

ABSTRACT
The seven human sirtuins are a family of ubiquitously expressed and evolutionarily conserved NAD+-dependent deacylases/mono-ADP ribosyltransferases that regulate numerous cellular and organismal functions, including metabolism, cell cycle, and longevity. Here, we report the discovery that all seven sirtuins have broad-range antiviral properties. We demonstrate that small interfering RNA (siRNA)-mediated knockdown of individual sirtuins and drug-mediated inhibition of sirtuin enzymatic activity increase the production of virus progeny in infected human cells. This impact on virus growth is observed for both DNA and RNA viruses. Importantly, sirtuin-activating drugs inhibit the replication of diverse viruses, as we demonstrate for human cytomegalovirus, a slowly replicating DNA virus, and influenza A (H1N1) virus, an RNA virus that multiplies rapidly. Furthermore, sirtuin defense functions are evolutionarily conserved, since CobB, the sirtuin homologue in Escherichia coli, protects against bacteriophages. Altogether, our findings establish sirtuins as broad-spectrum and evolutionarily conserved components of the immune defense system, providing a framework for elucidating a new set of host cell defense mechanisms and developing sirtuin modulators with antiviral activity.
IMPORTANCE
We live in a sea of viruses, some of which are human pathogens. These pathogenic viruses exhibit numerous differences: DNA or RNA genomes, enveloped or naked virions, nuclear or cytoplasmic replication, diverse disease symptoms, etc. Most antiviral drugs target specific viral proteins. Consequently, they often work for only one virus, and their efficacy can be compromised by the rapid evolution of resistant variants. There is a need for the identification of host proteins with broad-spectrum antiviral functions, which provide effective targets for therapeutic treatments that limit the evolution of viral resistance. Here, we report that sirtuins present such an opportunity for the development of broad-spectrum antiviral treatments, since our findings highlight these enzymes as ancient defense factors that protect against a variety of viral pathogens.
INTRODUCTION
Human sirtuins form a family of seven (SIRT1 to 7) NAD+-dependent enzymes that are expressed in a wide range of tissues. Their NAD+ requirement ties their activity to the metabolic state of cells. Sirtuins have diverse subcellular localizations (1) to the nucleus (SIRT1, SIRT6, and SIRT7), cytoplasm (SIRT2), and mitochondria (SIRT3, SIRT4, and SIRT5) and have been shown to impact numerous cell functions, including metabolism, cell cycle, apoptosis, stress response, DNA repair, and gene expression (2–5). While they are predominantly known as lysine deacetylases (6, 7), recent studies have discovered that sirtuins have a range of enzymatic functions, such as ADP ribosylation for SIRT4 (8) and SIRT6 (9), desuccinylation and demalonylation for SIRT5 (10, 11), and hydrolysis of long-chain fatty acyl lysine for SIRT6 (12). As such, sirtuins are sensors of extra- and intracellular environmental changes, playing important roles in the maintenance of human health and disease prevention. Nevertheless, knowledge regarding sirtuin functions in response to viral infections has remained limited and primarily focused on SIRT1. During HIV-1 infection, SIRT1 was shown to regulate the function of the HIV-1 Tat protein (13), which, conversely, inhibits SIRT1 deacetylase activity (14). SIRT1 was also reported to impact additional viruses, including vesicular stomatitis virus (15) and Kaposi’s sarcoma-associated herpesvirus (KSHV) (16). However, although viruses are known to rely on numerous cellular mechanisms modulated by SIRT1, the other human sirtuins have not been yet studied in the context of viral infection.
Here, we have shown that inhibition of each of the seven sirtuins by small interfering RNA (siRNA) knockdown enhances the growth of diverse human viruses, as does treatment with a SIRT1 antagonist. In contrast, drugs that activate SIRT1 inhibit the production of viral progeny. Furthermore, knockout or overexpression of the Escherichia coli sirtuin, CobB, modulates the growth of bacteriophages. These results lead to the conclusion that sirtuins are broad-spectrum, evolutionarily conserved viral restriction factors.
RESULTS
The seven human sirtuins exhibit broad-spectrum antiviral properties.
To investigate the roles of the seven mammalian sirtuins during viral infection, we designed an siRNA screen to test the consequences of knocking down the expression of each sirtuin mRNA on virus yields (Fig. 1A). We first assessed their impact on the widespread human pathogen human cytomegalovirus (HCMV), which is a major cause of birth defects and opportunistic infections in immunosuppressed individuals and a possible cofactor in certain cancers (17). Human fibroblasts were transfected with siRNA for each sirtuin and then infected with HCMV (AD169 strain, BADinUL99GFP). The titers of viral progeny in culture supernatants were determined by infectious center assay of human fibroblasts, providing a functional readout. The knockdown and infection were timed to achieve the maximal knockdown of sirtuins during the period of peak viral replication and egress. Knockdown efficiencies were validated by quantitative PCR (qPCR) assay (Fig. 1B), and the knockdown of the essential HCMV immediate early protein IE2 was used as a positive control and validation of transfection efficiency (Fig. 1C). When the validated siRNAs were used to knock down each of the sirtuins, the HCMV titer increased compared to results with a control nontargeted siRNA (siNT) (Fig. 1C, red bars), suggesting that this entire family of enzymes normally functions to antagonize HCMV infection. The increase in virus titers was further confirmed using a second siRNA for each sirtuin (Fig. 1C, gray bars).
FIG 1 .

Human SIRTs exhibit broad-range antiviral activity against DNA and RNA viruses. (A) Schematic representation of the siRNA assay. t1, time after siRNA transfection at which cultures were infected with a test virus; t2, time after infection with a test virus when cell-free virus was harvested in order to determine its titer. (B) Knockdown efficiency of sirtuin-specific siRNAs used to test the full set of viruses. MRC5 cells were transfected with indicated siRNAs, and RNA was analyzed by RT-qPCR 96 h later. Results were normalized to the expression level of β-actin for SIRT1-6 or GAPDH for SIRT7. (C to F) Sirtuin knockdown enhanced virus yields. MRC5 cells were transfected with siRNAs targeting each individual sirtuin (siSIRT1 to -7), as well as a nontargeting siRNA (siNT) control, and then infected at the indicated times (t1) after knockdown with HCMV (0.5 IU/cell) (C), HSV-1 (1 IU/cell) (D), Ad5 (5 IU/cell) (E), or influenza virus WSN (0.01 TCID50/cell) (F). Infectious virus in culture supernatants was quantified at indicated times (t2) by the methods indicated in panel A. As a control, the efficiency of siRNA-mediated knockdown was monitored in HCMV-infected cells following knockdown of an essential immediate early gene product (siIE2). For all experiments, means ± SD (n = 3) are shown; **, P < 0.01; *, P < 0.05; weakly significant (ws), P < 0.1; nonsignificant (ns), P > 0.1.
