

Abstract
Linear conjugated oligothiophenes of variable length and different substitution pattern are ubiquitous in technologically advanced optoelectronic devices, though limitations in application derive from insolubility, scarce processability and chain-end effects. This study describes an easy access to chiral cyclic oligothiophenes constituted by 12 and 18 fully conjugated thiophene units. Chemical oxidation of an “inherently chiral” sexithiophene monomer, synthesized in two steps from commercially available materials, induces the formation of an elliptical dimer and a triangular trimer endowed with electrosensitive cavities of different tunable sizes. Combination of chirality with electroactivity makes these molecules unique in the current oligothiophenes literature. These macrocycles, which are stable and soluble in most organic solvents, show outstanding chiroptical properties, high circularly polarized luminescence effects and an exceptional enantiorecognition ability.
Keywords: chirality, circularly polarized luminescence, enantioselectivity, macrocycles, oligothiophenes
We have recently reported the example of a new approach to chiral electroactive oligothiophenes, consisting in the electro-oligomerization of monomers (R)-1 a and (S)-1 a in which chirality results from an internal torsion in the conjugated backbone (from here the appellative of inherently chiral monomers).[1] According to this design, the same conjugated system responsible for the electro-optical properties of these materials is also responsible for molecular chirality, implying that electrochemical, chiroptical and enantiorecognition properties are strictly interdependent (Scheme 1).
Scheme 1.
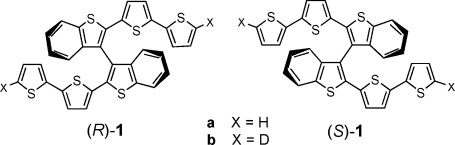
The inherently chiral enantiomeric monomers employed in the present research.
The conducting films resulting from electro-oxidation of enantiopure (R)-1 a and (S)-1 a, constitute oligomers from the dimer (largely prevailing) to the pentamer (present in traces). They display impressive chiroptical activity, reversible potential-driven variations of the circular dichroism (CD) signal and outstanding enantioselection ability towards a chiral ferrocenylamine selected as an electrochemical probe.
Here we report the results obtained when performing the chemical oxidation of racemic 1 a and its enantiopure antipodes with FeCl3 excess, in CHCl3 solution, under stirring, at RT, according to a procedure currently employed for preparing polythiophenes from electron-rich monomers when regioselectivity problems are not involved.[2]
The preliminary experiments have been carried out on racemic (±)-1 a.[3] Extraction with THF in a Soxhlet apparatus of the crude reaction product to remove some insoluble iron-containing materials, gives an orange residue (50 % yields in weight), composed of dimers (67 %), trimers (27 %), tetramers (2 %) and pentamers (less than 1 %), on the basis of peak intensities in laser desorption ionization (LDI) mass spectrum (see the Supporting Information section SI 1). High resolution LDI (HR-LDI) experiments show that the molecular weights and the isotopic patterns of all the components of the mixture are in agreement with formulas having two hydrogen atoms less than the expected values for linear oligomers (see the Supporting Information sections SI 2 and SI 3), as if the thiophene terminals of the oligomers had undergone a FeCl3-promoted intramolecular coupling.
To prove this hypothesis, we submitted to the same FeCl3 treatment the dideuterated compound (±)-1 b, prepared by quenching with D2O the dianion of (±)-1 a, obtained in turn by lithiation of the latter with nBuLi excess, in the presence of N,N,N′,N′-tetramethylethylenediamine, in THF solution, at −78 °C (see the Supporting Information section SI 4). We found (HR-LDI, see the Supporting Information section SI 5) that the deuterium had been totally lost, thus proving the cyclic structure of the oligomers. These MS experiments did not give evidence of the formation of open chain compounds, which are largely present (1:1) in the electrochemically prepared material on ITO electrodes (HR-LDI, see the Supporting Information section SI 6).
These results demonstrate that the FeCl3 oxidation of monomer 1 a generates a series of cyclic oligothiophenes, constituted by 12, 18, 24 and so on, conjugated thiophene units. When produced from enantiopure starting materials, all of them must be inherently chiral macrocycles constituted by a single Dn symmetric stereoisomer.
