

Summary
Aspartylglucosaminuria (AGU) is a lysosomal storage disease caused by a metabolic disorder of lysosomes to digest Asn-linked glycoproteins. The specific enzyme linked to AGU is a lysosomal hydrolase called glycosylasparaginase. Crystallographic studies revealed that a surface loop blocks the catalytic center of the mature hydrolase. Autoproteolysis is thus required to remove this P-loop and open up the hydrolase center. Nonetheless, AGU mutations result in misprocessing of their precursors and are deficient in hydrolyzing glycoasparagines. To understand the catalytic and structural consequences of AGU mutations, we have characterized two AGU models, one corresponding to a Finnish allele and the other found in a Canadian family. We also report a 2.1 Å-resolution structure of the latter AGU model. The current crystallographic study provides the first structure of an AGU mutant. It reveals substantial conformation changes at the defective autocleavage site of the AGU mutant, which is trapped as an inactive precursor.
Introduction
Aspartylglucosaminuria (AGU) is a genetic disease caused by the failure of lysosomes to process the protein-to-carbohydrate linkage of Asn-linked glycoproteins (Aula et al., 2001). Such a disorder results in accumulation of glycoasparagines in the lysosomes of virtually all cell types, with severe clinical symptoms involving the central nervous system, skeletal abnormalities and connective tissue lesions. AGU has been reported worldwide, with close to 30 different alleles being characterized so far (Hreidarsson et al., 1983; Mononen et al., 1993; Opladen et al., 2014; Saarela et al., 2004). Due to a founder effect, AGU is enriched in Finland. However, the majority of AGU alleles are found outside Finland with sporadic AGU causing mutations (Aronson, 1999; Hreidarsson et al., 1983; Ikonen et al., 1991; Opladen et al., 2014).
AGU mutations occur in the gene of a well-known lysosomal enzyme glycosylasparaginase (GA), which is also known as aspartylglucosaminidase (Aronson, 1999; Mononen et al., 1993). GA is an amidase that cleaves the Asn-linked glycoprotein. Like all other members of the N-terminal nucleophile (Ntn) hydrolase family, GA is synthesized as an enzymatically inactive precursor (Brannigan et al., 1995). An obligatory processing step for activating GA is thus an intramolecular or cis- autoproteolysis to cleave the single-chain polypeptide precursor into the heavy (α) and light (β) subunits, and to expose the critical nucleophile of the amidase at the newly generated N-terminus of the β subunit. This type of site-specific autoproteolysis of polypeptide precursor is also required to activate many other critical enzymes (Dembek et al., 2012; Paulus, 2000). Molecular characterizations of AGU causing mutations revealed problems in autoproteolytic processing of their precursors. Thus AGU molecules are misprocessed and are retained at the pre-autoproteolysis stage as single-chain precursors, inactive for glycoprotein processing (Aronson, 1999; Saarela et al., 2001).
To understand the molecular details of the disease, a detailed characterization of AGU causing mutations is essential. In addition, high-resolution crystallographic analyses of AGU molecules are critical for a precise understanding of the structural consequences of AGU mutations. However, a major obstacle for structural and biochemical studies of AGU is the difficulty to obtain purified and active human GA in sufficient quantity (Heiskanen et al., 1994). Due to failed attempts to over-express active recombinant human GA, the protein material used to determine the only available structure of human GA, in its autoproteolyzed form, was purified from human blood leukocytes (Oinonen et al., 1995; Tikkanen et al., 1996b). Furthermore, efforts over more than a decade to crystallize diffraction quality crystals of human GA precursor using a variety of expression systems have been unsuccessful (Saarela, 2004). To the contrary, both the precursor and autoproteolyzed forms of Flavobacterium GA have been purified at sufficiently high quantity and quality for structural, biochemical and biophysical studies (Guo et al., 1998; Xu et al., 1999).
All data indicate that, from bacteria to eukaryotes, GAs are conserved in primary sequences and tertiary structures, and utilize the same cis-autoproteolysis mechanism to activate their hydrolase activities. Molecular and biochemical studies have revealed a mechanistic relationship between human and bacterial GAs. Both utilize the same mechanism for autoactivation through intramolecular autoproteolysis (Saarela, 2004; Xu et al., 1999). They also share the same hydrolysis mechanism for processing glycoasparagines (Liu et al., 1998; Tikkanen et al., 1996a). It has also been shown that the glycosylation of GA that occurs in mammalian cells is not absolutely required for either the autoproteolysis or hydrolase activity (Tikkanen et al., 1995); deletion of the N-glycan in one of the subunits could be tolerated. The bacterial and human GA share highly significant sequence homology, with an overall 36% identity and additional 53% conserved similarity. Furthermore, structural studies have provided direct evidence that these two enzymes share a conserved αββα structure (Oinonen and Rouvinen, 2000). More importantly, they have an essentially identical autoproteolytic center (Guo et al., 1998; Oinonen et al., 1995), with 100% identical residues forming the autocatalytic site and a rmsd of 0.6 Å for all these conserved active site residues. Altogether, these data confirm the suitability of the bacterial enzyme as a model to analyze the consequences of mutations in AGU patients at the atomic level.