Given the impact on HCMV titers by all SIRT enzymes, we next tested whether sirtuin levels have a broad effect on diverse DNA and RNA viruses. We used the validated siRNA for each sirtuin (Fig. 1B) to analyze two additional DNA viruses, herpes simplex virus 1 (HSV-1) strain KOS 1.1 and adenovirus type 5 (Ad5), and an RNA virus, influenza virus A/WSN (H1N1). Strikingly, the knockdown of each sirtuin resulted in increased virus titers for all tested viruses, except in the case of SIRT3 and SIRT7 in HSV-1 infection (Fig. 1D to F). Overall, the effects of sirtuin knockdowns on virus replication were most significant for HCMV and influenza virus H1N1 infections, with up to 10-fold increases in virus titers, suggesting a more active role for sirtuins during these infections. The impact on HSV-1 and Ad5 infections was significant but less prominent, with increases in virus production ranging from 1.5- to 3-fold. The siRNA results also indicate that distinct sirtuin classes can differentially impact the replication of various viruses. For example, SIRT1 knockdown led to increased virus titers for both DNA and RNA viruses. In contrast, knockdown of mitochondrial sirtuins had the most robust effects on HCMV and less impact on influenza virus A/WSN replication.
Sirtuin-modulating drugs impact the growth of diverse viruses.
To determine if the impact of sirtuin knockdown on virus replication is driven by sirtuin enzymatic activity, the small-molecule EX-527, an inhibitor of SIRT1 deacetylation activity (18), was tested for an effect on HCMV yield. EX-527 inhibited SIRT1 activity (Fig. 2A) without reducing cell viability (Fig. 2B and I). Treatment with the SIRT1 inhibitor mirrored the siRNA results, leading to a dose-dependent increase in the HCMV titer (Fig. 2A). Furthermore, EX-527 did not increase virus titers in a SIRT1 knockdown background (Fig. 2C), confirming its specific dependence on SIRT1 activity and levels. This experiment indicates that sirtuins have antiviral properties mediated via their enzymatic activities. We therefore predicted that sirtuin agonists would inhibit virus replication. To test our hypothesis, we treated HCMV-infected fibroblasts with the known sirtuin activator resveratrol. The potency of resveratrol treatment for SIRT1 activity (Fig. 2D) and maintenance of cell viability (Fig. 2E and I) was confirmed. Earlier work showed that resveratrol treatment significantly reduced HCMV (19) and influenza A virus (20) titers, although its effect was not previously connected to sirtuin activation. We also found that resveratrol inhibited the production of HCMV progeny (Fig. 2D), and we showed that SIRT1 knockdown leads to a partial but substantial rescue of resveratrol-induced viral inhibition (Fig. 2F), confirming the specific involvement of sirtuin levels and activities. The fact that the rescue was partial suggests that resveratrol may also activate other sirtuins or impact other cellular processes required for viral replication. To further confirm that the observed inhibition of virus titer is associated with sirtuin activity, we next tested CAY10602, a more potent agonist of SIRT1 (Fig. 2G), shown to modulate SIRT1 functions (21). CAY10602 had a more profound effect on virus replication, triggering the reduction of HCMV titers to undetectable levels without affecting cell viability (Fig. 2G to I).
FIG 2 .

Sirtuin-modulating drugs impact HCMV replication. (A) SIRT1 antagonist EX-527 enhances HCMV replication. MRC5 cells were infected with HCMV (0.5 IU/cell) and treated with EX-527 or vehicle control (DMSO [dimethyl sulfoxide]) at 2 hpi. Cells were harvested at 96 hpi, and virus yield was determined. SIRT1 deacetylase activity was assessed using a direct fluorescent (Fluor-de-Lys) assay. (B) EX-527 does not alter the morphology of HCMV-infected cells assessed by phase microscopy. (C) EX-527 increases HCMV replication in a SIRT1-dependent manner. Cells were transfected with an siRNA targeting SIRT1 (siSIRT1) or a control nontargeting siRNA (siNT), and the infection, treatment (EX-527 or DMSO), and virus titer measurements were performed as for panel A. (D) Resveratrol inhibits HCMV replication. MRC5 cells, infected with HCMV (2 IU/cell), were treated with resveratrol at 2 hpi. Virus yields and SIRT1 deacetylase activity were assessed as with panel A. (E) Resveratrol does not alter the morphology of HCMV-infected cells. (F) SIRT1 knockdown partially rescues resveratrol-induced viral inhibition. Cells were transfected with siSIRT1 or siNT and analyzed as for panel D. (G) CAY10602 inhibits HCMV replication. Cells were infected and analyzed as for panel A. (H) CAY10602 does not alter the morphology of HCMV-infected cells. (I) MRC5 cells were treated with sirtuin modulators or DMSO for 96 h. Cell viability was assessed using a colorimetric tetrazolium reduction assay. For all experiments, means ± SD (n = 3) are shown; **, P < 0.01; *, P < 0.05; ns, nonsignificant.
As a further confirmation of the impact of sirtuin agonists on virus replication, we monitored by Western blotting the expression levels of viral proteins representative of different stages of the HCMV life cycle. While the level of an immediate early protein (IE1) was not changed following sirtuin activation, expression of early (pUL26) and late (pUL99) viral proteins decreased following treatment with resveratrol and, to an even greater extent, with the activator CAY10602 (Fig. 3A). As expected, an opposite effect on late viral protein accumulation was observed following treatment with the inhibitor EX-527 (Fig. 3B).
FIG 3 .