Oligothiophene macrocycles are not only aesthetically charming but operationally active molecules. They idealize conducting polymers without ends, offering electrosensitive cavities of different sizes for selective inclusion of guest molecules. The α-cyclo[n]thiophenes have been investigated by Bäuerle and co-workers who studied both their synthetic accessibility through different strategies and their physicochemical and electro-optical properties.[4] In the present case, the oligothiophene macrocycles display non-interrupted π systems, even though not fully α-conjugated. Combination of chirality with electroactivity makes these cavities unique in the current library of oligothiophenes.
In this context, we considered as primary target the isolation of the more abundant oligomers present in the mixture resulting from the FeCl3 oxidation of both the enantiopure antipodes of 1 a, since the oxidation of racemic (±)-1 a is expected to afford diastereomeric mixtures of cyclic oligomers produced by homochiral or heterochiral couplings: a pair of D2 symmetric enantiomers and a C2h symmetric meso compound in the case of the dimer 2 and two diastereomeric racemates, one D3 and one C2 symmetric, in the case of the trimer 3.
Accurate column chromatography enabled us to isolate the enantiopure dimer (S,S)-(−)-2 and the (S,S,S)-(+)-3 trimer from (S)-(+)-1 a and, specularly, the (R,R)-(+)-2 dimer and the (R,R,R)-(−)-3 trimer from (R)-(−)-1 a, in 15 and 8 % isolation yields, calculated on 1 a, respectively, for dimers and trimers (Figure 1). The enantiomeric purity of the four compounds was checked and demonstrated by analytical HPLC on a chiral stationary phase (see the Supporting Information section SI 7).
Figure 1.
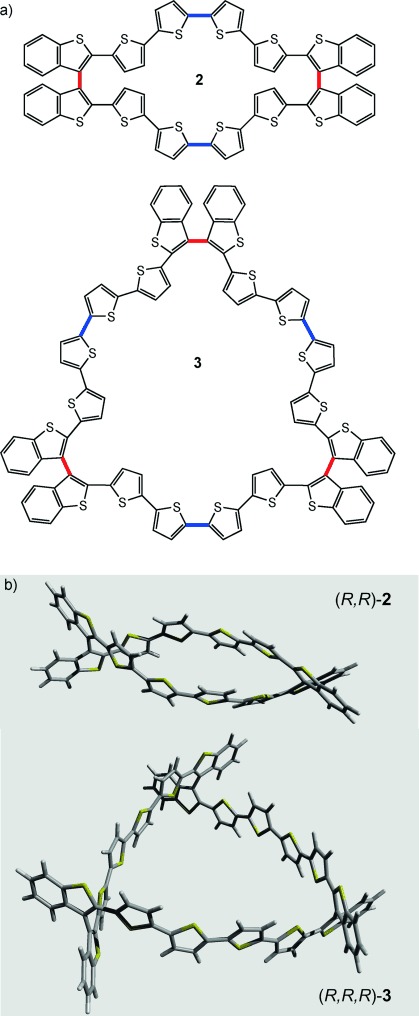
a) Planar molecular formulas of cyclic dimer 2 and trimer 3 evidencing connectivity. Bonds in red highlight the 3,3′-bithiophene junctions corresponding to the stereogenic axes, while bonds in blue indicate the oligomerization sites. b) Calculated structures of (R,R)-2 and (R,R,R)-3.
The 1H and 13C NMR spectra of enantiopure cyclodimers 2 and trimers 3 unequivocally proved the D2 and D3 symmetry of the molecules. According to stereotopism rules, only four coupled signals for the benzothiophene ring protons and four doublets for the protons in β-position of the two thiophene rings directly connected to it are observed in the 1H NMR spectra, while only sixteen signals are present in the 13C NMR spectra (see the Supporting Information section SI 8).