In this study, we have carried out biochemical analyses of two model mutants with AGU alleles. Autoproteolysis activities of these mutant proteins are dramatically diminished, with a t1/2 ranging from hours to days, in contrast to be a spontaneous process (< 1 min) for the wild type GA. Furthermore, we have determined a crystallographic structure of one AGU mutation found in a Canadian family (Coulter-Mackie, 1999). The crystallographic snapshot at a resolution of 2.1 Å has provided a structural basis of the AGU disease caused by a missense mutation.
Results and Discussion
Construction and Biochemical Characterization of AGU Mutants
To study the functional and structural consequence of AGU missense mutations while overcoming the difficulty of expressing human GA, we have resorted to a model enzyme. As discussed above, Flavobacterium GA appears to be an excellent model to study the structural consequences of AGU mutations. In this study, we have generated two AGU model enzymes. One is a Finnish allele that carries a mutation of a nucleotide C to T and thus changing the residue 234 of human GA from a threonine to an isoleucine (Saarela et al., 2001). The other mutant is a Canadian allele that carries a missense mutation with a change of a nucleotide from G to A and thus changing the residue 203 of human GA from a glycine to an aspartic acid (Coulter-Mackie, 1999). Based on structural and homology alignments (Guo et al., 1998), Gly172 and Thr203 of Flavobacterium GA is equivalent to the Gly203 and Thr234 of the human counterpart, respectively. We have thus generated two Flavobacterium model mutants: one carries a Gly to Asp missense mutation at bacterial residue 172 (called G172D mutant) and another has a Thr to Ile missense mutation at bacterial residue 203 (called T203I mutant). Precursor proteins of these mutants have been purified to homogeneity. As shown in Figure 1, the wild-type GA autocleaves spontaneously and thus is purified entirely as a mature form with α and β subunits. In contrast, both AGU mutant proteins are defective to some degree in autoproteolysis. The G172D mutant was purified as a single-chain precursor and remains as a precursor even after 16 hours incubation at 37°C (Figure 1A), whereas the T203I mutant can be purified as a single-chain precursor, but about half of the precursor form undergoes autocleavage within 8 hours of incubation at 37°C (Figure 1B).
Figure 1. Autoproteolysis and hydrolysis activities of AGU mutants.

(A) Autoproteolysis analysis of G172D mutant. Autoproteolysis of purified G172D precursor protein was initiated by incubating the precursor at 37°C in a solution of 20 mM Tris, pH 7.4, 50 mM NaCl, and 1 mM EDTA. Aliquots were then removed at various time points, as indicated, and analyzed by SDS-PAGE. The precursor (α-β), autocleaved subunits (α and β) and wild- type GA (WT) are marked. Lane M is a mixture of molecular weight markers.
(B) Autoproteolysis analysis of T203I mutant. Purified T203I precursor protein was analyzed the same as in (A).
(C) Enzyme hydrolysis assays. Purified AGU mutant or wild-type GA proteins were incubated at 37 °C for 0–7 days to measure their hydrolase activities. See Experimental Procedures for detailed assay conditions. Activity of wild-type GA is normalized to 100%. Data are averages of 3 repeats ± standard deviation.
To ask whether the mature form of the AGU mutants generated by the in vitro autoprocessing are active hydrolases, we performed a time-course experiment and analyzed the level of hydrolase activity. Wild-type and mutant proteins were incubated at 37°C to initiate the autoproteolysis, and samples were taken at various time points to measure the activity of hydrolyzing a glycoasparagine (Figure 1C). As expected from the gel analysis of autoproteolysis (Figure 1A), the G172D mutant essentially has no hydrolase activity throughout the in vitro autoactivation. On the other hand, the T203I mutant protein showed a small but significant level of hydrolase activity, ~7% of the wild-type activity at the initial time point. This detectable hydrolase activity further increased to about 10% of the wild-type level after 1 day in vitro autoprocessing, correlated well with a concurrent increase of the autocleaved form (Figure 1B). This increase in activity is even more significant when considering that the wild-type enzyme actually showed a continuous drop of hydrolase activity over time, likely due to enzyme denaturation during the thermal incubation. It is possible that the T203I mutant enzyme is more thermally stable than the wild-type enzyme, and its hydrolase activity was further elevated by the thermal incubation. Nonetheless a more likely explanation for the increase of hydrolysis activity of the T203I mutant protein is that more active hydrolase was being generated through autocleavage during the in vitro thermal incubation. Thus eventhough the T203I mature enzyme is not as efficient as the wild-type enzyme, it still has some hydrolase activity to process glycoasparagines. These data suggest that an in vitro activation of autoproteolysis can be applied to enhance the hydrolase activity of the AGU mutant. It is worth noting that an enhancement of autoprocessing to just partially restore GA hydrolase activity in lysosomes might be beneficial for AGU patients.