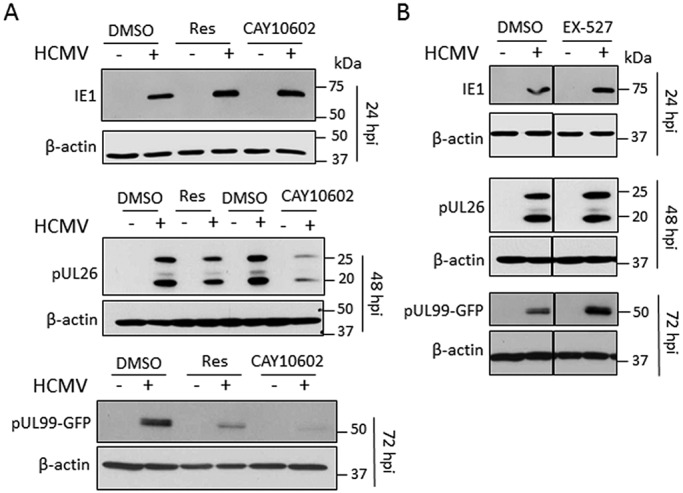
Sirtuin-modulating drugs impact virus protein expression. (A) HCMV early and late protein levels are reduced upon treatment with the sirtuin activators resveratrol (Res) and CAY10602. Cells were infected with HCMV (10 IU/cell) and treated with the indicated activator or DMSO at 2 hpi. Immediate early (IE1), early (pUL26), and late (pUL99-GFP) viral protein levels were assessed by Western blotting at 24 hpi, 48 hpi, and 72 hpi, respectively. β-Actin was used as a loading control. (B) HCMV late viral protein levels are increased upon treatment with the SIRT1 inhibitor EX-527. Cells were infected with HCMV (10 IU/cell) and treated with EX-527 (10 µM) or DMSO at 2 hpi. Viral protein levels were assessed as for panel A.
Next, we probed whether the modulation of sirtuin activity with drugs also altered the yield of influenza virus A/PR8 following infection of MDCK cells. Our findings mirrored those observed for HCMV, since the SIRT1 inhibitor EX-527 increased A/PR8 titers (Fig. 4A), while the sirtuin agonist resveratrol reduced virus titers (Fig. 4B) without compromising the viability of MDCK cells (Fig. 4C). Therefore, sirtuin activity constitutes a core host defense mechanism against both DNA and RNA viruses. Importantly, our results show that a single drug, targeting a host factor, can effectively inhibit a wide range of viruses.
FIG 4 .
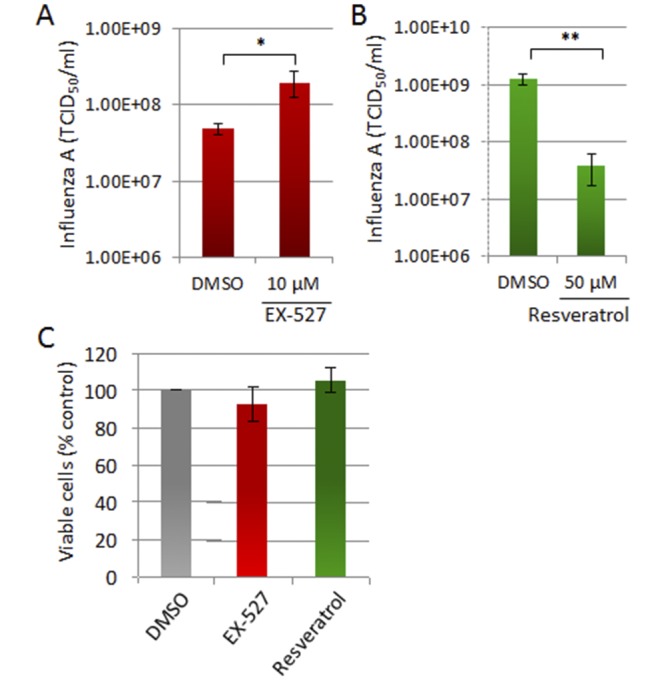
Sirtuin-modulating drugs impact influenza virus H1N1 virus replication. (A) EX-527 enhances influenza A virus replication. MDCK cells were infected with influenza A virus PR8 (0.001 TCID50/cell) and treated with EX-527 at 1 hpi. Virus yield was determined at 24 hpi. (B) Resveratrol inhibits influenza A virus replication. MDCK cells were infected with PR8 (0.1 TCID50/cell) and treated with resveratrol (50 µM) at 1 hpi. Virus yields were determined as for panel A. MDCK cells were treated with sirtuin modulators or DMSO for 24 h. Cell viability was assessed using a colorimetric tetrazolium reduction assay. For all experiments: **, P < 0.01; *, P < 0.05.
Sirtuin defense functions are evolutionarily conserved.
Because sirtuins are known to be evolutionarily conserved, we next asked whether their antipathogen functions emerged early on as an antibacteriophage defense mechanism in bacteria. We investigated the sirtuin homologue in E. coli, CobB (22), and its effect on the infection with the enterobacterium phage T4 (T4Δrl phage) (23). CobB overexpression was achieved using an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible CobB-expressing plasmid (Fig. 5A), without altering the cell growth rate over the time frame tested (Fig. 5B). The overexpression of CobB led to a significant reduction in T4Δrl phage titers during infection (Fig. 5C). Furthermore, CobB overexpression also delayed (by ~4.5 min) the cell lysis triggered by T4Δrl phage infection (Fig. 5D). We next tested the impact of CobB knockout. Cell growth remained consistent in the wild-type (WT) and knockout backgrounds (Fig. 5E). As expected, knockout of CobB resulted in increased T4Δrl phage plaque size (Fig. 5F) and burst size (Fig. 5G), and this effect was reversed by the IPTG-induced expression of CobB (Fig. 5G). To expand our studies, a second bacterial virus was studied, the virulent mutant of the enterobacterium λ phage (λvir) (24). Similar to our results for T4Δrl phage, the λvir burst size was substantially increased in the CobB knockout background compared to that in wild-type cells (Fig. 5H). Altogether, these results establish the CobB deacetylase as a host defense factor against bacteriophages.
FIG 5 .

The bacterial sirtuin CobB restricts phage replication in E. coli. (A) Induction of CobB RNA accumulation in wild-type and CobB knockout cells. CobB RNA levels were assayed by RT-qPCR in MC4100-wt and MC4100-ΔCobB cells and in cells containing a CobB expression plasmid (wt-pCobB) before and after 15 min of IPTG induction and were determined by RT-qPCR. Results (means ± SD; n = 3) were normalized to the expression level of cysG. (B) CobB overexpression does not alter the rate of bacterial growth. MC4100-wt cells containing a CobB expression plasmid were induced with IPTG (0.25 mM) for 15 min. Cells were diluted 10-fold, and cell growth was monitored by absorbance (OD600); means ± SD, n = 3. (C) T4ΔrI phage titers are reduced in E. coli overexpressing CobB. MC4100-wt cells containing the ASKA(−) CobB plasmid (wt-pCobB) were induced with IPTG (0.25 mM) for 15 min. Cells were diluted 10-fold and infected with T4-ΔrI (0.1 PFU/cell). Phage titers were measured at the indicated times by plaque assay; means ± SD are shown (n = 3). (D) E. coli lysis is delayed during T4-ΔrI infection upon CobB overexpression. CobB expression was induced with IPTG as for panel A, cells were infected with T4-ΔrI (2 PFU/cell), and bacterial growth was monitored by absorbance (OD600); means ± SD are shown (n = 3). (E) CobB deletion does not alter the rate of bacterial growth. MC4100-wt and MC4100-ΔCobB growth rates were monitored by absorbance (OD600); means ± SD are shown (n = 4). (F) T4ΔrI phage plaque size is larger in the CobB knockout background. MC4100-wt and MC4100-ΔCobB were infected with T4 phage (T4-ΔrI). Images of agar plates were captured at 16 hpi. (G) T4-ΔrI burst size is increased in a CobB knockout background. MC4100-wt and MC4100-ΔCobB cells with or without the ASKA(−) CobB plasmid were induced and infected as for panel A. Burst size was determined at 75 min postinfection; means ± SD are shown (n = 3). (H) λvir burst size is increased in a CobB knockout background. Wild-type (MC4100-wt) and CobB knockout (MC4100-ΔCobB) E. coli cells were infected with λvir (0.1 PFU/cell). Burst size was determined at 60 min after infection. For all experiments, means ± SD are shown (n = 3); ***, P < 0.001; **, P < 0.01; *, P < 0.05; ns, nonsignificant.