A picture of the shapes of homochiral dimers 2 and trimers 3 (Figure 1, http://users.unimi.it/ChiralCycles) was obtained by density functional theory (DFT) calculations in the generalized gradient approximation, using the Perdew–Burke–Ernzerhof functional to handle exchange-correlation effects (see the Supporting Information section SI 9).[5]
The cavities are characterized by different internal sizes and shapes, small and elliptical in the case of 2 (the major and minor axis lengths are about 17.7 and 5.8 Å, respectively) and quite large, equilateral triangular, in the case of 3 (side of the triangle: ∼17.9 Å long; geometrical height: ∼17.0 Å). It is interesting to note how bent outwards are the two facing sequences of the six α-linked thiophene units in dimers 2 under the constraint of the atropisomeric scaffolds, with consequent conjugation loss.
From the spectroscopic point of view, both 2 and 3 display absorption maxima significantly red-shifted with respect to that observed for the monomeric unit 1 a; this is attributable to the spin allowed π–π* transition (λmax=423 nm, ε=5.3×104 m−1 cm−1, for 2; λmax=447 nm, ε=1.3×105 m−1 cm−1 for 3 and λmax=372 nm, ε=4.8×104 m−1 cm−1 for 1 a in CH2Cl2 solution; Figure 2 a). This feature is in agreement with the extended π-conjugated system involving the six α-linked thiophene units. However, it is interesting to note that the absorption maximum of 2 is slightly blue-shifted with respect to that of the corresponding linear α-sexithiophene (λmax=445 nm, ε=5.0×104 m−1 cm−1).[6] This is mainly due to the constraint of the atropisomeric scaffolds, which bends outwards the two sequences of the six α-linked thiophene units with consequent conjugation loss. A sterically more relaxed situation characterizes trimers 3, where the rings of the α-sexithiophene sequences are coplanar, with remarkable gain in conjugation extent. However, the broad features of the absorption spectra indicate that 2 and 3 still maintain conformational flexibility in the ground state (see the Supporting Information section SI 10).
Figure 2.
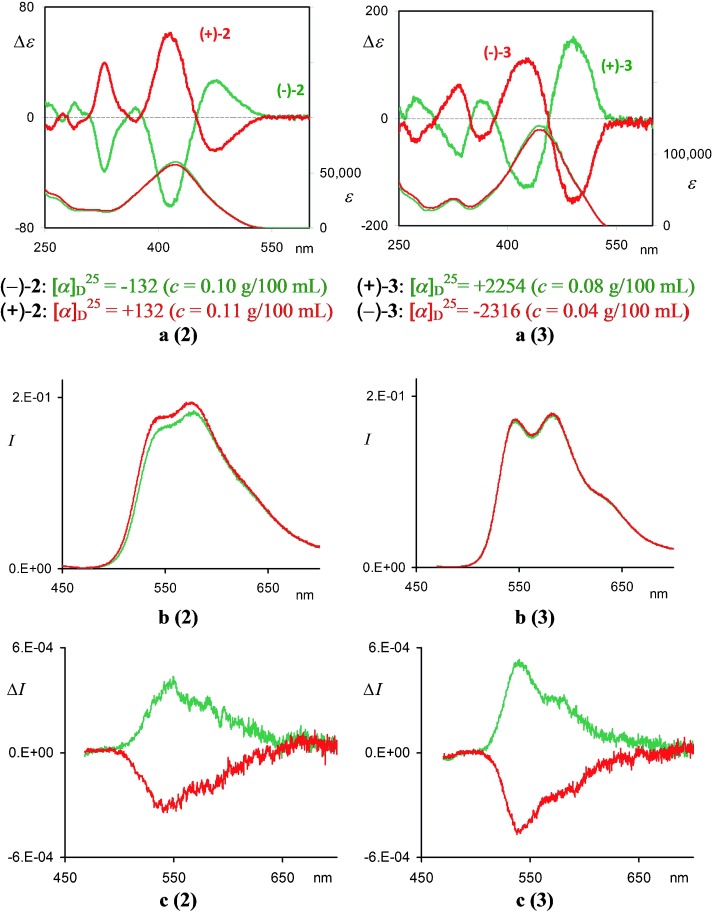
a (2), a (3): CD and UV spectra and [α]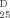 (CHCl3) of (S,S)-(−)-2 and (R,R)-(+)-2 (6.0×10−6 m), and of (S,S,S)-(+)-3 and (R,R,R)-(−)-3 (3.0×10−6 m). b (2), b (3): Emission spectra of (−)-2 (green) and (+)-2 (6.0×10−6 m) and of (+)-3 and (−)-3 (3.0×10−6 m) in aerated toluene solution at room temperature. Emission spectra were corrected for emission spectral response (detector and grating) by standards correction curves. c (2), c (3): CPL of (−)-2 and (+)-2 and CPL of (+)-3 and (−)-3 under the same conditions. Spectra of enantiomers with S stereogenic axes configuration are in green, with R configuration in red.