Crystal Preparation to Capture AGU Mutants at the Pre-Autoproteolysis Stage
To determine the pre-autoproteolysis structure, AGU precursor would need to be stabilized up to several days for crystal growth. However, we found that both the purified G172D and T203I mutant precursors undergo autoproteolysis to the mature forms during the several days required for crystallization. Glycine has been used as a reversible inhibitor of autoproteolysis to stabilize precursors of active GA proteins for crystallization (Wang and Guo, 2010; Xu et al., 1999). To find an analogous inhibitor for the AGU-causing variants, we screened a library of glycine-like small molecules, which led to the identification that L-aspartic acid β-hydroxamate (β-AHA) was able to stabilize the G172D precursor for up to several days for crystallization. In the presence of the β-AHA, crystals containing the G172D precursor were obtained for structure determination. However, proteolysis could be resumed by removing β-AHA from the G172D samples through dialysis. On the other hand, despite vigorous efforts, no effective inhibitor was yet found to be able to stabilize the precursors of the T203I mutant for crystallization.
The Structures of the Precursor and the Mature Form Were Found in a Single Crystal
Structures of the G172D mutant have been determined by X-ray crystallography and refined to 2.1 Å resolution. Crystallographic statistics are summarized in Table 1. The crystal has a triclinic unit cell and contains two molecules in the asymmetric unit. These two molecules form a bi-molecular assembly through the same interfaces as previously observed in the structures of precursor homodimers (Xu et al., 1999) or the equivalent α2β2 heterotetramers of autocleaved enzymes (Guo et al., 1998; Oinonen et al., 1995). However, unique to this G172D crystal form, one molecule was in its precursor form with a bound β-AHA, and the other molecule was in the mature/autocleaved form (with α & β subunits) and without any sign of β-AHA binding. The space group and cell constants of the G172D crystal (P1, a = 45.8, b = 50.6, c = 61.2Å, α = 86.3, β = 91.0, γ = 107.8°) are closely related to those of the precursor crystals (P1, a = 46.3, b = 52.8, c = 62.4Å, α = 80.8, β = 90.5, γ = 105.1°), whereas crystals of autocleaved enzymes were typically grown in a monoclinic space group (P21, a = 46.2, b = 97.3, c = 61.8Å, β = 90.3) (Cui et al., 1999). Thus, it is possible that the G172D crystal was initially grown with precursor homodimers, and one of the two precursors was able to autocleave in the crystal. It has been well documented that GA crystals retain both the autoproteolysis and hydrolysis activities (Oinonen et al., 1995; Wang and Guo, 2007, 2010). The difference in autocleavage of the two molecules in the G172D crystal appears to be due to differences in crystal contacts of a surface loop near the active site that in turn alter binding affinity and precursor stabilization by β-AHA (see below). Thus in this report, we are studying both the structures of the precursor and the mature form of the G172D mutant.
Table 1.
Crystallographic data collection and refinement statistics
| G172D Crystal | |
|---|---|
| Resolution (Å)a | 48.1–2.1 (2.16–2.10) |
| Space Group | P1 |
| Cell Dimensions: | |
| a, b, c (Å) | 45.8 50.6 61.2 |
| α, β, γ (°) | 86.3 91.0 107.8 |
| No. of molecules per asymmetric unit | 2 |
| I/sigma-I | 14.2 (9.5) |
| Completeness (%) | 90.0 (90.3) |
| No. of reflections | 103,797 (8,672) |
| Unique reflections | 27,448 (2,257) |
| Rsym (%)b | 5.2 (9.8) |
| Structure refinement | |
| Rworkc | 0.1920 |
| Rfreed | 0.2568 |
| R.m.s. deviationse | |
| Bond-lengths (Å) | 0.0165 |
| Bond-angles (°) | 1.7604 |
| Ramachandran plot | |
| Most favored regions (%) | 96.7 |
| Additional allowed regions (%) | 3.3 |
| Average B-factors (Å2) | |
| Main chain | 18.6 |
| Side chain | 21.5 |
| Water | 24.3 |
Numbers in parenthesis refer to the outermost resolution bin.
Rsym = ΣhΣi|Ihi−Ih|/ΣhΣiIhi for the intensity (I) of i observation of reflection h.
Rwork = Σ|Fobs − Fcalc|/Σ|Fobs|, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respective.
Rfree was calculated as Rwork, but with 5% of the amplitudes chosen randomly and omitted from the start of refinement.
R.m.s. deviations are deviations from ideal geometry.
Structural Comparisons Between the G172D Precursor and its Autocleaved Form
The overall structure of the G172D precursor in complex with the β-AHA is shown in Figure 2. The G172D precursor folds into an αββα sandwich, with a surface loop (denoted precursor P-loop, containing residues 139–151) located next to the autocleavage site. When the G172D precursor is compared to its mature counterpart, a small but significant difference in conformation is found at the surface loop containing the mutated residue Asp172, where the main-chain of the loop has shifted by more than 3 Å (Supplemental Figure S1). As a result, the mature G172D enzyme has adopted a wider opening near the substrate site. There are two additional disordered surface loops in the mature form structure, both are located outside of the catalytic center. Excluding these disordered loops, the precursor structure of the G172D mutant is very similar to its autocleaved form. The rmsd between two structures is 0.83 Å for all main-chain atoms of common residues (residues 3–44, 56–138, 152–170, and 174–295). The slightly high rmsd is due to small differences in loops and small movements of flanking helices (containing residues 31–45 and residues 126–136, shifted by about 1.8 Å). However, these small conformational shifts appear to be related to loop disorder, possibly as a result of differences in crystal contacts. There is no other major conformational differences observed between the G172D precursor and its autocleaved structure.