DISCUSSION
siRNA-mediated knockdown of individual human sirtuins increased the yield of HCMV, HSV-1, Ad5, and influenza virus H1N1 in cultured cells (Fig. 1C to F). The production of HCMV and influenza virus was enhanced by treatment with the SIRT1 antagonist EX-527 (Fig. 2A and 4A), whereas yields were reduced by SIRT1 activators (Fig. 2D and G and 4B). These observations reveal that the sirtuins constitute a broadly acting family of viral restriction factors.
The bacterial sirtuin CobB, a protein deacetylase that is closely related to mammalian sirtuins in its structure (22), impacted the growth of bacteriophages T4 and λ (Fig. 5). Thus, it appears that the sirtuin system is one of the earliest forms of cellular antiviral immunity to arise. The CRISPR (clustered regularly interspaced short palindromic repeats) system (25), which protects bacteria and archaea from bacteriophages, is also ancient, but it has not yet been found in eukaryotes. RNA interference (RNAi) (26) and Toll-like receptors (TLRs) (27) arose more recently, beginning with invertebrates; and the interferon system is a vertebrate invention (28).
How broad is the spectrum of sirtuin antiviral activity? The four viruses we tested are very different in terms of their biology (DNA versus RNA genomes, enveloped versus naked particles, rapid versus slow replication), so the fact that each of them was modulated by multiple members of the sirtuin family suggests that the family will prove to target a wide range of viruses. In addition to the viruses studied here, SIRT1 has been reported to protect against additional viruses, but in many cases the evidence is based primarily on sensitivity to resveratrol. Mechanisms underlying resveratrol activity must be interpreted cautiously, because it is known to target proteins other than sirtuins (29). As noted above, however, inhibition of two bacteriophages by the bacterial sirtuin homologue CobB argues that sirtuins arose as an antiviral defense early in evolution. As a consequence, it is quite possible that many viruses beyond those that we have tested will also prove susceptible to their action.
Are any viruses resistant to the antagonistic effects of sirtuins? HIV and hepatitis B virus (HBV) appear to leverage SIRT1 activity to their advantage. The HIV Tat protein binds to the TAR motif at the 5′ end of HIV RNAs to enable the continued elongation of transcription. After transcripts are completed, Tat is released and deacetylated by SIRT1 to allow recycling of Tat and continued HIV mRNA elongation (13). Furthermore, Tat binding blocks the ability of SIRT1 to deacetylate and inhibit the NF-κB p65 subunit, activating proinflammatory genes controlled by the transcription factor and possibly contributing to the chronic immune activation that accompanies HIV infection (14). Thus, SIRT1 allows multiple cycles of Tat action, and the perturbation of SIRT1 function through its interaction with Tat might contribute to the pathogenesis of HIV infection. For HBV, SIRT1 mRNA and protein are upregulated in cell lines expressing viral gene products (30), and SIRT1 associates with the viral covalently closed circular DNA (cccDNA) (30, 31), the template for viral transcription. Consistent with this localization, the HBV core promoter is upregulated in a SIRT1-dependent manner. SIRT1 function is altered by the HBV X protein, which interacts with SIRT1, blocking its ability to modulate the activity of one of its cellular interaction partners, β-catenin (32). Thus, SIRT1 appears to be targeted by the Tat and X proteins and to support the activation of HBV transcription and the elongation of HIV transcripts. Nevertheless, given its many targets, SIRT1 might mediate distinct antagonistic effects toward these viruses, and, of course, HIV and HBV might prove to be sensitive to the action of one or more of the remaining six sirtuins.
How do SIRTs influence virus replication? Since sirtuins display a range of subcellular localizations and enzymatic activities, we expect that the mechanisms involved in their antiviral functions will be diverse. Consistent with this view, the specific siRNAs that most effectively increased yields varied across the viruses tested (Fig. 1C to F), arguing that different SIRT activities, and therefore different SIRT-mediated antiviral mechanisms, influenced the four viruses to a greater or lesser extent.
The metabolic regulatory activities shared by multiple sirtuins might underlie key aspects of their antiviral activity. All of the sirtuins require NAD+ for activity, tightly linking their function to the metabolic status of cells, and multiple sirtuins have been shown to directly influence cellular metabolic homeostasis (2–4). For example, in various cell types, SIRT1 has been shown to inhibit glycolysis by deacetylating phosphoglycerate mutase 1 (PGAM-1) (33), to inhibit fatty acid synthesis by deacetylating serum response element binding protein 1c (SREBP1c) and targeting it for degradation (34) while inducing fatty acid oxidation by activating peroxisome proliferator-activated receptor α (PPARα) (35), and to promote reverse cholesterol transport by deacetylating and activating the liver X nuclear receptor (LXR) (36). Similarly, SIRT6, which is induced by SIRT1 (37), represses glycolysis by serving as a corepressor for hypoxia-inducible factor 1α (HIF-1α) (38) and reducing glucose uptake (39), and it inhibits SREBP1 and SREBP2 to reduce cholesterol biosynthesis (40). Further, the mitochondrial SIRT3 promotes fatty acid oxidation by deacetylating long-chain-specific acyl coenzyme A dehydrogenase (LCAD) (41), and SIRT4 blocks the utilization of glutamine as a carbon source to drive the tricarboxylic acid (TCA) cycle by inhibiting glutamate dehydrogenase (GDH) (8). All of these sirtuin activities would be expected to antagonize HCMV replication, because HCMV induces and requires active glycolysis, very long chain fatty acid synthesis, continued mitochondrial oxidative phosphorylation, and intracellular cholesterol to generate infectious virions (42–45). Numerous viruses in addition to HCMV perturb their host cell’s metabolism, e.g., HSV-1 (46) and hepatitis C virus (47), so sirtuin activities designed to maintain metabolic homeostasis might well be central to the broad-spectrum antiviral activities of sirtuins.