(CHCl3) of (S,S)-(−)-2 and (R,R)-(+)-2 (6.0×10−6 m), and of (S,S,S)-(+)-3 and (R,R,R)-(−)-3 (3.0×10−6 m). b (2), b (3): Emission spectra of (−)-2 (green) and (+)-2 (6.0×10−6 m) and of (+)-3 and (−)-3 (3.0×10−6 m) in aerated toluene solution at room temperature. Emission spectra were corrected for emission spectral response (detector and grating) by standards correction curves. c (2), c (3): CPL of (−)-2 and (+)-2 and CPL of (+)-3 and (−)-3 under the same conditions. Spectra of enantiomers with S stereogenic axes configuration are in green, with R configuration in red.
Films of enantiopure and racemic dimer 2 and trimer 3, drop-casted on screen printed electrodes are electroactive and conducting, affording reversible and repeatable oxo-reduction cycles (see the Supporting Information section SI 11). The cyclic voltammetry (CV) of enantiopure 2 displays a peculiarly sharp oxidation peak which is lost in racemic 2, pointing to a better supramolecular organization. In all cases the activation of the redox sites strongly depends on the scan rate, in accordance with slow doping/undoping processes similar to those displayed by oligothiophene films.
The chiroptical manifestations (optical rotation and circular dichroism) of the antipodes of 2 and 3 are outstanding, as expected, since produced by extended inherently dissymmetric chromophores (Figure 2 a).
Preliminary experiments on enantiopure 2 and 3 were focused: 1) to evaluate their circularly polarized luminescence (CPL) activity; 2) to check their enantiorecognition capacity in front of classical electrosensitive chiral probes.
Fluorescence and CPL simultaneous measurements were carried out in a toluene solution, at RT (λexc=450 nm for 2 and 470 nm for 3). Upon optical excitation, both 2 and 3 display intense orange-yellow vibrationally structured emission at λem=545, 573 nm and 542, 580 nm, respectively (Figure 2 b). These structured emissions are consistent with the planar quinoid-like structure proposed for the singlet excited-state of oligothiophenes.[7] The Stokes shift observed for 2 (Δλmax=122 nm), which is larger than that observed for 3 (Δλmax=96 nm), suggests that the ground state of the latter better matches the quinoid-like geometry of the excited state requiring a lesser conformational reorganization. This feature is in agreement with the more planar conformation of the α-sexithiophene sequences in 3, as indicated by DFT calculations and absorption features. The extent of backbone rigidity in the excited state has a significant impact on the photoluminescence quantum yields (ΦF), which are found to be 16 and 37 % for 2 and 3, respectively. The more accessible nonradiative deactivation pathways, favored by torsional vibration motions and responsible for the lower quantum yields observed for 2, match the high steric constrain of this species, both in ground and excited states.
The CPL emission is notable (Figure 2 c), with an anisotropy g factor close to 3×10−3, comparable with other inherently chiral molecules like helicenes[8] and higher than polythiophenes with chiral side chains.[9] These properties suggest possible applications of these materials in chiroptical devices (see the Supporting Information section SI 12).