Figure 2. Overal Structure of the G172D precursor in complex with β-AHA.
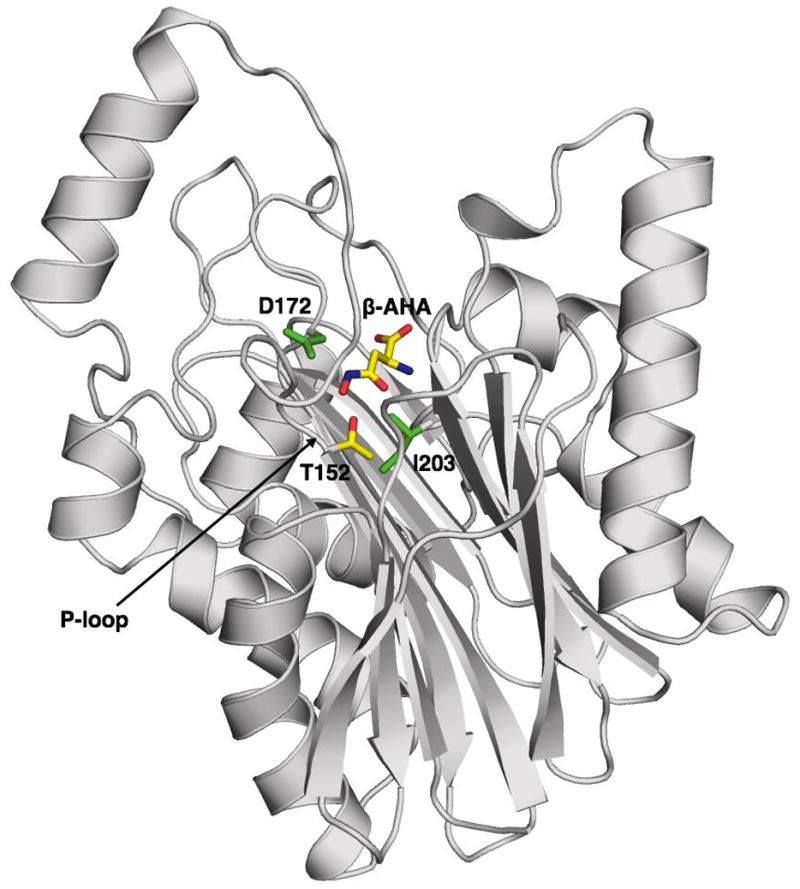
Ribbon representation of the G172D precursor structure, with the bound β-AHA molecule shown as a stick model. The arrow points to the defective scissile peptide bond between Asp151 and Thr152 in the P-loop. The β-AHA molecule and side chain of the autoproteolysis key residue T152 are shown by atom type: yellow for carbon atoms, blue for nitrogen atoms, red for oxygen atoms. Side chains of the two separate AGU mutations studied, Asp172 and Ile203, are shown in green.
Structural Comparisons with GA Wild-Type Enzyme and Active Precursors
Comparing to previously reported GA wild-type structure, the G172D mature enzyme has an essentially identical structure (see Supplemental Information). Likewise, no gross conformational differences were observed when the structure of the G172D precursor was compared to previously published GA active precursors (PDB code 9GAC) (Xu et al., 1999). The rmsd between these two precursor structures is 0.82 Å for all main-chain atoms of common 284 residues (including surface loops). However, some localized conformational changes are found as a result of the single residue change at residue 172. In the active precursor structure, residues 46–51 are in an unstructured loop. But in the G172D precursor, these same residues join residues 31–45 (previously denoted as aH2 helix in ref. (Guo et al., 1998)) to form a continuous α-helix with one additional turn (Figure 3). Interestingly, no electron density was observed for these same residues in the mature G172D structure, suggesting dynamic flexibility or static disorder in this region of the mutant. The extended α-helix in the G172D precursor structure appears to be stabilized by a new salt-bridge between side chain of Asp172 and side chain of Arg56 as well as the binding of β-AHA. Furthermore, near the autocleavage site, there are also dramatic conformational changes. As shown in Figure 3, the bulky side chain of Asp172 in the mutant precursor pushes the P-loop and the defective scissile bond to take a different conformation. As a result of these conformational changes, side chains of Ser50 in these two structures are pointing to almost opposite directions, with side-chain atom Oγ being displaced by as much as 11.1 Å (Figure 3). Similarly, side-chain hydroxyl oxygen of Tyr53 also moves by as much as 14.3 Å and is pointed toward opposite sides of the helix. These conformational changes result in a more relaxed backbone at the defective scissile bond (details below).
Figure 3. Structural comparison of the autocleavage sites in the G172D mutant and an active GA Precursor.
