non-appendicular secondary peritonitis. Crit. Care 15:R227. 10.1186/cc10467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang B, Fu E, Cao Y, Zhong Y, Fu G, Tian X, Li S. 2013. Effect of leptin receptor mutation on the development of chronic bronchitis. Asia Pac. J. Public Health 25(4 Suppl):80S–87S. 10.1177/1010539513497218. [DOI] [PubMed] [Google Scholar]
- 16. Asgharpour A, Gilchrist C, Baba D, Hamano S, Houpt E. 2005. Resistance to intestinal Entamoeba histolytica infection is conferred by innate immunity and Gr-1+ cells. Infect. Immun. 73:4522–4529. 10.1128/IAI.73.8.4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chadee K. 1989. Human neutrophils activated by interferon-y and tumour necrosis factor-α kill Entamoeba histolytica trophozoites. In Vitro 274:270–274. [DOI] [PubMed] [Google Scholar]
- 18. Guo X, Roberts MR, Becker SM, Podd B, Zhang Y, Chua SC, Myers MG, Duggal P, Houpt ER, Petri WA. 2011. Leptin signaling in intestinal epithelium mediates resistance to enteric infection by Entamoeba histolytica. Mucosal Immunol. 4:294–303. 10.1038/mi.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang SK, Eckmann L, Panja A, Kagnoff MF. 1997. Differential and regulated expression of C-X-C, C–C, and C-chemokines by human colon epithelial cells. Gastroenterology 113:1214–1223. 10.1053/gast.1997.v113.pm 9322516. PubMed. [DOI] [PubMed] [Google Scholar]
- 20. Sitaraman S, Liu X, Charrier L, Gu LH, Ziegler TR, Gewirtz A, Merlin D. 2004. Colonic leptin: source of a novel pro-inflammatory cytokine involved in inflammatory bowel disease. FASEB J. 18:696–698. 10.1096/fj.03-0422fje. [DOI] [PubMed] [Google Scholar]
- 21. Song F, Ito K, Denning TL, Kuninger D, Papaconstantinou J, Gourley W, Klimpel G, Balish E, Hokanson J, Ernst PB. 1999. Expression of the neutrophil chemokine KC in the colon of mice with enterocolitis and by intestinal epithelial cell lines: effects of flora and proinflammatory cytokines. J. Immunol. 162:2275–2280. [PubMed] [Google Scholar]
- 22. Ohtsuka Y, Lee J, Stamm DS, Sanderson IR. 2001. MIP-2 secreted by epithelial cells increases neutrophil and lymphocyte recruitment in the mouse intestine. Gut 49:526–533. 10.1136/gut.49.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ogunwobi OO, Beales IL. 2007. The anti-apoptotic and growth stimulatory actions of leptin in human colon cancer cells involves activation of JNK mitogen activated protein kinase, JAK2 and PI3 kinase/Akt. Int. J. Colorectal Dis. 22:401–409. 10.1007/s00384-006-0181-y. [DOI] [PubMed] [Google Scholar]
- 24. Bruno A, Conus S, Schmid I, Simon HU. 2005. Apoptotic pathways are inhibited by leptin receptor activation in neutrophils. J. Immunol. 174:8090–8096. 10.4049/jimmunol.174.12.8090. [DOI] [PubMed] [Google Scholar]
- 25. Papathanassoglou E, El-haschimi K, Li XC, Matarese G, Strom T, Mantzoros C. 2006. Leptin receptor expression and signaling in lymphocytes: kinetics during lymphocyte activation, role in lymphocyte survival, and response to high fat diet in mice. J. Immunol. 176:7745–7752. 10.4049/jimmunol.176.12.7745. [DOI] [PubMed] [Google Scholar]
- 26. Azuma T, Suto H, Ito Y, Ohtani M, Dojo M, Kuriyama M, Kato T. 2001. Gastric leptin and Helicobacter pylori infection. Gut 49:324–329. 10.1136/gut.49.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ubags ND, Vernooy JH, Burg E, Hayes C, Bement J, Dilli E, Zabeau L, Abraham E, Poch KR, Nick JA, Dienz O, Zuñiga J, Wargo MJ, Mizgerd JP, Tavernier J, Rincón M, Poynter ME, Wouters EF, Suratt BT. 2014. The role of leptin in the development of pulmonary neutrophilia in infection and acute lung injury. Crit. Care Med. 42:e143–e151. 10.1097/CCM.0000000000000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moshyedi AK, Josephs MD, Abdalla EK, Mackay SL, Edwards CK, Copeland EM, Moldawer LL. 1998. Increased leptin expression in mice with bacterial peritonitis is partially regulated by tumor necrosis factor alpha. Infect. Immun. 66:1800–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Siegl D, Annecke T, Johnson BL, Schlag C, Martignoni A, Huber N, Conzen P, Caldwell CC, Tschöp J. 2014. Obesity-induced hyperleptinemia improves survival and immune response in a murine model of sepsis. Anesthesiology 121:98–114. 10.1097/ALN.0000000000000192. [DOI] [PubMed] [Google Scholar]
- 30. Zarkesh-Esfahani H, Pockley AG, Wu Z, Hellewell PG, Weetman AP, Ross RJ. 2004. Leptin indirectly activates human neutrophils via induction of TNF-alpha. J. Immunol. 172:1809–1814. 10.4049/jimmunol.172.3.1809. [DOI] [PubMed] [Google Scholar]
- 31. Montecucco F, Bianchi G, Gnerre P, Bertolotto M, Dallegri F, Ottonello L. 2006. Induction of neutrophil chemotaxis by leptin: crucial role for p38 and Src kinases. Ann. N. Y. Acad. Sci. 1069:463–471. 10.1196/annals.1351.045. [DOI] [PubMed] [Google Scholar]
- 32. Caldefie-Chezet F, Poulin A, Vasson MP. 2003. Leptin regulates functional capacities of polymorphonuclear neutrophils. Free Radic. Res. 37:809–814. 10.1080/1071576031000097526. [DOI] [PubMed] [Google Scholar]
- 33. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. 2010. Neutrophil kinetics in health and disease. Trends Immunol. 31:318–324. 10.1016/j.it.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Caldefie-Chezet F, Poulin A, Tridon A, Sion B, Vasson MP. 2001. Leptin: a potential regulator of polymorphonuclear neutrophil bactericidal action? J. Leukoc. Biol. 69:414–418. [PubMed] [Google Scholar]
- 35. Rodríguez L, Graniel J, Ortiz R. 2007. Effect of leptin on activation and cytokine synthesis in peripheral blood lymphocytes of malnourished infected children. Clin. Exp. Immunol. 148:478–485. 10.1111/j.1365-2249.2007.03361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Amebic trogocytosis of splenocytes is not enhanced by reduced leptin signaling.
Splenocytes isolated from WT and ob/ob mice (4 mice per group) were incubated with E. histolytica trophozoites and compared for their ability to resist amebic trogocytosis and cell killing using imaging flow cytometry. Cell death was assessed with fluorescent live/dead staining after incubation with amebae for 40 min or, as a control, incubation without amebae for 40 min (A). Amebic trogocytosis was assessed based on the percentage of amebae that had ingested fluorescently labeled splenocyte cell parts in 40 min (B). Four mice were analyzed per group, and technical replicates from each mouse were collected; the mean values of the technical replicates are plotted. The mean values and standard deviations for each group are shown. Download