

Abstract
In a three-step protocol involving regioselective enzymatic acylation, per-O-trimethylsilylation, and a one-pot α-glycosidation/deprotection sequence, cholesteryl 6-O-tetradecanoyl-α-D-glucopyranoside (α-CAG) of Helicobacter pylori is afforded starting from glucose in an overall yield of 45%. The production of CAG can be scaled to make purified quantities available to the bilogical community for the first time.
It is increasingly clear that membrane-bound cholesterol plays an important regulatory role in pathogenic processes. For example, cholesterol has been shown to modulate glycolipid conformations, which can either inhibit or facilitate recognition processes involving raft-associated proteins.1 There is also recent evidence that cholesterol may directly mask glycosphingolipids such as monosialotetrahexosylganglioside (GM1) and thereby protect the carbohydrate from recognition by toxin-derived proteins including cholera toxin.2 When one considers these findings in the context of the discovery that many pathogenic bacteria produce enzymes that require extrusion of cholesterol from host cells, an intriguing interplay of cholesterol transport emerges.
Nowhere is the game more evident than in Helicobacter pylori infection.3,4 It is estimated that over 50% of the world’s population is infected with H. pylori4 a bacterium that resides in the mucosa of the stomach. Although H. pylori infection often goes undetected, chronic infection can lead to gastric ulcers or even cancer.5-11 Imbedded in the cell wall of H. pylori is a glycosyl transferase that combines uridine diphosphate glucose with host derived cholesterol to make cholesteryl-α-D-glucopyranoside (CG).8,9 Recent studies suggest that through a still unknown mechanism, H. pylori is able to sense cholesterol rich regions in host epithelial tissue and extract cholesterol from the host membrane for incorporation into α-CG and two other CGs modified at the C-6 hydroxyl of glucose.12 One analog is acetylated with tetradecanoic acid and is referred to as α-CAG (1) (Figure 1), the other is phosphorylated and abbreviated as α-CPG. Together these three glycolipids make up approximately 25% of the lipid content of H. pylori and are a distinguishing feature of these bacteria.8,9
Figure 1.
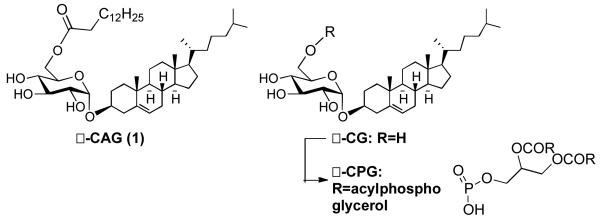
Structures of H. pylori cholesteryl glycosides.
To date, biological studies involving CG, CAG and CPG have mostly relied upon natural sources, which furnish a mixture of all three glycolipids making it difficult to determine the independent roles of each consituent.10,11 To facilitate glycolipidomic profiling studies, we have initiated a synthetic campaign directed towards a streamlined synthesis of cholesteryl glycosides. An earlier reported synthesis of α-CG required a series of 8 steps starting from free glucose and the key step used Lewis acid promoted glycosylation to yield 75% of a 1:1 α:β mixture of CG.13 However, no known synthetic procedure has generated α-CAG and a more direct route to cholesteryl glycosides would be advantageous.
For the past few years, we have been developing one-pot glycosylation strategies that capitalize on the unique reactivity of glycosyl iodides.14 We have found that trimethylsilyloxy protecting groups increase the reactivity of glycosyl iodides to the extent that even deactivated or hindered acceptors can be efficiently glycosidated.15 The overall reaction typically involves reacting a per-O-trimethylsilylated sugar with iodotrimethylsilane (TMSI) to generate the iodide in situ, followed by addition of an acceptor and acid aqueous work-up to remove the silyl protecting groups. This transient protecting group stategy is efficient but it is not without complications when trying to couple less reactive acceptors, such as cholesterol. In these cases, we find that a side reaction occurs and the acceptor becomes silylated, which leads to diminished yields. We reasoned that transilylation may be occurring from iodide attack on the primary C-6 trimethylsilyl group generating TMSI in situ, which subsequently reacts with cholesterol (Figure 2).
Figure 2.
