

Abstract
Almost 40 years from when it was first reported that UVB radiation exposure would modulate immune signaling, the photoimmunology field is still trying to understand the mechanisms by which UVB initiates inflammatory responses and modulates immune recognition. This commentary focuses on the ability of Toll-like receptors (TLRs), specifically TLR4 (Ahmad et al., 2014) and ligands such as damage-associated molecular patterns (DAMPs) released from injured cells to stimulate innate immune signaling and inflammatory cytokine production following UVB irradiation.
In many individuals, even brief exposure to intense sunlight can cause marked tissue injury. In fact, repeated exposures to ultraviolet (UV) radiation at the earth's surface is a causative factor in many diseases of the skin, including premature aging and the development of skin cancer. How the skin responds and repairs injury from UV is therefore an area of great importance, and research into this field has been marked by several important observations.
In the late 1970s, work done by Kripke (1974) demonstrated that UVB is able to trigger profound immunosup-pression in mice. Her studies showed that UVB-induced tumors were highly antigenic and that they were usually rejected by untreated syngeneic recipients, unless the recipients had been exposed to UVB. Further studies demonstrated that the systemic immunologic alteration was secondary to a direct effect of UVB on the skin. Although this work was seminal to the field, it did not identify the cells and signals that mediate UVB-induced immunosuppression.
In 1983, De Fabo and Noonan (1983) proposed that urocanic acid acted as a skin chromophore in the stratum corneum by demonstrating that it has the capacity to induce systemic immunosuppression. Many investigators have since confirmed the relevance of cis-UCA to immunosuppression. Cis-UCA, an imidazole derivative, is produced by the isomerization of trans-UCA following UV exposure, and it interacts with immune cells both locally and systemically to promote T-suppressor function, impairing CHS through the induction of TNF-α. The exact mechanism, and whether cis-UCA initiates immunosuppression locally or systemically, still remains unclear.
In addition to cis-UCA, nucleotide bases of both DNA and RNA are the chromophores that absorb radiation in the UVB and UVC (200–290 nm) ranges. Damage to DNA, especially in the form of cyclobutane pyrimidine dimers, induces both immunosuppressive and carcinogenic outcomes. Mice injected with liposomes containing the DNA repair enzyme, T4 endonuclease, had reduced UV-induced immunosuppression. In contrast, patients with xeroderma pigmentosum have both DNA repair defects and higher incidences of skin cancer. Collectively, these studies demonstrate that DNA damage is involved in UV-induced immunosuppression, but many questions remain regarding how UV damage is recognized.
Lack of TLR4 activity promotes resistance to UVB-induced immunosuppression
In this issue, Ahmad et al. (2014) demonstrated that the products of the DNA repair gene XPA (xeroderma pigmentosum complementation group A) were significantly lower in skin and dendritic cells of wild-type mice with functional toll-like receptor 4 (TLR4), compared with TLR4 −/− mice after UVB exposure. This suggests that the repair of UVB-induced DNA damage is regulated in part by TLR4. Moreover, lack of TLR4 activity increased the amounts of IL-12 and IL-23 synthesized following UVB exposure. IL-12 and IL-23 promote the induction of IFN-γ-producing CD8+ TC1 cells and IL-17 CD8+ TC17 cells, respectively, both of which are known to serve as effector cells for CHS responses and to stimulate the synthesis of DNA repair enzymes and reduce CPDs through the NER pathway, suggesting a mechanism for TLR4-induced immunosuppression (Ahmad et al., 2014). This is an extension of their earlier study demonstrating that TLR4 is required for the generation of IL-10-secreting T-regulatory cells that are an essential component of UV-induced immunosuppression (Lewis et al., 2011). Both of these studies stem from an earlier observation that the Lps locus, the same region where the TLR4 gene has been mapped, is critical for the ability of UV radiation to impair DNFB-specific contact hypersensitivity in mice (Yoshikawa and Streilein, 1990). Why TLR4, a pattern recognition receptor (PRR) that recognizes lipopolysaccharides from Gram negative bacteria, has this function is currently unknown. However, an explanation for these observations may lie in recent advances in the understanding of PRRs and their many ligands.
The role of PAMPs, MAMPs, and DAMPs following UVB exposure
TLRs activate aspects of the innate immune system and act through the recognition of non-self molecules known as pathogen-associated molecular patterns (PAMPs) or microbe-associated molecular patterns (MAMPs). However, TLRs and other PPRs are not restricted to detection of only a single chemical entity. Many have the capacity to be activated by a diverse repertoire of compounds found both on microbes and in the host. It is now apparent that the same sensing mechanism that detect microbes can detect damaged molecules derived from the host, called damage-associated molecular patterns (DAMPs). There are several DAMPs that are candidates for activating TLR4, including oxidized lipids, heat-shock proteins, S100 family proteins, HMGB1, and hyaluronic acid (Figure 1). Thus, one explanation for how TLR4 participates in recognition of UV injury is that injury to self generates one or more of these DAMPs to trigger TLR4. Another potential explanation is that UV injury to the epidermis alters the distribution of the skin microbiome across the epidermis (Nakatsuji et al., 2013). Such a change in the penetration of bacteria across the stratum corneum and epidermis could directly activate TLR4 in deeper cells. Further investigations are needed to determine which of the TLR4 ligands are critical for TLR4-dependent UV responses.