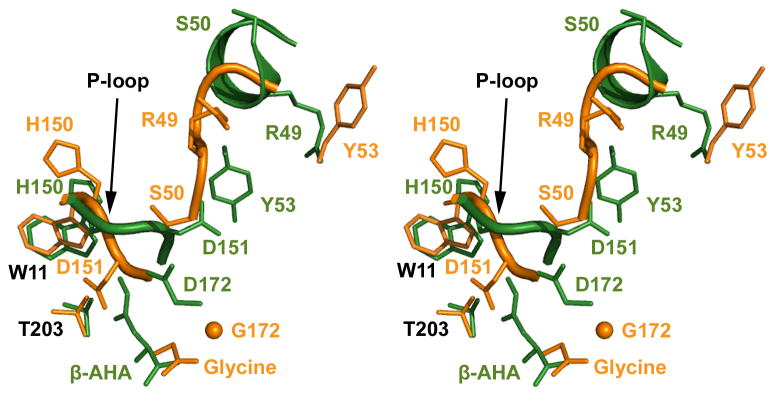
Shown is a stereo view of the superimposition of the autocatalytic site residues in the G172D precursor (green) and an active GA precursor model published previously (orange; ref. (Xu et al., 1999)), with black labels for well-superimposed residues. Side-chain atoms of GA are shown as stick models, and main chain traces of the P-loop flanking the scissile bond, residues 149–152, are shown as tubes. Also shown are bound inhibitors (a β-AHA or a glycine). Note the mutation of side chain at residue 172 from a glycine (an orange sphere for Cα) to an aspartic acid (green side chain). Residues 46–51 are in an unstructured loop in the active GA precursor (orange tube), but fold into an α-helix (green ribbon) in the G172D mutant.
Comparing β-AHA Binding with Glycine Binding
The binding site of β-AHA to the G172D precursor is located near the autocleavage site at the same cavity as previously observed for the glycine inhibitor bound to an active GA precursor (Xu et al., 1999). The electron density map around the scissile peptide bond of the G172D mutant in complex with β-AHA is shown in Figure 4A. This cavity is a partial substrate site and forms around the conserved residues Arg180, Asp183, and Gly204 that directly interact with the aspartate moiety of the GA substrates in the mature/autocleaved enzyme (Guo et al., 1998; Oinonen et al., 1995). In the precursor of active GA, this cavity is only accessible to small molecules such as glycine through a narrow port. However, as noted above, the G172D precursor adopts a wider opening for this cavity due to a conformational shift of a surface loop containing the mutated side chain of Asp172. This would explain how the G172D precursor could bind the larger molecule β-AHA. Binding modes of both inhibitors to the small cavities are similar. They utilize their α-carboxylate ends to form salt bridges with the conserved residue Arg180 (Figure 4B). At the same time, their α-amino groups interact with the conserved residues Asp183 and Gly204 via hydrogen bonds. These conserved interactions are similar to those observed in the enzyme/substrate or enzyme/product complexes (Oinonen et al., 1995; Wang and Guo, 2007).
Figure 4. The structure of β-AHA as bound to the G172D precursor.
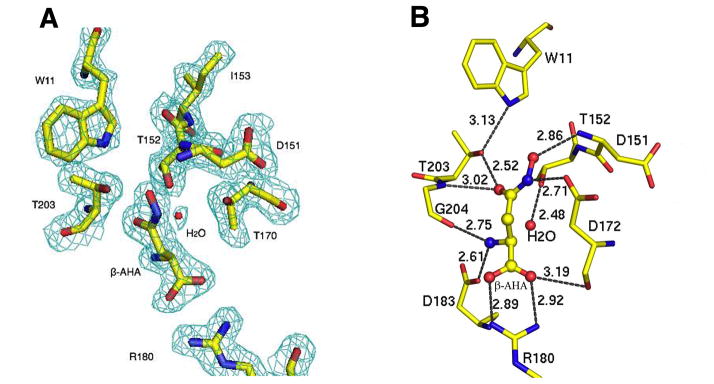
(A) The cyan electron density corresponds to a 2Fo-Fc type map at 2.1 Å resolution contoured at the 1.2-σ level. The defective scissile peptide bond is located between Asp151 and Thr152. Bound β-AHA and side chains of a few key active site residues are shown by atom type: yellow for carbon atoms, blue for nitrogen atoms, red for oxygen atoms. A bound waster molecule is shown as a red sphere.
(B) Hydrogen-bond network at the autocleavage site with the bound β-AHA inhibitor. Active site residues of the G172D precursor are shown as stick model, whereas β-AHA inhibitor is shown as a ball-and-stick model. Dash lines indicate the hydrogen-bond interactions with distances in angstroms (Å).
The Autoproteolytic Center of the G172D Precursor
Near the scissile peptide bond (Asp151-Thr152), the G172D mutant precursor exhibits substantial conformational differences when compared to that of an active GA precursor (Figure 3) (Wang and Guo, 2010; Xu et al., 1999). As shown in Figure 3, mutation at residue 172, from a glycine to an aspartic acid, alters the main-chain trace near the scissile peptide bond by about 3.4 Å. Furthermore, packing of side chains also changed dramatically so that the side chains of Asp151 pointed roughly in opposite directions, with Oδ1 shifts by as much as 8.4 Å. This conformational change breaks a key interaction between the side chains of residues Asp151 and Thr152, which has been demonstrated to be critical in holding the conformational strains important for driving autoproteolysis (Qian et al., 2003), through a “twist-and-break” mechanism (Wang and Guo, 2010; Xu et al., 1999). As a result, the structural constraints observed in an active GA precursor are no longer present in the G172D precursor. Thus a backbone relaxation near the defective scissile peptide bond appears to be the cause of trapping AGU molecules at the non-productive precursor stage.