Tests of the enantiorecognition capacity of the enantiopure macrocycles were performed on gold screen printed electrodes in (BMIM)PF6, employing (S)-(−)- and (R)-(+)-N,N-dimethyl-1-ferrocenylethylamine, (S)-4 and (R)-4, as chiral probes. The same probes had been previously employed for evaluating the enantiodiscrimination properties of the electrochemically prepared materials (Figure 3).[1]
Figure 3.
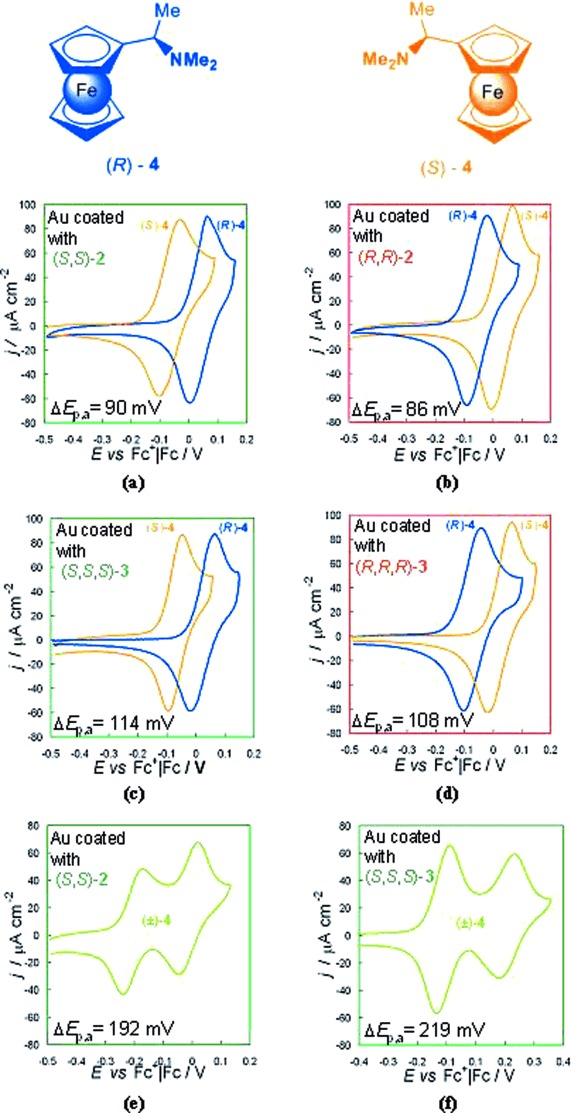
Enantiorecognition tests with enantiopure (S)-4 (dark yellow curves), enantiopure (R)-4 (blue curves) and (±)-4 (green curves) on gold electrodes coated with enantiopure films of (S,S)-2 [green frame, (a), (e)], (R,R)-2 [red frame, (b)], (S,S,S)-3 [green frame, (c), (f)]; (R,R,R)-3 [red frame (d)].
On bare gold electrodes (R)-4 and (S)-4 result in the same CV pattern, a diffusive reversible peak at a formal potential very similar to that of ferrocene. Outstanding enantiorecognition is achieved when the electrodes are coated with the enantiomers of 2 and 3.
In particular, the electrodes coated with the enantiomeric cyclodimers (R,R)-(+)-2 and (S,S)-(−)-2 result in specular separation of enantiopure (R)-4 and (S)-4 probes, with a Epa separation of approximately 90 mV;⋅a slightly larger Epa separation (∼110 mV) is obtained with the electrodes coated with enantiomeric cyclotrimers (R,R,R)-(−)-3 and (S,S,S)-(+)-3.
It is worthwhile noting that, unlike the performances of the films obtained by 1 a electro-oligomerization,[1] the CV peak morphology resembles a reversible diffusional process (distance of 60–70 mV between forward and backward peak even at low scan rates) rather than a solid state one (specular forward and backward peaks). This could point either to a different mode in the approach of the probe molecule to the gold surface within the film and/or to the fact that the reaction occurs on the film surface, slightly charged at the potential at which the electron transfer takes place.