Figure 1. Innate immune signaling through damage-associated molecular patterns (DAMPs) in UVB-exposed skin.
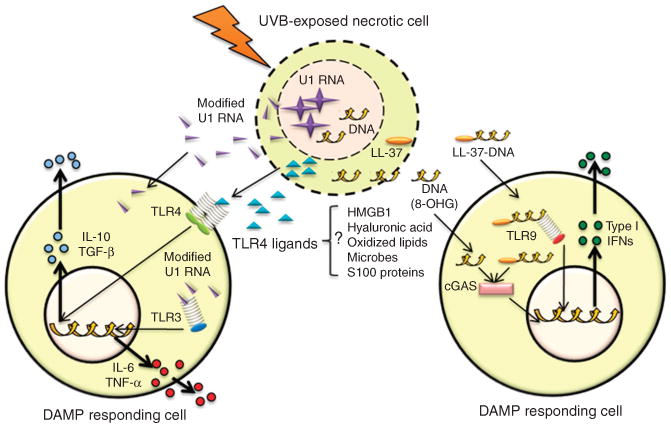
UVB-modified nucleic acids, as well as lipids and proteins, may act as DAMPs to stimulate innate immune signaling in responding cells. DAMPs are released from necrotic cells and taken up by adjacent cells to activate membrane and cytosolic innate immune sensors such as TLRs and cGAS. This initiates downstream signaling cascades that lead to the upregulation of cytokines involved in the inflammatory and immune responses of UVB exposure. cGAS, cGMP-AMP-synthase; TGF, transforming growth factor; TLR, Toll-like receptor; TNF, tumor necrosis factor.
In addition to TLR4, other TLRs also contribute to the recognition of UVB injury. We recently applied next-generation RNA sequencing to demonstrate that U1 RNA, an abundant self-noncoding RNA released from necrotic cells, is altered following UVB irradiation and is detected by TLR3 (Bernard et al., 2012; Figure 1). TLR3 was best known as a PRR responsible for detecting double-stranded RNA from viruses. The release of U1 RNA from UVB-damaged necrotic cells was found to act as a DAMP to partially initiate the acute inflammatory sunburn response. Confirming this result were observations that TLR3 −/− mice have diminished the production of TNF-α and IL-6 following UVB exposure. TLR3 −/− mice also maintained a normal capacity to become sensitized to cutaneous antigens after UVB exposure, suggesting that, in addition to the participation of TLR4 in response to UV, the capacity of TLR3 to detect damaged self-RNA is also a critical step in UVB-mediated immunosuppression. Furthermore, the relative roles of TLR3 and TLR4 may depend on the cell type studied. For example, keratinocytes are highly sensitive to TLR3 ligands but resistant to TLR4, whereas most bone marrow–derived cells respond to both.
There is evidence to suggest that other DAMPs are produced after UV injury. UVB induces nucleic acid modifications such as RNA–RNA cross-linking and the oxidation of guanine to 8-oxo-7,8-dihydroguanosine (8-oxoG) and 8-hydroxyguanosine (8-OH-dG). However, little is known about how these modifications contribute to the inflammatory and immunosuppressive effects of UVB exposure or how they may be recognized. Recently, 8-OH-dG has also been suggested to act as a DAMP (Gehrke et al., 2013). 8-OH-dG is released from dying cells and taken up by immune cells, where it accumulates in the cytosol and activates cGMP-AMP-synthase (cGAS) (Figure 1). cGAS signaling is potentiated by a second messenger cyclic dinucleotide c[G(2050)pA(3050)p], which binds to and activates the adaptor molecule STING, resulting in the activation of cytosolic kinases IKKε and TBK1 and eliciting a type 1 IFN response. This effect was most potent with oxidized DNA, as nondamaged DNA is mostly degraded in the cytosol by 3′ repair exonuclease1 (TREX1). Moreover, self-DNA released by dying cells forms a degradation-resistant complex with the endogenous antimicrobial peptide LL-37 in neutrophils and epithelial cells where it is transported into plasmacytoid dendritic cells (Lande et al., 2007) and monocytes (Chamilos et al., 2012). Interestingly, the LL-37-DNA complex is sensed by TLR9 in plasmacytoid dendritic cells and by cytosolic DNA sensors and STING and TBK1 in monocytes to elicit a type 1 IFN response (Figure 1).