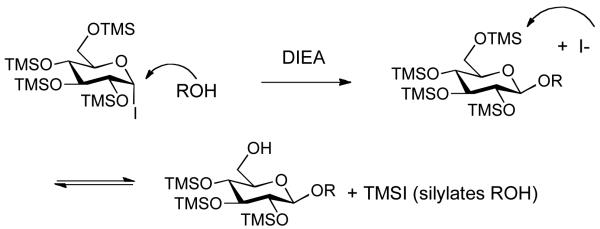
Unwanted acceptor transilylation side reaction.
In targeting α-CAG, we decided to explore a chemoenzymatic synthesis wherein the C-6 acyl group would first be incorporated using a lipase enzyme (Figure 3). We were hopeful that per-O-silylation of the resulting ester would proceed uneventfully, and that glycosyl iodide glycosylation would proceed more smoothly with an acyl group at C-6 rather than the more labile silyl group.
Figure 3.

Retrosynthesis of α-CAG.
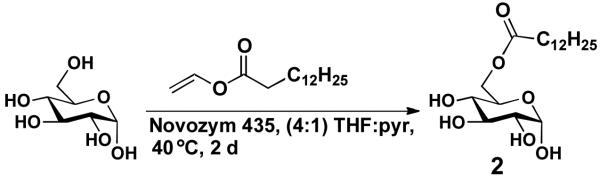
Previous work with lipase B from Candida antarctica provided powerful precedent for our strategy. In one study, Schmid and co-workers reacted glucose with a fatty acid donor in acetone or 2-butanone under reduced pressure using a soxhlet extractor for the removal of water.16 Under these conditions, high yields of 6-O-acyl glucose analogs were obtained. Later, these researchers investigated similar transformations on solid support.17 Presently, resin bound lipase B from Candida antarctica (Novozym 435) is commercially available and we decided to explore using this form of the enzyme to effect a similar transformation. Employing vinyl esters has proven advantageous, as the release of acetaldehyde acts as an internal water scavenger and alleviates the need to azeotrope. Novozym 435 has also been shown to work well in a variety of organic solvents, but portrays poor to no activity in DMF or pyridine.17 The enzyme activity however can be maintained at a high level by combining pyridine with a co-solvent. Earlier work illustrating that enzyme activity is inversely proportional to the ratio of pyridine to co-solvent18 led us to use a 4:1 ratio of tetrahydrofuran (THF) to pyridine, which gave decent solubility properties of free glucose. After testing this solvent system with relatively large amounts of enzyme (1 μmol of glucose per mg of enzyme, Table 1, entry 1), optimization of the enzymatic acylation reaction was carried out varying the amount of vinyl ester and the quantity of enzyme.
Table 1.
Regioselective Acylation using Novozym 435.
| |||
|---|---|---|---|
|
| |||
| Entry | Novozym 435 | Vinyl Ester (Equiv.) | % Yield |
| 1 | 250 mg | 6 | 88 |
| 2 | 50 mg | 6 | 87 |
| 3 | 25 mg | 6 | 94 |
| 4 | 10 mg | 6 | 90 |
| 5 | 3 mg | 6 | 93 |
| 6 | 10 mg | 3 | 89 |
| 7 | 10 mg | 2 | 23 |
| 8 | 10 mg | 1 | 31 |
Experiments showed that the amount of enzyme could be significantly reduced without affecting the yield. Eventually we found that approximately 90 μmol of glucose per mg of enzyme could be converted to 6-O-tetradecanoyl-D-glucopyranoside (2) with high yields (Table 1, entry 5). In contrast, the amount of vinyl ester employed was impactful (Table 1, entry 7 & 8). At least three equiv. of vinyl tretradecanoate were required to achieve efficient conversion (Table 1, entry 6). Upon scaling up to >250 mg, the enzymatic regioselective acylation became quantitative. It is important to note that the enzyme reaction requires sufficient mixing and must be void of all water to proceed efficiently. Thus, glucose was dried in a vacuum oven for 48 h prior to use and the reaction was carried out on a microplate thermoshaker at 40 °C.