Finally, the enantioselection performance in the case of experiments with the (±)-4 racemic probe is spectacular:⋅the electrodes coated with (S,S)-(−)-2 result in a 190 mV Epa separation;⋅also in this case, the electrodes coated with enantiopure trimer (S,S,S)-(+)-3 perform slightly better than those coated with the corresponding dimer, resulting in an impressive 210 mV Epa separation.
In conclusion, a very simple strategy with high preparative value was discovered for achieving inherently chiral electroactive oligothiophene macrocycles, soluble in organic solvents and therefore easily processable, starting from inexpensive monomers. This combination of properties makes the new molecules an outstanding entry in the library of dissymmetric organic cavities. The preparative enantioseparation of the monomer racemate is not a severe limitation given the capacity of modern chiral technologies to resolve racemates at the level of several kilograms. It is also probable that the crude complex mixture of stereoisomeric cyclo-oligomers resulting from the FeCl3 oxidation of racemic 1 a could find application in fields where chirality is not important, like energy and optoelectronics.
The formation of cyclic systems from 1 a looks general for open-blade scissor-shaped molecules (directional bond strategy),[10] and we are convinced that the oxidative oligomerization and cyclization steps would occur on the FeCl3 surface. Computed DFT cyclization energetics was found to be only moderately endothermic (+94.5 and 46.9 kJ mol−1, for homodimer 2 and homotrimer 3, respectively, if referenced to gas-phase H2), thereby suggesting that cyclization is indeed energetically feasible under these reaction conditions, where the removed hydrogen atoms are likely to adsorb on the oxidant microparticles.
Acknowledgments
This work was supported with the contribution of Fondazione Cariplo, grant no. 2011-0417. The authors gratefully acknowledge the support of Mr. Pasquale IIliano (NMR), Ms. Elisa Lo Bello (preparation of 1 b), Dr. Giuseppe Mazzeo (CD experiments), Sergio Menta (chiral HPLC), Prof. A. Gennaro (scientific discussion), Dr. Ilaria Libani (drafting and supervision of patent MI2014A000948-23/05/2014).
References
- [1].Sannicolò F, Arnaboldi S, Benincori T, Bonometti V, Cirilli R, Dunsch L, Kutner W, Longhi G, Mussini PR, Panigati M, Pierini M, Rizzo S. Angew. Chem. 2014;126:2661–2665. doi: 10.1002/anie.201309585. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2014;53:2623–2627. doi: 10.1002/anie.201309585. [DOI] [PubMed] [Google Scholar]
- [2].Kenning DD, Rasmussen SC. Macromolecules. 2003;36:6298–6299. [Google Scholar]
- [3]a.Sannicolò F, Rizzo S, Benincori T, Kutner W, Noworyta K, Sobczak JW, Bonometti V, Falciola L, Mussini PR, Pierini M. Electrochim. Acta. 2010;55:8352–8364. [Google Scholar]
- [3]b.Pietrzyk A, Kutner W, Chitta R, Zandler ME, D′Souza F, Sannicolò F, Mussini PR. Anal. Chem. 2009;81:10061–10070. doi: 10.1021/ac9020352. [DOI] [PubMed] [Google Scholar]
- [4]a.Zhang F, Götz G, Mena-Osteritz E, Weil M, Sarkar B, Kaim W, Bäuerle P. Chem. Sci. 2011;2:781–784. [Google Scholar]