Low Level of cis-Autoprocessing Activity of AGU Mutant
Both AGU mutants do however have a low activity of autoproteolysis to generate an active hydrolase. This is demonstrated by an activity of about 4–10% of wild-type activity in the hydrolase assay after 2–7 days’ incubation at 37 °C (Figure 1C). During the process of crystallization that took 5–10 days at 4 °C, one of the two molecules of the G172D mutant in the P1 unit cell had undergone autoproteolysis. Differences in crystal contacts appear to play a role on the effectiveness of β-AHA inhibition. Apparently, for the residual autoproteolysis to occur, the P-loop near the autocleavage site of AGU mutants would have to change from the defective conformation (green trace in Figure 3) to an active conformation, presumably similar to the twisted conformation observed in an active precursor (orange trace in Figure 3). Interestingly, for the precursor structures determined so far (Qian et al., 2003; Wang and Guo, 2010; Xu et al., 1999), the P-loop regions all have significantly higher B factor, or even lack electron density for part of the loop, indicating dynamic or static disorder of this region. Thus it is plausible that a thermal motion or flexibility of the P-loop would result in a small fraction of the AGU precursors to adopt an active conformation for autoproteolysis. Nonetheless, such an active conformation would have steric conflicts with the bound β-AHA molecules (Figure 3). This would explain why no density of β-AHA was found in the active site for the autocleaved molecule in the crystal.
Conclusion
Here we report mutational and biochemical studies on two AGU mutant models. We have also presented the first high resolution structure of an AGU model to study the structural consequences of a disease-causing mutation. None of these two missense mutations of AGU molecules are located close enough to the catalytic center for a direct involvement in the catalysis (Figure 2). Thus a loss of a functional group directly required for the autoproteolysis is not the cause of AGU deficiency. Instead, as demonstrated by the structure of the G172D mutant, it is the indirect effects of the mutation on the conformation that propagate to the defective scissile peptide bond. As shown in Figure 3, mutation at residue 172 has altered the backbone trace flanking the scissile peptide bond at residues 151–152, as well side-chain packing and secondary structure. These localized conformational changes in turn result in an unproductive precursor trapped at the pre-autoproteolysis stage, which is incapable of accommodating and processing glycoasparagines. Nonetheless, small molecules had been shown to be able to stimulate autoprocessing of GA mutants with autoproteolysis-defect similar to AGU molecules (Guan et al., 1998). As shown in Figure 1C, in vitro autoprocessing of AGU mutants could increase the activity of hydrolyzing glycoasparagines. Thus, in principle, the misprocessed AGU precursors represent excellent targets for designing small molecules to ameliorate the AGU disorder. There have been successful precedents of identifying small-molecules to reduce protein misfolding, to stabilize native state, and/or to activate trans-proteolytic processing of proenzyme (Petrassi et al., 2005; Ray et al., 2005; Wolan et al., 2009). The AGU precursor structure reported here provides a solid foundation for structure-based design of small molecules to rescue AGU mutants by activating cis-autoproteolysis, in hope to further develop into therapeutics to alleviate the suffering of AGU patients.
Experimental Procedures
Hydrolase Activity Assay
Hydrolase activity was assayed by a method modified from a previously described approach (Liu et al., 1998). Details are in Supplemental Information.
Protein Purification and Crystallization
Wild-type GA and AGU mutant proteins were over-expressed and purified using previously published protocols (Cui et al., 1999). Crystals of the G172D mutant in complex with β-AHA were obtained with the crystallization condition of 0.2M NaCl, 0.1M Bis-Tris pH 6.5, 25% PEG 3350, and a protein concentration of 2mg/ml.
Data Collection and Processing
Before flash-cooling in liquid nitrogen for data collection, crystals were soaked in a reservoir solution supplemented with 20% glycerol. X-ray data were collected at 100 K using the beamline X29 at the National Synchrotron Light Source (NSLS), Brookhaven National Laboratory. The data were processed with the HKL2000 (Otwinowski and Minor, 1997) and the CCP4 suite (Collaborative Computational Project, 1994). The crystal has P1 symmetry with two protein molecules in the asymmetric unit.
Structure Determination and Refinement
The structures of G172D mutant were determined by the molecular replacement method, using the previously published structure of the D151N mutant structure (PDB code 1P4K) (Qian et al., 2003). To avoid model bias, the initial MR phases were calculated by omitting the P-loop residues 148–158, which include the scissile peptide bond at residues 151–152. Molecular replacement was performed with Molrep and refinements were carried out with the Refmac program, with 5% of the total reflection data excluded from the refinement cycles and used to calculate the free R-factor (Rfree). This partial model was further subjected to rigid body and restrained refinements, with model rebuilding done with COOT (Emsley and Cowtan, 2004) to obtain the final structure. X-ray data collection, processing, and structure refinement statistics are summarized in Table 1. Figures were made with the program PyMOL (Delano Scientific).
Protein Data Bank accession codes
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with an ID code 4R4Y.
Supplementary Material
Figure S1, related to Figure 2. Main-chain conformational difference between the G172D precursor and its autocleaved Form.
Bonds are shown by atom type: yellow for carbon atoms, blue for nitrogen atoms, red for oxygen atoms. A 3 Å deviation in a loop’s main-chain atoms, at residues 171–172, is highlighted by an open arrow.