Viruses generally evolve to resist cellular antiviral responses. For example, many viruses antagonize aspects of the interferon response (48). Are some viruses resistant to sirtuin antiviral activities? In this regard, the yields of HSV-1 and Ad5 were increased to a lesser extent than those of HCMV and influenza virus A/WSN by treatment with sirtuin-specific siRNAs (Fig. 1C to F). Since sirtuin activity is dependent on NAD+ levels, this observation fits with the reported reduction in NAD+ during HSV-1 infection (46) and may represent a viral resistance mechanism designed to escape the effects of sirtuins. In contrast, NAD+ levels do not change significantly during HCMV infection (46), so it is conceivable that sirtuins contribute to the slower replication of HCMV in comparison with its relative, HSV-1. Further, SIRT3 and SIRT7 showed no significant antiviral activity against HSV-1 infection. This raises the possibility that their antiviral effects are blocked during HSV-1 infection, possibly by a viral protein.
In conclusion, we have discovered that the sirtuin enzymes are evolutionarily conserved antiviral host factors. The bacterial CobB sirtuin has antibacteriophage functions, and the seven human sirtuins exhibit broad-range defensive properties against DNA and RNA viruses. Our findings point to sirtuins as ancient antiviral defense factors that protect against a variety of pathogens. This discovery predicts that a single host factor, a sirtuin, can be modulated to inhibit numerous types of viral infections, thereby offering the opportunity for broad-spectrum antiviral treatments that do not allow rapid evolution of resistant viruses.
MATERIALS AND METHODS
Viral infections.
Infections with HCMV AD169 BADinUL99GFP (49), HSV-1 KOS 1.1 (50), adenovirus type 5 (Ad5) (wt300) (51), and influenza A (H1N1) viruses, A/WSN/1933 (A/WSN) and A/Puerto Rico/8/1934 (A/PR8) (52), were performed in human MRC5 embryonic lung fibroblasts (HCMV, HSV-1, and Ad5) or canine MDCK kidney epithelial cells (A/WSN and PR8). Bacteriophage λvir (virulent, clear-plaque mutant) (24) or bacteriophage T4Δrl (rapid lysis mutant which produces large plaques) (23) (a kind gift from Ry Young, University of Texas, Dallas) was propagated and studied in E-coli K-12 MC4100 cells (53).
Sirtuin siRNA screen.
Knockdowns of each of the seven mammalian sirtuins were generated via transfecting MRC5 cells with siRNAs (Sigma) (Table 1) as described previously (44). An siRNA targeting HCMV IE2 (siIE2) (54) was used to assess transfection efficiency. Efficiency of knockdown was validated by qPCR analysis using an ABI 7900 HT real-time PCR system with SYBR green PCR master mix (Applied Biosystems) and appropriate primers (Table 2). To assay the effect of sirtuin knockdown on virus replication, cells were treated with sirtuin-specific siRNAs and then processed as follows: infect with HCMV (0.5 IU/cell) at 24 h after knockdown and harvest cell-free progeny at 96 h postinfection (hpi); infect with HSV-1 (1.0 IU/cell) or influenza virus A/WSN (0.01 50% tissue culture infective dose [TCID50]) at 72 h after knockdown and harvest cell-free progeny at 24 hpi; infect with Ad5 (5 IU/cell) at 72 h after knockdown and harvest cell-free progeny at 48 hpi. Virus yields were determined by infectious center assay using antibodies for HCMV IE1 (1B12) (55) in MRC5 cells, HSV-1 ICP4 (56) in MRC5 cells, and Ad5 E2A-72K (B6-8) (57) in HeLa cells or by TCID50 for influenza virus A/WSN in MDCK cells.
TABLE 1 .
siRNAs used in this studya
| siRNA | Sequence (sense) (from Sigma) or description |
|---|---|
| siSirt1_1 | 5′ CUGUGAAGCUGUACGAGGA [dT][dT] 3′ |
| siSirt1_2 | 5′ GAAGUACAAACUUCUAGGA [dT][dT] 3′ |
| siSirt2_1 | 5′ CUACUCCUGCGCUGCUACA [dT][dT] 3′ |
| siSirt2_2 | 5′ CGCGUUUCUUCUCCUGUAU [dT][dT] 3′ |
| siSirt3_1 | 5′ CUCAAAGCUGGUUGAAGCU [dT][dT] 3′ |
| siSirt3_2 | 5′ GACAAGACCUCAUGCCUGA [dT][dT] 3′ |
| siSirt4_1 | 5′ GGGAUCAUCCUUGCAGGUA [dT][dT] 3′ |
| siSirt4_2 | 5′ CUUUGAGCACCUGGGAGAA [dT][dT] 3′ |
| siSirt5_1 | 5′ CAGCAUCCCAGUUGAGAAA [dT][dT] 3′ |
| siSirt5_2 | 5′ GAGAUCCAUGGUAGCUUAU [dT][dT] 3′ |
| siSirt6_1 | 5′ CUGUCCAUCACGCUGGGUA [dT][dT] 3′ |
| siSirt6_2 | 5′ CUCACUUUGUUACUUGUUU [dT][dT] 3′ |
| siSirt7_1 | 5′ GGGAGUACGUGCGGGUGUU [dT][dT] 3′ |
| siSirt7_2 | 5′ CCCUGAAGCUACAUGGGAA [dT][dT] 3′ |
| siIE2 | 5′ AAACGCAUCUCCGAGUUGGAC [dT][dT] 3′ |
| siNT | siRNA universal negative control 1 (Sigma) |
DNA bases within RNA oligonucleotides are shown in brackets.
TABLE 2 .