- [4]b.Zhang F, Bäuerle P. J. Am. Chem. Soc. 2007;129:3090–3091. doi: 10.1021/ja070083k. [DOI] [PubMed] [Google Scholar]
- [4]c.Bäuerle P, Ammann M, Wilde M, Götz G, Mena-Osteritz E, Rang A, Schalley C. Angew. Chem. 2007;119:367–372. doi: 10.1002/anie.200602652. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:363–368. [Google Scholar]
- [4]d.Casado J, Zgierski MZ, Fuhrmann G, Bäuerle P, Lopez-Navarrete T. J. Chem. Phys. 2006;125:044518–044519. doi: 10.1063/1.2217746. [DOI] [PubMed] [Google Scholar]
- [4]e.Kaiser A, Bäuerle P. Top. Curr. Chem. 2005;249:127–201. [Google Scholar]
- [4]f.Casado J, Hernandez V, Ruiz Delgado MC, Lopez Navarrete JT, Fuhrmann G, Bäuerle P. J. Raman Spectrosc. 2004;35:592–599. [Google Scholar]
- [4]g.Fuhrmann G, Debaerdemaeker T, Bäuerle P. Chem. Commun. 2003:948–949. [PubMed] [Google Scholar]
- [4]h.Fuhrmann G, Krömer J, Bäuerle P. Synth. Met. 2001;119:125–126. [Google Scholar]
- [4]i.Krömer J, Rios-Carreras I, Fuhrmann G, Musch C, Wunderlin M, Debaerdemaeker T, Mena-Osteritz E, Bäuerle P. Angew. Chem. 2000;112:3623–3628. [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2000;39:3481–3486. [PubMed] [Google Scholar]
- [4]j.Mishra A, Ma C-Q, Segura JL, Bäuerle P. In: Thiophene-Based Materials, Vol. 1. Perepichka IF, Perepichka D, editors. Chichester, UK: Wiley; 2009. pp. 74–97. [Google Scholar]
- [5]a.Perdew JP, Burke K, Ernzerhof M. Phys. Rev. Lett. 1996;77:3865. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- [5]b.Perdew JP, Burke K, Ernzerhof M. Phys. Rev. Lett. 1997;78:1396. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- [6].Sears WM, MacKinnon CD, Kraft TM. Synth. Met. 2011;161:1566–1574. [Google Scholar]
- [7].Takayanagi M, Gejo T, Hanazaki I. J. Phys. Chem. 1994;98:12893–12898. [Google Scholar]
- [8].Abbate S, Longhi G, Lebon F, Castiglioni E, Superchi S, Pisani L, Fontana F, Torricelli F, Caronna T, Villani C, Sabia R, Tommasini M, Lucotti A, Mendola D, Mele A, Lightner DA. J. Phys. Chem. C. 2014;118:1682–1695. [Google Scholar]
- [9].Meskers SCJ, Peeters E, Langeveld-Voss BMW, Janssen RAJ. Adv. Mater. 2000;12:589–594. [Google Scholar]
- [10]a.Caricato M, Coluccini C, Dondi D, Vander Griend DA, Pasini D. Org. Biomol. Chem. 2010;8:3272–3280. doi: 10.1039/c004379f. [DOI] [PubMed] [Google Scholar]
- [10]b.Coluccini C, Dondi D, Caricato M, Taglietti A, Baiocchi M, Pasini D. Org. Biomol. Chem. 2010;8:1640–1649. doi: 10.1039/b920867d. [DOI] [PubMed] [Google Scholar]
- [10]c.Kim TW, Lah MS, Hong J-I. Chem. Commun. 2001:743–744. [Google Scholar]
- [10]d.Chang S-Y, Jang H-Y, Jeong K-S. Chem. Eur. J. 2003;9:1535–1541. doi: 10.1002/chem.200390176. [DOI] [PubMed] [Google Scholar]
- [10]e.Hua J, Lin W. Org. Lett. 2004;6:861–864. doi: 10.1021/ol036111v. [DOI] [PubMed] [Google Scholar]; Jiang H, Lin W. J. Am. Chem. Soc. 2003;125:8084–8085. doi: 10.1021/ja034402t. [DOI] [PubMed] [Google Scholar]
- [10]f.Jiang H, Hu A, Lin W. Chem. Commun. 2003:96–97. [PubMed] [Google Scholar]
- [10]g.Lee SJ, Hu A, Lin W. J. Am. Chem. Soc. 2002;124:12948–12949. doi: 10.1021/ja028099s. [DOI] [PubMed] [Google Scholar]
- [10]h.Arena CG, Drommi D, Faraone F, Graiff C, Tiripicchio A. Eur. J. Inorg. Chem. 2001:247–255. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.