

Abstract
Background and Objectives
This study evaluates the efficacy and safety of a sufentanil sublingual tablet system (SSTS) for the management of postoperative pain following open abdominal surgery.
Methods
At 13 hospital sites in the United States, patients following surgery with pain intensity of greater than 4 on an 11-point numerical rating scale were randomized to receive SSTS dispensing a 15-μg sufentanil tablet sublingually with a 20-minute lockout or an identical system dispensing a placebo tablet sublingually. Pain intensity scores were recorded at baseline and for up to 72 hours after starting study drug. The primary end point was time-weighted summed pain intensity difference (SPID) over 48 hours. Secondary end points included SPID and total pain relief (TOTPAR) for up to 72 hours and patient and health care provider global assessments of the method of pain control.
Results
Summed pain intensity difference over 48 hours was significantly higher in the SSTS group than in the placebo group (least squares mean [SEM], 105.60 [10.14] vs 55.58 [13.11]; P = 0.001). Mean SPID and TOTPAR scores were significantly higher in the SSTS group at all time points from 1 hour (SPID) or 2 hours (TOTPAR) until 72 hours (P < 0.05). In the SSTS group, patient global assessment and health care provider global assessment ratings of good or excellent were greater than placebo at all time points (P < 0.01). Safety parameters, including adverse events and vital signs, were similar for SSTS and placebo.
Conclusions
These results suggest that SSTS is effective and safe for the management of postoperative pain in patients following open abdominal surgery.
Administration of opioids to postoperative patients using intravenous patient-controlled analgesia (IV PCA) results in lower pain scores and higher patient satisfaction compared with nurse-administered modalities.1,2 However, the requirement of a patent IV line and tethering of the patient to an IV PCA pump mounted on an IV pole results in risk of infection, reduced mobility, and analgesic gaps due to IV catheter infiltration or IV tubing obstruction.3,4 The programming of the pump, which is required to be completed by the nurse to set up the device, can result in dosing errors.5,6 The sufentanil sublingual tablet system (SSTS; Zalviso; AcelRx Pharmaceuticals, Redwood City, California), currently under review by the FDA, is a preprogrammed, noninvasive patient-activated bedside system to allow patients to manage moderate to severe pain in a hospital setting. The systemic uptake of sublingual sufentanil is rapid because of its high lipophilicity, and the resultant pharmacokinetics demonstrates a blunted peak plasma level and longer plasma half-time than IV administered sufentanil.7 The device has a preprogrammed 20-minute lockout interval and uses a radiofrequency identification (RFID) thumb tag to allow only the patient to operate the device (Fig. 1). Upon setup of the system, completed without a need for programming decisions, the nurse inserts a small cartridge containing 40 sufentanil tablets (approximately a 2-day supply) into the dispenser tip, which is then locked into the controller base, and the system is tethered to the bedside or other secure location. The controller base has a graphic user interface screen that facilitates patient training by the nurse and displays setup instructions and system data for authorized health care professionals. Phase 2 dose-finding studies in patients following major surgery demonstrated that sufentanil 15 μg per tablet was the optimal dosage strength, resulting in high patient satisfaction and a similar adverse event profile to lower dosage strengths.7 Sufentanil 15 μg dosed sublingually is equivalent to 3 to 4 mg IV morphine based on 300 to 400 potency factor and 60% bioavailability of sublingual sufentanil. Therefore, this sufentanil dose, available every 20 minutes, reflects an approximately equianalgesic dose to the standard 1 mg morphine on-demand every 6 minutes with typical IV PCA settings.
FIGURE 1.
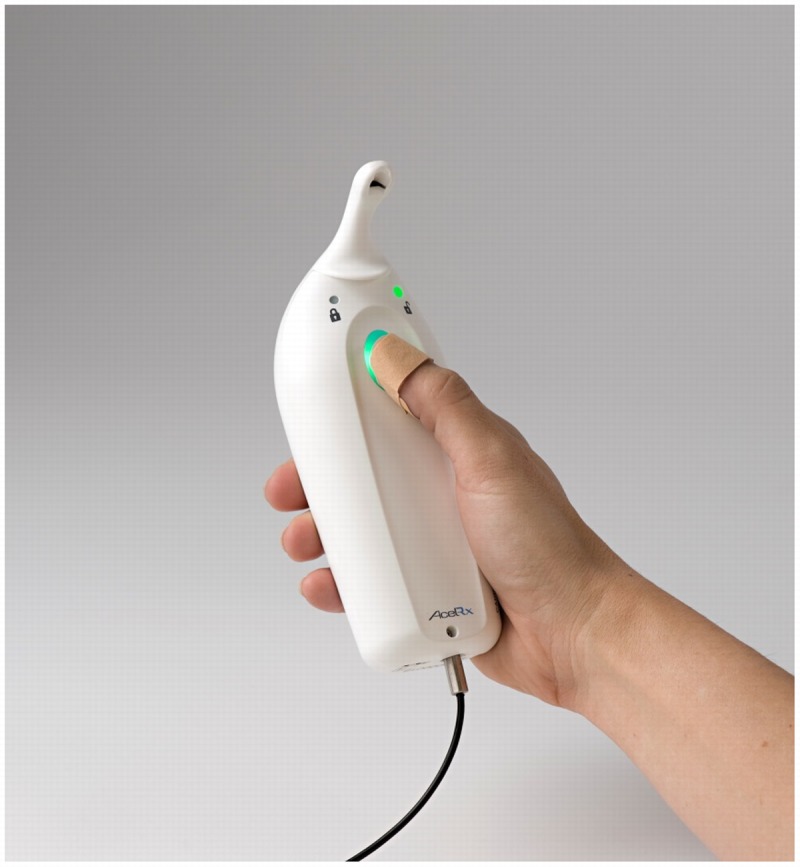
Sufentanil sublingual tablet system with RFID patient thumb tag and security tether attached to the bottom of the controller.