These studies give credence to the notion that activation of innate immunity by DAMPs is important in altering adaptive immunity following UVB exposure. It also extends the paradigm that the innate and adaptive immune systems modulate each other. UVB activates other aspects of the innate immune system such as stimulating antimicrobial peptide production and conferring resistance to some bacterial infections. In addition, UVB-induced 8-oxoG RNA can act as an autoantigen and promote autoadjuvant activity through TLR activation of B cells. Therefore, it may be more appropriate to specify how immune responses are modulated following UVB exposure instead of generalizing that UVB induces immunosuppression.
Clinical Implications.
This report suggests the use of TLRs and cGAS as new targets in treating UVB-induced DNA damage and inflammation and photosensitive disorders such as lupus erythematosus.
This work may also have implications for cancer biology owing to the many associations of inflammation and immunosuppression with tumor cell initiation and neoplastic transformation.
However, the complex nature of carcinogenesis may make it difficult to predict the outcome of inhibiting or activating these MAMP, PAMP, and DAMP sensors.
Depending on the stage of carcinogenesis, it may be important to either limit inflammation and reduce immunosuppression or stimulate an inflammatory response to enhance immune surveillance.
Activation of TLR4 has been associated with the immune escape of coloncancer cells and the promotion of hepatocellular, gastric, and breastcancers (Tang and Zhu, 2012; Yuan et al., 2013), and thus it is attractive tospeculate that activation of TLR4 by the skin microbiome may increase skintumor formation.
In addition, Ahmad et al. (2014) demonstrated that inhibiting TLR4 promotes DNA damage repair.
This may be important for reducing tumor burden or may increase tumor burden if damaged cells are not repaired properly and undergo mitosis.
Further work to characterize the clinical implications of DAMPS and innate immune sensors in the response to UVB exposure is therefore warranted.
Footnotes
Conflict of Interest: The authors state no conflict of interest.
References
- Ahmad I, Simanyi E, Guroji P, et al. Toll-Like Receptor-4 deficiency enhances repair of ultraviolet radiation induced cutaneous DNA damage by nucleotide excision repair mechanism. J Invest Dermatol. 2014;134:1710–7. doi: 10.1038/jid.2013.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard JJ, Cowing-Zitron C, Nakatsuji T, et al. Ultraviolet radiation damages self non-coding RNA and is detected by TLR3. Nat Med. 2012;18:1286–90. doi: 10.1038/nm.2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamilos G, Gregorio J, Meller S, et al. Cytosolic sensing of extracellular self-DNA transported into monocytes by the antimicrobial peptide LL37. Blood. 2012;120:3699–707. doi: 10.1182/blood-2012-01-401364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Fabo EC, Noonan FP. Mechanism of immune suppression by ultraviolet irradiation in vivo. I. Evidence for the existence of a unique photoreceptor in skin and its role in photoimmunology. J Exp Med. 1983;158:84–98. doi: 10.1084/jem.158.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrke N, Mertens C, Zillinger T, et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity. 2013;39:482–95. doi: 10.1016/j.immuni.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Kripke ML. Antigenicity of murine skin tumors induced by ultraviolet light. J Natl Cancer Inst. 1974;53:1333–6. doi: 10.1093/jnci/53.5.1333. [DOI] [PubMed] [Google Scholar]
- Lande R, Gregorio J, Facchinetti V, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–9. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- Lewis W, Simanyi E, Li H, et al. Regulation of ultraviolet radiation induced cutaneous photoimmunosuppression by toll-like receptor-4. Arch Biochem Biophys. 2011;508:171–7. doi: 10.1016/j.abb.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chiang HI, Jiang SB, et al. The microbiome extends to subepidermal compartments of normal skin. Nat Commun. 2013;4:1431. doi: 10.1038/ncomms2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Zhu Y. TLR4 signaling promotes immune escape of human colon cancer cells by inducing immunosuppressive cytokines and apoptosis resistance. Oncol Res. 2012;20:15–24. doi: 10.3727/096504012x13425470196092. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, Streilein JW. Genetic basis of the effects of ultraviolet light B on cutaneous immunity. Evidence that polymorphism at the Tnfa and Lps loci governs susceptibility. Immunogenetics. 1990;32:398–405. doi: 10.1007/BF00241633. [DOI] [PubMed] [Google Scholar]
- Yuan X, Zhou Y, Wang W, et al. Activation of TLR4 signaling promotes gastric cancer progression by inducing mitochondrial ROS production. Cell Death Dis. 2013;4:e794. doi: 10.1038/cddis.2013.334. [DOI] [PMC free article] [PubMed] [Google Scholar]