DNA sequences of primers used for qPCR analysis
| Gene product | Primer sequence (from Integrated DNA Technologies)a |
|---|---|
| Sirtuin 1 | Fwd: 5′ ACAGGTTGCGGGAATCCAAAGG 3′ |
| Rev: 5′ CCTAGGACATCGAGGAACTACCTG 3′ | |
| Sirtuin 2 | Fwd: 5′ ACCCGCTAAGCTGGATGAAAGAG 3′ |
| Rev: 5′ AGTCTTCACACTTGGGCGTCAC 3′ | |
| Sirtuin 3 | Fwd: 5′ ACATCGATGGGCTTGAGAGAGTG 3′ |
| Rev: 5′ CAGAGGCAAAGGTTCCATGAGC 3′ | |
| Sirtuin 4 | Fwd: 5′ ACAGGGTCCTGTGCTTGGATTG 3′ |
| Rev: 5′ CTTGGAAACGCTCTTGCAGCAC 3′ | |
| Sirtuin 5 | Fwd: 5′ TGGCTCGGCCAAGTTCAAGTATG 3′ |
| Rev: 5′ AAGGTCGGAACACCACTTTCTGC 3′ | |
| Sirtuin 6 | Fwd: 5′ TGTGGAAGAATGTGCCAAGT 3′ |
| Rev: 5′ CTTAGCCACGGTGCAGAG 3′ | |
| Sirtuin 7 | Fwd: 5′ GCGTCTATCCCAGACTACCG 3′ |
| Rev: 5′ GTGATGCTCATGTGGGTGA 3′ | |
| GAPDH | Fwd: 5′ CGACAGTCAGCCGCATCTTT 3′ |
| Rev: 5 ′ GGCAACAATATCCACTTTACCAGAG 3′ | |
| β-Actin | Fwd: 5′ TCCTCCTGAGCGCAAGTACTC 3′ |
| Rev: 5′ CGGACTCGTCATACTCCTGCTT 3′ | |
| CobB | Fwd: 5′ ATGCCATTGTTGCCAGTTTCCG 3′ |
| Rev: 5′ ATCCATGCCGAGTGGCATTTCG 3′ | |
| CysG | Fwd: 5′ TTGTCGGCGGTGGTGATGTC 3′ |
| Rev: 5′ ATGCGGTGAACTGTGGAATAAACG 3′ |
Fwd, forward; Rev, reverse.
Modulation of sirtuin activity.
MRC5 fibroblasts (HCMV, HSV-1, and Ad5) or MDCK cells (influenza virus A/PR8) were infected at the input multiplicities described above. At 2 hpi, culture media were changed to fresh media supplemented with one of the following drugs: resveratrol (a sirtuin activator; Sigma), CAY10602 (a sirtuin activator; Cayman), or EX-527 (a SIRT1 inhibitor; Cayman), and the viral titer was determined at various times after infection as described above. The impact of drug treatment on SIRT1 deacetylation activity was assessed using the direct fluorescence assay (Cayman) according to the manufacturer’s specifications. Cell viability following treatment was assessed by CellTiter 96 AQueous One solution assay (Promega) and by phase microscopy (Nikon Eclipse TE200). The effects of treatments on HCMV proteins were analyzed by Western blotting. Cells were collected at 24, 48, and 72 hpi (3 IU/cell) and lysed in radioimmunoprecipitation assay (RIPA)-light buffer (50 mM Tris-HCl, pH 8.0, 1% NP-40, 0.1% SDS, 150 mM NaCl, 0.1% Triton X-100, and 5 mM EDTA) with protease inhibitor cocktail (Sigma). Protein concentrations were determined by Bradford assay, and proteins were resolved by SDS-PAGE and transferred to a polyvinylidene fluoride membrane. The antibodies used were anti-IE1 (clone 1B12) (55), anti-UL26 (7H-19) (58), anti-UL99 (10B4-29) (49), and anti-β-actin–horseradish peroxidase (HRP) (Abcam).
Bacteriophage experiments.
MC4100-wt-pCobB and MC4100-ΔCobB-pCobB cells were generated by transformation of MC4100-wt (53) (wild type) and MC4100-ΔCobB (CobB knockout) with the CobB-His6 ASKA(−) plasmid (59). The CobB knockout strain was generated by transferring a ΔcobB::kan insertion-deletion allele obtained from the Keio collection (60) to the E. coli K-12 laboratory strain MC4100 through generalized transduction. The kanamycin resistance marker was then cured by FLP-mediated recombination in the presence of the plasmid pCP20 (61), yielding a 34-bp FLP recombination target (FRT) site “scar” in place of the CobB open reading frame (ORF). Culture cells were grown at 37°C. Where appropriate, chloramphenicol (100 µg/ml; Fisher Scientific) was added to the culture medium. Isopropyl β-d-1-thiogalactopyranoside (IPTG) (Ambion) induction was performed by adding IPTG (0.25 mM) to E. coli at an optical density at 600 nm (OD600) of 0.5, vigorous shaking for 15 min at 37°C. To ensure that cell toxicity associated with CobB overexpression is minimized, IPTG was diluted 1:10 after induction. Cell growth was monitored by measuring the OD600 every 5 min using a Bio-Tek Synergy HT microplate reader with Gen5 software. For kinetic lysis assay, cells treated with IPTG were infected with T4-ΔrI (2 PFU/cell), and cell growth was monitored as described above. Lysis time was determined by nonlinear curve fitting and calculating the midpoint of the lysis curve using the software program GraphPad Prism 6. CobB mRNA levels were compared between three bacterial strains: MC4100-wt, MC4100-ΔCobB, and MC4100-wt-pCobB. Bacterial cultures inoculated from single colonies from each strain were grown overnight at 37°C in 5 ml Luria broth. The next day, cells were synchronized in equal volumes by dilution to an OD600 of 0.05 and further incubated at 37°C until reaching an OD600 of 0.6. CobB expression from the ASKA plasmid was induced with IPTG (0.25 mM) for 15 min, and the MC4100-wt and MC4100-ΔCobB cultures were incubated at 37°C for 3 h. Cells were collected by centrifugation, and RNA in the cell pellet was stabilized using RNAprotect bacterial reagent (Qiagen), following the manufacturer’s protocol. RNA was extracted using an RNeasy minikit (50) (Qiagen) and treated with DNase I from the same kit to degrade DNA trace amounts. cDNA was synthesized using a RETROscript kit (Invitrogen), and reverse transcription-qPCR (RT-qPCR) analysis was performed as described above, using primers listed in Table 2.
λvir and T4ΔrI phage infections were carried out at 15 min of IPTG induction, and titers were measured by plaque assay at indicated time points postinfection. λvir burst size was determined as described previously (62). Briefly, the relevant E. coli strains were grown in triplicate to mid-exponential phase in rich medium containing 0.2% maltose to induce expression of the λ receptor LamB and incubated with λvir in 10 mM MgSO4 for 15 min. Infective centers were isolated by centrifugation, incubated in rich medium at 37°C for 1 h, and then plated for plaques overnight at 37°C. Burst size was established by determining the ratio of initial PFU number (“prelysis”) to final PFU number (“postlysis”). T4ΔrI phage burst size was determined essentially as described elsewhere (63).