The objectives of the present study were to evaluate the efficacy and safety of SSTS for the management of postoperative pain in adult patients who had undergone open abdominal surgery. The study hypothesis was that sublingually administered sufentanil using the SSTS device would allow patients to titrate rapidly to acceptable levels of analgesia and would result in a good safety profile and ease of use for both patients and nurses.
METHODS
This phase 3, randomized, placebo-controlled, double-blind study was conducted at 13 hospitals in the United States between March 2012 and January 2013 (clinicaltrials.gov; NCT01539642). The study protocol and statement of informed consent were approved by a centralized institutional review board (Copernicus Group, Research Triangle Park, North Carolina) or the local institutional review board at each study site. All patients provided written informed consent before undergoing any study procedures. Male and nonpregnant female patients were eligible for inclusion if they were at least 18 years old, American Society of Anesthesiologists physical status I to III, and scheduled to undergo open abdominal surgery (including open abdominal surgeries that were laparoscopic assisted, such as partial colectomies) under general anesthesia or spinal anesthesia that did not include intrathecal opioids. Fully laparoscopic surgeries were not included. Patients were excluded if they were opioid tolerant (use of >15 mg oral morphine equivalent per day within the past 3 months); had documented sleep apnea, alcohol or drug abuse, or a need for outpatient oxygen therapy; or had any medical condition that would interfere with pain assessments. The use of any drug that may affect postoperative pain levels, such as gabapentanoids, steroids, or anti-inflammatory drugs were not allowed intraoperatively or postoperatively. Therefore, patients with a chronic pain condition necessitating treatment with these agents were also excluded from the study.
During surgery, IV opioids were allowed as needed for analgesia, but the use of any regional anesthetic technique to provide postoperative analgesia, such as epidural, peripheral nerve block, or local anesthetic wound infiltration was prohibited. Following surgery, IV morphine, hydromorphone, or fentanyl could be administered as needed to keep the patient comfortable in the postanesthesia care unit (PACU). Antiemetic prophylaxis and treatment was allowed per standard hospital protocol at each site.
Patients were randomized 2:1 to receive SSTS 15 μg or an identical system containing placebo tablets (“placebo system”) using an interactive web-response system. The sponsor, investigator, other study center personnel, and patients were blinded to treatment group assignment. Patients were randomized in the PACU provided they continued to meet entrance criteria and had a respiratory rate of 8 to 24 breaths/min, oxygen saturation greater than 95%, were able to answer questions and follow commands, and had no vomiting that was unresponsive to standard treatment.
Following randomization, but before receiving study drug, patients were required to have a pain intensity of less than 5 at some point while in the PACU to demonstrate their pain was able to be managed, have been discharged or were ready for discharge from the PACU, and lastly have a pain intensity that escalated back greater than 4 just before the first dose of study drug. Pain intensity was based on an 11-point numerical rating scale (NRS), where 0 = no pain and 10 = worst possible pain. When these conditions were met and the patient requested medication for pain, baseline vital signs, oxygen saturation, and pain intensity were assessed before the patient self-administered the first dose of study drug. The SSTS or placebo system was used for 48 hours because that is the typical duration of use for IV PCA following surgery. If patients continued to require strong opioid analgesia following 48 hours, then sites had the option to extend the study up to 72 hours; however, a completer was considered any patient finishing 48 hours in the study. Patients were educated about proper use of the SSTS device by study personnel using patient-training screens displayed on the system both during the initial patient screening process and before the first dose of study drug.
In order to maintain patients in both arms of the study as long as possible to minimize missing data, inadequate analgesia was treated using 2 mg IV morphine. Morphine dosing was allowed after only 10 minutes had passed following study drug dosing and not more than once per hour throughout the study. Patients who required additional analgesia beyond this were discontinued from the study because of inadequate analgesia and could receive any standard analgesic available at the clinical site. Patients who had oxygen saturation levels that could not be maintained at 95% or greater with or without the use of supplemental oxygen, respiratory rate less than 8 breaths/min, or excessive sedation were not allowed to have access to study drug or morphine until their vital signs had improved.