Figure S2, related to Figure 2. G172D mature structure folds similarly to the wild-type GA hydrolase.
C-α traces are shown as yellow and blue for the G172D mature form and the wild-type GA, respectively. Termini of the α and β subunits are also indicated.
Acknowledgments
We thank Dr. Howard Robinson for assistance on data collection, Siavash Mostafavi for assistance on protein purification and enzyme assays, and Dr. Matthew Nugent for critical reading of the manuscript. This work was supported by Grant DK075294 from the NIH.
Abbreviations
- AGU
aspartylglucosaminuria
- β-AHA
L-aspartic acid β-hydroxamate (β-aspartic acid hydroxamate)
- GA
glycosylasparaginase
- Ntn
N-terminal nucleophile
- rmsd
root mean square deviation/displacement
Footnotes
Author Contributions
L.S. completed the structure determination and analyses.
D.L. collected the x-ray data and carried out the initial crystallographic study.
S.P. carried out the biochemical studies.
L.S. and H.-C. G. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aronson NNJ. Aspartylglycosaminuria: biochemistry and molecular biology. Biochim Biophys Acta. 1999;1455:139–154. doi: 10.1016/s0925-4439(99)00076-9. [DOI] [PubMed] [Google Scholar]
- Aula P, Jalanko A, Peltonen L. Aspartylglucosaminuria. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, Kinzler KW, editors. The Metabolic and Molecular Bases of Inherited Disease. New York, USA: McGraw-Hill; 2001. pp. 3535–3550. [Google Scholar]
- Brannigan JA, Dodson G, Duggleby HJ, Moody PCE, Smith JL, Tomchick DR, Murzin AG. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature. 1995;378:416–419. doi: 10.1038/378416a0. [DOI] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project N. 4) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Coulter-Mackie MB. A novel exonic mutation in the aspartylglucosaminidase gene causes exon skipping. J Inherit Metab Dis. 1999;22:682–683. doi: 10.1023/a:1005563004678. [DOI] [PubMed] [Google Scholar]
- Cui T, Liao PH, Guan C, Guo HC. Purification and crystallization of precursors and autoprocessed enzymes of Flavobacterium glycosylasparaginase: an N-terminal nucleophile hydrolase. Acta Crystallogr D Biol Crystallogr. 1999;55:1961–1964. doi: 10.1107/s0907444999011798. [DOI] [PubMed] [Google Scholar]
- Dembek M, Reynolds CB, Fairweather NF. Clostridium difficile cell wall protein CwpV undergoes enzyme-independent intramolecular autoproteolysis. J Biol Chem. 2012;287:1538–1544. doi: 10.1074/jbc.M111.302463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Guan C, Liu Y, Shao Y, Cui T, Liao W, Ewel A, Whitaker R, Paulus H. Characterization and functional analysis of the cis-autoproteolysis active center of glycosylasparaginase. J Biol Chem. 1998;273:9695–9702. doi: 10.1074/jbc.273.16.9695. [DOI] [PubMed] [Google Scholar]
- Guo HC, Xu Q, Buckley D, Guan C. Crystal structures of Flavobacterium glycosylasparaginase: an N-terminal nucleophile hydrolase activated by intramolecular proteolysis. J Biol Chem. 1998;273:20205–20212. doi: 10.1074/jbc.273.32.20205. [DOI] [PubMed] [Google Scholar]
- Heiskanen T, Tollersrud OK, Zhao M, Peltonen L. Large-scale purification of human aspartylglucosaminidase: utilization of exceptional sodium dodecyl sulfate resistance. Protein Expr Purif. 1994;5:205–210. doi: 10.1006/prep.1994.1032. [DOI] [PubMed] [Google Scholar]
- Hreidarsson S, Thomas GH, Valle DL, Stevenson RE, Taylor H, McCarty J, Coker SB, Green WR. Aspartylglucosaminuria in the United States. Clin Genet. 1983;23:427–435. doi: 10.1111/j.1399-0004.1983.tb01977.x. [DOI] [PubMed] [Google Scholar]
- Ikonen E, Aula P, Gron K, Tollersrud O, Halila R, Manninen T, Syvanen AC, Peltonen L. Spectrum of mutations in aspartylglucosaminuria. Proc Natl Acad Sci U S A. 1991;88:11222–11226. doi: 10.1073/pnas.88.24.11222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Guan C, Aronson NNJ. Site-directed mutagenesis of essential residues involved in the mechanism of bacterial glycosylasparaginase. J Biol Chem. 1998;273:9688–9694. doi: 10.1074/jbc.273.16.9688. [DOI] [PubMed] [Google Scholar]
- Mononen I, Fisher KJ, Kaartinen V, Aronson NNJ. Aspartylglycosaminuria: protein chemistry and molecular biology of the most common lysosomal storage disorder of glycoprotein degradation. FASEB J. 1993;7:1247–1256. doi: 10.1096/fasebj.7.13.8405810. [DOI] [PubMed] [Google Scholar]
- Oinonen C, Rouvinen J. Structural comparison of Ntn-hydrolases. Protein Sci. 2000;9:2329–2337. doi: 10.1110/ps.9.12.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oinonen C, Tikkanen R, Rouvinen J, Peltonen L. Three-dimensional structure of human lysosomal aspartylglucosaminidase. Nat Struct Biol. 1995;2:1102–1108. doi: 10.1038/nsb1295-1102. [DOI] [PubMed] [Google Scholar]