ACKNOWLEDGMENTS
This work was supported by NIH grants DP1DA026192 and R21AI102187 and HFSPO award RGY0079/2009-C to I.M.C., NIH grants AI87672 and AI97382 to T.S., and an NSF graduate research fellowship for H.G.B.
E.K., T.S., and I.M.C. conceived and designed the study. E.K., H.G.B., D.P.R., and Y.V.M. performed experiments. All authors discussed and interpreted the results and commented on the manuscript.
E.K., T.S., and I.M.C. are shareholders of Forge Life Science, which has licensed sirtuin-related technology from Princeton University.
Footnotes
Citation Koyuncu E, Budayeva HG, Miteva YV, Ricci DP, Silhavy TJ, Shenk T, Cristea IM. 2014. Sirtuins are evolutionarily conserved viral restriction factors. mBio 5(6):e02249-14. doi:10.1128/mBio.02249-14.
REFERENCES
- 1. Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. 2005. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 16:4623–4635. 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Michan S, Sinclair D. 2007. Sirtuins in mammals: insights into their biological function. Biochem. J. 404:1–13. 10.1042/BJ20070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Choi JE, Mostoslavsky R. 2014. Sirtuins, metabolism, and DNA repair. Curr. Opin. Genet. Dev. 26C:24–32. 10.1016/j.gde.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haigis MC, Sinclair DA. 2010. Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5:253–295. 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tsai YC, Greco TM, Cristea IM. 2014. Sirtuin 7 plays a role in ribosome biogenesis and protein synthesis. Mol. Cell Proteomics 13:73–83. 10.1074/mcp.M113.031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV, Jr, Weissman S, Verdin E, Schwer B. 2007. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27:8807–8814. 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rauh D, Fischer F, Gertz M, Lakshminarasimhan M, Bergbrede T, Aladini F, Kambach C, Becker CF, Zerweck J, Schutkowski M, Steegborn C. 2013. An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat. Commun. 4:2327. 10.1038/ncomms3327. [DOI] [PubMed] [Google Scholar]
- 8. Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L. 2006. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126:941–954. 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 9. Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. 2011. SIRT6 promotes DNA repair under stress by activating PARP1. Science 332:1443–1446. 10.1126/science.1202723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H. 2011. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334:806–809. 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, Luo H, Zhang Y, He W, Yang K, Zwaans BM, Tishkoff D, Ho L, Lombard D, He TC, Dai J, Verdin E, Ye Y, Zhao Y. 2011. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics 10:M111.012658. 10.1074/mcp.M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, Lin H. 2013. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496:110–113. 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, Dorr A, Hetzer-Egger C, Henklein P, Frye R, McBurney MW, Hruby H, Jung M, Verdin E, Ott M. 2005. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 3:e41. 10.1371/journal.pbio.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kwon HS, Brent MM, Getachew R, Jayakumar P, Chen LF, Schnolzer M, McBurney MW, Marmorstein R, Greene WC, Ott M. 2008. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe 3:158–167. 10.1016/j.chom.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Campagna M, Herranz D, Garcia MA, Marcos-Villar L, González-Santamaría J, Gallego P, Gutierrez S, Collado M, Serrano M, Esteban M, Rivas C. 2011. SIRT1 stabilizes PML promoting its sumoylation. Cell Death Differ. 18:72–79. 10.1038/cdd.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Q, He M, Zhou F, Ye F, Gao SJ. 2014. Activation of Kaposi’s sarcoma-associated herpesvirus (KSHV) by inhibitors of class III histone deacetylases: identification of sirtuin 1 as a regulator of the KSHV life cycle. J. Virol. 88:6355–6367. 10.1128/JVI.00219-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 325:417–470. [DOI] [PubMed] [Google Scholar]
- 18. Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS, Huber LJ. 2006. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell. Biol. 26:28–38. 10.1128/MCB.26.1.28-38.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Evers DL, Wang X, Huong SM, Huang DY, Huang ES. 2004. 3,4′,5-Trihydroxy-trans-stilbene (resveratrol) inhibits human cytomegalovirus replication and virus-induced cellular signaling. Antiviral Res. 63:85–95. 10.1016/j.antiviral.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 20. Palamara AT, Nencioni L, Aquilano K, De Chiara G, Hernandez L, Cozzolino F, Ciriolo MR, Garaci E. 2005. Inhibition of influenza A virus replication by resveratrol. J. Infect. Dis. 191:1719–1729. 10.1086/429694. [DOI] [PubMed] [Google Scholar]
- 21. Nayagam VM, Wang X, Tan YC, Poulsen A, Goh KC, Ng T, Wang H, Song HY, Ni B, Entzeroth M, Stünkel W. 2006. SIRT1 modulating compounds from high-throughput screening as anti-inflammatory and insulin-sensitizing agents. J. Biomol. Screen. 11:959–967. 10.1177/1087057106294710. [DOI] [PubMed] [Google Scholar]
- 22. Zhao K, Chai X, Marmorstein R. 2004. Structure and substrate binding properties of cobB, a Sir2 homolog protein deacetylase from Escherichia coli. J. Mol. Biol. 337:731–741. 10.1016/j.jmb.2004.01.060. [DOI] [PubMed] [Google Scholar]
- 23. Burch LH, Zhang L, Chao FG, Xu H, Drake JW. 2011. The bacteriophage T4 rapid-lysis genes and their mutational proclivities. J. Bacteriol. 193:3537–3545. 10.1128/JB.00138-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eisen HA, Fuerst CR, Siminovitch L, Thomas R, Lambert L, Pereira da Silva L, Jacob F. 1966. Genetics and physiology of defective lysogeny in K12 (lambda): studies of early mutants. Virology 30:224–241. 10.1016/0042-6822(66)90098-5. [DOI] [PubMed] [Google Scholar]
- 25. Marraffini LA, Sontheimer EJ. 2010. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 11:181–190. 10.1038/nrg2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cullen BR, Cherry S, tenOever BR. 2013. Is RNA interference a physiologically relevant innate antiviral immune response in mammals? Cell Host Microbe 14:374–378. 10.1016/j.chom.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 27. Ferrandon D, Imler JL, Hetru C, Hoffmann JA. 2007. The Drosophila systemic immune response: sensing and signalling during bacterial and fungal infections. Nat. Rev. Immunol. 7:862–874. 10.1038/nri2194. [DOI] [PubMed] [Google Scholar]
- 28. Roberts RM, Liu L, Guo Q, Leaman D, Bixby J. 1998. The evolution of the type I interferons. J. Interferon Cytokine Res. 18:805–816. 10.1089/jir.1998.18.805. [DOI] [PubMed] [Google Scholar]