After obtaining 2, the remaining glucosyl hydroxyl groups were protected using chlorotrimethylsilane (TMSCl) in pyridine and catalytic 4-N,N-dimethylaminopyridine (DMAP) to afford 6-O-tetradecanoyl-per-O-trimethylsilyl glucopyranoside (3) in quantitative yield (Scheme 1). The one-pot glycosidation protocol relies upon in situ generation of the α-iodide (4) using iodotrimethylsilane (TMSI) in chloroform. Initial formation of 4 was monitored via proton nuclear magnetic resonance (1H NMR) for the disappearance of the anomeric proton in compound 3 at δ 5.17 ppm and the appearance of the anomeric proton in 4 δ 6.73 ppm. To our surprise, 3 reacted very slowly with TMSI taking approximately 5 h in deuterated chloroform to generate 4. The long reaction time necessary to form 4 could be attributed to aggregation and micelle formation preventing significant exposure of the anomeric position. Consistent with this hypothesis, the 1H NMR signals were broadened in deuterated chloroform. Upon complete conversion of 3 to generate 4, the one-pot glycosidation/deprotection procedure was carried out using microwave assistance at 110 °C for 2 h followed by deprotection with Dowex50WX8-200 acidic resin in methanol to afford 1 (Scheme 1). Interestingly, the reaction also proceeded in the absence of tetrabutylammonium iodide (TBAI) to give the desired α-linked product in approximately the same yield although the stereochemical outcome was only 6:1 in favor of the α-product rather than 8:1 in the presence of TBAI.
Scheme 1.

Stereoselective synthesis of α-CAG and β-CAG.
Glycolipid bioactivity is often correlated with anomeric steroechemistry. Thus, stereoselective synthetic methods capable of preparing highly pure anomers is necessary for the advancement of glycolipid research. With this in mind, we investigated the possibility of expanding the chemoenzymatic approach to include the synthesis of β-CAG, which has distinct biological properties relatvie to α-CAG and has been shown to increase phagocytosis of bacteria.5,8 Much to our delight, activation of the glycosyl iodide (4) with silver triflate afforded β-CAG in 57% yield (1:6 α:β), after aqueous work-up and purification. Taken together, these results illustrate a powerful chemoenzymatic platform that can be applied toward creating libraries of compounds likely to be useful in both biological studies and glycolipidomic profiling.
In summary, the presence of α-cholesteryl glycosides in H. pylori was first reported in 1995.8 Shortly thereafter, α-cholesteryl glucosyl transferase was identified as the enzyme responsible for making CG.19,20 While this enzyme is imbedded in the cell wall, presumably it loads UDP-Glc from the cytoplasm of H. pylori, which is then combined with extracellular cholesterol depleted from host membrane.12 CG appears to be the substrate for an enzyme that transfers a tetradecanoic acid donor to the C-6 hydroxyl to form α-CAG but that enzyme and the exact acyl donor are yet to be identified.20 Curiously, lyso-phosphoglycerolipid biosynthesis is correlated with increased α-CAG lipid content suggesting that at least some of the fatty acid donor may derive from phosphoglycerolipids.21 Moreover, H. pylori isolated from individuals with ulcer disease have increased levels of lyso-phosphoglycerolipids.22 Understanding the specific role of α-CAG in this process and H. pylori infection in general has been hindered due to difficulties in isolating ample quantities of pure material from natural sources.
To address this unmet need, herein we have communicated the first total synthesis of α-CAG using a three-step chemoenzymatic synthesis. The regioselective enzymatic acylation was carried out on free glucose in organic solvent and required relatively small amounts of enzyme compared to earlier reported procedures. The glycosylation was carried out under microwave conditions to shorten reaction times from one or two days to just hours.15 The glycosylation does not require TBAI to afford α-linked product, but TBAI does enhance the α-selectivity. In contrast, activation of the iodide with AgOTf affords β-CAG. The synthesis of both α- and βCAG can now be achieved in reasonably large quantities (>250 mg batches) making these compounds available to the biological community. It is our hope that access to these important biomarkers will facilitate studies directed toward understanding the roles of cholesteryl glycosides in bacterial infections.
Supplementary Material
Acknowledgments
This work was supported by NIH R01GM090262, NSF CHE-0196482, NSF CRIF Program (CHE-9808183), and NSF OSTI 97-24412. Grants for 600 and 800MHz instruments include NSF DBIO 722538 and NIH PR1973.