- Opladen T, Ebinger F, Zschocke J, Sengupta D, Ben-Omran T, Shahbeck N, Moog U, Fischer C, Burger F, Haas D, et al. Aspartylglucosaminuria: unusual neonatal presentation in qatari twins with a novel aspartylglucosaminidase gene mutation and 3 new cases in a Turkish family. J Child Neurol. 2014;29:36–42. doi: 10.1177/0883073812469049. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Paulus H. Protein splicing and related forms of protein autoprocessing. Annu Rev Biochem. 2000;69:447–496. doi: 10.1146/annurev.biochem.69.1.447. [DOI] [PubMed] [Google Scholar]
- Petrassi HM, Johnson SM, Purkey HE, Chiang KP, Walkup T, Jiang X, Powers ET, Kelly JW. Potent and selective structure-based dibenzofuran inhibitors of transthyretin amyloidogenesis: kinetic stabilization of the native state. J Am Chem Soc. 2005;127:6662–6671. doi: 10.1021/ja044351f. [DOI] [PubMed] [Google Scholar]
- Qian X, Guan C, Guo HC. A dual role for an aspartic acid in glycosylasparaginase autoproteolysis. Structure. 2003;11:997–1003. doi: 10.1016/s0969-2126(03)00150-3. [DOI] [PubMed] [Google Scholar]
- Ray SS, Nowak RJ, Brown RHJ, Lansbury PTJ. Small-molecule-mediated stabilization of familial amyotrophic lateral sclerosis-linked superoxide dismutase mutants against unfolding and aggregation. Proc Natl Acad Sci U S A. 2005;102:3639–3644. doi: 10.1073/pnas.0408277102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarela J. PhD Thesis. University of Helsinki; Helsinki, Finland: 2004. Characterization of aspartylglucosaminidase activation and aspartylglucosaminuria mutations. [Google Scholar]
- Saarela J, Laine M, Oinonen C, Schantz C, Jalanko A, Rouvinen J, Peltonen L. Molecular pathogenesis of a disease: structural consequences of aspartylglucosaminuria mutations. Hum Mol Genet. 2001;10:983–995. doi: 10.1093/hmg/10.9.983. [DOI] [PubMed] [Google Scholar]
- Saarela J, von Schantz C, Peltonen L, Jalanko A. A novel aspartylglucosaminuria mutation affects translocation of aspartylglucosaminidase. Hum Mutat. 2004;24:350–351. doi: 10.1002/humu.9276. [DOI] [PubMed] [Google Scholar]
- Tatusova TA, Madden TL. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999;174:247–250. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- Tikkanen R, Enomaa N, Riikonen A, Ikonen E, Peltonen L. Intracellular sorting of aspartylglucosaminidase: the role of N-linked oligosaccharides and evidence of Man-6-P-independent lysosomal targeting. DNA Cell Biol. 1995;14:305–312. doi: 10.1089/dna.1995.14.305. [DOI] [PubMed] [Google Scholar]
- Tikkanen R, Riikonen A, Oinonen C, Rouvinen R, Peltonen L. Functional analyses of active site residues of human lysosomal aspartylglucosaminidase: implications for catalytic mechanism and autocatalytic activation. EMBO J. 1996a;15:2954–2960. [PMC free article] [PubMed] [Google Scholar]
- Tikkanen R, Rouvinen J, Torronen A, Kalkkinen N, Peltonen L. Large-scale purification and preliminary x-ray diffraction studies of human aspartylglucosaminidase. Proteins. 1996b;24:253–258. doi: 10.1002/(SICI)1097-0134(199602)24:2<253::AID-PROT12>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Wang Y, Guo HC. Crystallographic snapshot of a productive glycosylasparaginase-substrate complex. J Mol Biol. 2007;366:82–92. doi: 10.1016/j.jmb.2006.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Guo HC. Crystallographic snapshot of glycosylasparaginase precursor poised for autoprocessing. J Mol Biol. 2010;403:120–130. doi: 10.1016/j.jmb.2010.08.038. [DOI] [PubMed] [Google Scholar]
- Wolan DW, Zorn JA, Gray DC, Wells JA. Small-molecule activators of a proenzyme. Science. 2009;326:853–858. doi: 10.1126/science.1177585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Buckley D, Guan C, Guo HC. Structural insights into the mechanism of intramolecular proteolysis. Cell. 1999;98:651–661. doi: 10.1016/s0092-8674(00)80052-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, related to Figure 2. Main-chain conformational difference between the G172D precursor and its autocleaved Form.
Bonds are shown by atom type: yellow for carbon atoms, blue for nitrogen atoms, red for oxygen atoms. A 3 Å deviation in a loop’s main-chain atoms, at residues 171–172, is highlighted by an open arrow.
Figure S2, related to Figure 2. G172D mature structure folds similarly to the wild-type GA hydrolase.
C-α traces are shown as yellow and blue for the G172D mature form and the wild-type GA, respectively. Termini of the α and β subunits are also indicated.