- 29. Baur JA, Ungvari Z, Minor RK, Le Couteur DG, de Cabo R. 2012. Are sirtuins viable targets for improving healthspan and lifespan? Nat. Rev. Drug Discov. 11:443–461. 10.1038/nrd3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ren JH, Tao Y, Zhang ZZ, Chen WX, Cai XF, Chen K, Ko BC, Song CL, Ran LK, Li WY, Huang AL, Chen J. 2014. Sirtuin 1 regulates hepatitis B virus transcription and replication by targeting transcription factor AP-1. J. Virol. 88:2442–2451. 10.1128/JVI.02861-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G, Fanciulli M, Raimondo G, Levrero M. 2009. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. U. S. A. 106:19975–19979. 10.1073/pnas.0908365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Srisuttee R, Koh SS, Kim SJ, Malilas W, Boonying W, Cho IR, Jhun BH, Ito M, Horio Y, Seto E, Oh S, Chung YH. 2012. Hepatitis B virus X (HBX) protein upregulates beta-catenin in a human hepatic cell line by sequestering SIRT1 deacetylase. Oncol. Rep. 28:276–282. 10.3892/or.2012.1798. [DOI] [PubMed] [Google Scholar]
- 33. Hallows WC, Yu W, Denu JM. 2012. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J. Biol. Chem. 287:3850–3858. 10.1074/jbc.M111.317404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Walker AK, Yang F, Jiang K, Ji JY, Watts JL, Purushotham A, Boss O, Hirsch ML, Ribich S, Smith JJ, Israelian K, Westphal CH, Rodgers JT, Shioda T, Elson SL, Mulligan P, Najafi-Shoushtari H, Black JC, Thakur JK, Kadyk LC, Whetstine JR, Mostoslavsky R, Puigserver P, Li X, Dyson NJ, Hart AC, Näär AM. 2010. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 24:1403–1417. 10.1101/gad.1901210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X. 2009. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 9:327–338. 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. 2007. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell 28:91–106. 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 37. Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, Vazquez-Ortiz G, Jeong WI, Park O, Ki SH, Gao B, Deng CX. 2010. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 12:224–236. 10.1016/j.cmet.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, Clish CB, Vaitheesvaran B, Iliopoulos O, Kurland I, Dor Y, Weissleder R, Shirihai OS, Ellisen LW, Espinosa JM, Mostoslavsky R. 2010. The histone deacetylase Sirt6 regulates glucose homeostasis via HIF1alpha. Cell 140:280–293. 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xiao C, Kim HS, Lahusen T, Wang RH, Xu X, Gavrilova O, Jou W, Gius D, Deng CX. 2010. SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J. Biol. Chem. 285:36776–36784. 10.1074/jbc.M110.168039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Elhanati S, Kanfi Y, Varvak A, Roichman A, Carmel-Gross I, Barth S, Gibor G, Cohen HY. 2013. Multiple regulatory layers of SREBP1/2 by SIRT6. Cell. Rep. 4:905–912. 10.1016/j.celrep.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 41. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV, Jr, Alt FW, Kahn CR, Verdin E. 2010. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464:121–125. 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. 2006. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2:e132. 10.1371/journal.ppat.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, Shenk T, Rabinowitz JD. 2008. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 26:1179–1186. 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koyuncu E, Purdy JG, Rabinowitz JD, Shenk T. 2013. Saturated very long chain fatty acids are required for the production of infectious human cytomegalovirus progeny. PLoS Pathog. 9:e1003333. 10.1371/journal.ppat.1003333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gudleski-O’Regan N, Greco TM, Cristea IM, Shenk T. 2012. Increased expression of LDL receptor-related protein 1 during human cytomegalovirus infection reduces virion cholesterol and infectivity. Cell Host Microbe 12:86–96. 10.1016/j.chom.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD. 2011. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 7:e1002124. 10.1371/journal.ppat.1002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Diamond DL, Syder AJ, Jacobs JM, Sorensen CM, Walters KA, Proll SC, McDermott JE, Gritsenko MA, Zhang Q, Zhao R, Metz TO, Camp DG, II, Waters KM, Smith RD, Rice CM, Katze MG. 2010. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 6:e1000719. 10.1371/journal.ppat.1000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Haller O, Kochs G, Weber F. 2007. Interferon, Mx, and viral countermeasures. Cytokine Growth Factor Rev. 18:425–433. 10.1016/j.cytogfr.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moorman NJ, Sharon-Friling R, Shenk T, Cristea IM. 2010. A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol. Cell. Proteomics 9:851–860. 10.1074/mcp.M900485-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hughes RG, Jr, Munyon WH. 1975. Temperature-sensitive mutants of herpes simplex virus type 1 defective in lysis but not in transformation. J. Virol. 16:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jones N, Shenk T. 1978. Isolation of deletion and substitution mutants of adenovirus type 5. Cell 13:181–188. 10.1016/0092-8674(78)90148-4. [DOI] [PubMed] [Google Scholar]
- 52. Taylor A, Sharp D, Beard D, Beard J, Dingle J, Feller A. 1943. Isolation and characterization of influenza A virus (PR8 strain). J. Immunol. 47:261–282. [Google Scholar]
- 53. Casadaban MJ. 1976. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J. Mol. Biol. 104:541–555. 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- 54. Wiebusch L, Truss M, Hagemeier C. 2004. Inhibition of human cytomegalovirus replication by small interfering RNAs. J. Gen. Virol. 85:179–184. 10.1099/vir.0.19453-0. [DOI] [PubMed] [Google Scholar]
- 55. Zhu H, Shen Y, Shenk T. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69:7960–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Showalter SD, Zweig M, Hampar B. 1981. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect. Immun. 34:684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Reich NC, Sarnow P, Duprey E, Levine AJ. 1983. Monoclonal antibodies which recognize native and denatured forms of the adenovirus DNA-binding protein. Virology 128:480–484. 10.1016/0042-6822(83)90274-X. [DOI] [PubMed] [Google Scholar]
- 58. Munger J, Yu D, Shenk T. 2006. UL26-deficient human cytomegalovirus produces virions with hypophosphorylated pp28 tegument protein that is unstable within newly infected cells. J. Virol. 80:3541–3548. 10.1128/JVI.80.7.3541-3548.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 12:291–299. 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- 60. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Clokie M, Kropinski A. 2009. Bacteriophages: methods and protocols, vol 1: Isolation, characterization, and interactions. Humana Press, New York, NY. [Google Scholar]
- 63. Wang IN. 2006. Lysis timing and bacteriophage fitness. Genetics 172:17–26. 10.1534/genetics.105.045922. [DOI] [PMC free article] [PubMed] [Google Scholar]