Footnotes
Electronic Supplementary Information (ESI) available: [Experimental details and characterisation data of all the new compounds]. See http://dx.doi.org/10.1039/b000000x/
Notes and references
- 1.Yahi N, Aulas A, Fantini J, J PLoS ONE. 2010;5:e9079. doi: 10.1371/journal.pone.0009079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lingwood D, Binnington B, Róg T, Vattulainen I, Grzybek M, Coskun Ü, Lingwood CA, Simons K. Nat. Chem. Biol. 2011;7:260–262. doi: 10.1038/nchembio.551. [DOI] [PubMed] [Google Scholar]
- 3.Kawakubo M, Ito Y, Okimura Y, Kobayashi M, Sakura K, Kasama S, Fukuda MN, Fukuda M, Katsuyama T, Nakayama J. Science. 2004;305:1003–1006. doi: 10.1126/science.1099250. [DOI] [PubMed] [Google Scholar]
- 4.Algood HMS, Cover TL. Clin. Microbiol. Rev. 2006;19:597–613. doi: 10.1128/CMR.00006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee SJ, Lee BI, Suh SW. Proteins. 2011;79:2321–2326. doi: 10.1002/prot.23038. [DOI] [PubMed] [Google Scholar]
- 6.Peek RM, Blaser MJ. Nat. Rev. Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi M, Lee H, Nakayama J, Fukuda M. Curr. Drug Metab. 2009;10:29–40. doi: 10.2174/138920009787048428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirai Y, Haque M, Yoshida T, Yokota K, Yasuda T, Oguma K. J. Bacteriol. 1995;177:5327–5333. doi: 10.1128/jb.177.18.5327-5333.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haque M, Hirai Y, Yokota K, Mori N, Jahan I, Ito H, Hotta H, Yano I, Kanemasa Y, Oguma K. J. Bacteriol. 1996;178:2065–2070. doi: 10.1128/jb.178.7.2065-2070.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wunder C, Churin Y, Winau F, Warnecke D, Vieth M, Lindner B, Zähringer U, Mollenkopf H-J, Heinz E, Meyer TF. Nat. Med. 2006;12:1030–1038. doi: 10.1038/nm1480. [DOI] [PubMed] [Google Scholar]
- 11.Beigier-Bompadre M, Moos V, Belogolova E, Allers K, Schneider T, Churin Y, Ignatius R, Meyer TF, Aebischer T. J. Infect. Dis. 2011;204:1339–1348. doi: 10.1093/infdis/jir547. [DOI] [PubMed] [Google Scholar]
- 12.Wang H-J, Cheng W-C, Cheng H-H, Lai CH, Wang W-C, W Mol. Microbiol. 2012;83:67–84. doi: 10.1111/j.1365-2958.2011.07910.x. [DOI] [PubMed] [Google Scholar]
- 13.Lee H, Wang P, Hoshino H, Ito Y, Kobayashi M, Nakayama J, Seeberger PH, Fukuda M. Glycobiology. 2008;18:549–558. doi: 10.1093/glycob/cwn037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kulkarni SS, Gervay-Hague J, J Org. Lett. 2008;10:4739–4742. doi: 10.1021/ol801780c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan Y, Bornscheur UT, Cao L, Schmid RD. Enzym. Microb. Technol. 1999;25:725–728. [Google Scholar]
- 16.Cao L, Bornscheuer UT, Schmid RD, D R. J. Mol. Catal. B: Enzym. 1999;6:279–285. [Google Scholar]
- 17.Danieli B, Luisetti M, Sampognaro G, Carrea G, Riva S. J. Mol. Catal. B: Enzym. 1997;3:193–201. [Google Scholar]
- 18.Lebrun AH, Wunder C, Hildebrand J, Churin J, Zähringer U, Lindner B, Meyer TF, Heinz E, Warnecke D. Biol. Chem. 2006;281:27765–72. doi: 10.1074/jbc.M603345200. [DOI] [PubMed] [Google Scholar]
- 19.Lee H, Kobayashi M, Wang P, Nakayama J, Seeberger PH, Fukuda M. Biochem. Biophys. Res. Commun. 2006;349:1235–1241. doi: 10.1016/j.bbrc.2006.08.145. [DOI] [PubMed] [Google Scholar]
- 20.Grille S, Zaslawski A, Thiele S, Plat J, Warnecke D. Prog. Lipid. Res. 2010;49:262–288. doi: 10.1016/j.plipres.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 21.Tannæs T, Grav HJ, Bukholm G. APMIS. 2000;108:349–356. doi: 10.1034/j.1600-0463.2000.d01-67.x. [DOI] [PubMed] [Google Scholar]
- 22.Tannæs T, Bukholm IK, Bukholm G. FEMS Immunol. Med. Microbiol. 2005;44:17–23. doi: 10.1016/j.femsim.2004.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.