Efficacy and Safety Assessments
Efficacy was assessed by patient reports of pain intensity (based on the 11-point NRS) and pain relief (based on a 5-point scale where 0 = no relief, 1 = a little relief, 2 = moderate relief, 3 = a lot of relief, 4 = complete relief). Patients recorded pain intensity and pain relief scores at 15, 30, 45, and 60 minutes; every hour until 12 hours; every 2 hours until 48 hours; and every 4 hours from 52 to 72 hours after the first dose of study drug. Pain intensity and pain relief scores were also obtained just prior to IV morphine dosing for inadequate analgesia. The primary efficacy end point was the time-weighted summed pain intensity difference (SPID) over 48 hours (SPID48). This cumulative pain intensity measure is recommended by regulatory agencies to demonstrate the efficacy of acute pain products. Secondary efficacy end points included SPID at each evaluation time point; total pain relief (TOTPAR), pain intensity difference (PID), and pain relief at each evaluation time point; the proportion of patients discontinuing the study or requiring additional opioid medication due to inadequate analgesia; and the patient global assessment (PGA) and health care professional global assessment (HPGA) of method of pain control at 24, 48, and 72 hours. The PGA and HPGA were assessed using a 4-point categorical scale, where 1 = poor, 2 = fair, 3 = good, and 4 = excellent.
Validated patient and nurse Ease-of-Care (EOC) questionnaires8,9 were completed to assess patient and nurse impressions of the SSTS. The patient EOC questionnaire has 23 questions, 21 of which are scored on a scale of 0 to 5 (where 0 = not at all and 5 = a very great deal) and summarized into 6 subscale scores (confidence with device, comfort with device, movement, dosing confidence, pain control, and knowledge/understanding) and a total EOC score. The other 2 questions (satisfaction with level of pain control and satisfaction with method of administration of pain medication) are scored on a 6-point scale (extremely dissatisfied to extremely satisfied) and combined into an overall satisfaction score. The nurse EOC questionnaire has 22 questions, 20 of which are scored on a scale of 0 to 5 (where 0 = not at all and 5 = a very great deal) and summarized into 2 subscale scores (time-consuming and bothersome) and a total EOC score. Two other questions (satisfaction with level of pain control and satisfaction with device) were scored on a 6-point scale (extremely dissatisfied to extremely satisfied) and combined into a total satisfaction score.
Safety assessments included spontaneously reported adverse events based on the Medical Dictionary for Regulatory Activities (medDRA version 11.0); clinical laboratory evaluations including alanine aminotransferase, aspartate aminotransferase, total bilirubin, creatinine, and blood urea nitrogen; vital signs; and continuous oxygen saturation monitoring. Patients were to be withdrawn from the study if the oxygen saturation could not be maintained at 95% or greater with or without supplemental oxygen, if the respiratory rate could not be maintained at 8 breaths/min or greater, or if excessive sedation occurred.
Statistical Analysis
The analyses of efficacy data were performed on the intent-to-treat population, defined as all randomized patients who received at least 1 dose of study medication. The pain intensity data collected after a patient received the first dose of study medication were included in the calculation of the primary efficacy end point, time-weighted SPID48. Pain intensity data collected within 1 hour after IV morphine dosing for inadequate analgesia were excluded from the derivation of the efficacy end points based on the pain assessment data. The pain intensity and pain relief scores collected just prior to each dose of rescue morphine was imputed for this 1-hour time interval. The last observation carried forward imputation method was used for any missing data points after termination because of reasons other than adverse event, and the worst observation carried forward imputation method was used for missing data points for patients who discontinued because of an adverse event. All statistical tests were 2-sided and were performed at the α = 0.05 significance level.
Demographics and baseline characteristics were compared by a 2-sample t test for numeric variables and the Fisher exact test for categorical data. A parallel lines analysis of covariance model was used for the analysis of the primary efficacy end point and continuous secondary efficacy end points. This model included treatment and center factors and baseline pain intensity as a covariate. The least squares mean of each treatment and its 95% confidence interval were constructed. Ordinal categorical data were analyzed using the Cochran-Mantel-Haenszel test of general association with modified ridit scores. Dichotomous outcome data were analyzed by a 2-sample Z test on 2 proportions between treatment groups. For time-to-event data, Kaplan-Meier product limit estimators of cumulative rates of patients reaching the event (ie, termination due to inadequate analgesia and time to take first rescue medication) at follow-up time points were calculated. A log-rank test was used to compare 2 treatment groups. The Fisher exact test was used to compare the incidence of adverse events between treatment groups.
Using an effect size of 0.55 for the primary efficacy end point, a sample size of 159 patients had 90% power to show statistical difference between 2 treatment groups. This calculation was based on a 2-sided 2-sample t test with a 2:1 sample size allocation ratio and a significance level of α = 0.05. Assuming a 10% nonevaluable rate, 180 patients were planned for randomization in this study.
RESULTS
Patient Disposition and Demographics
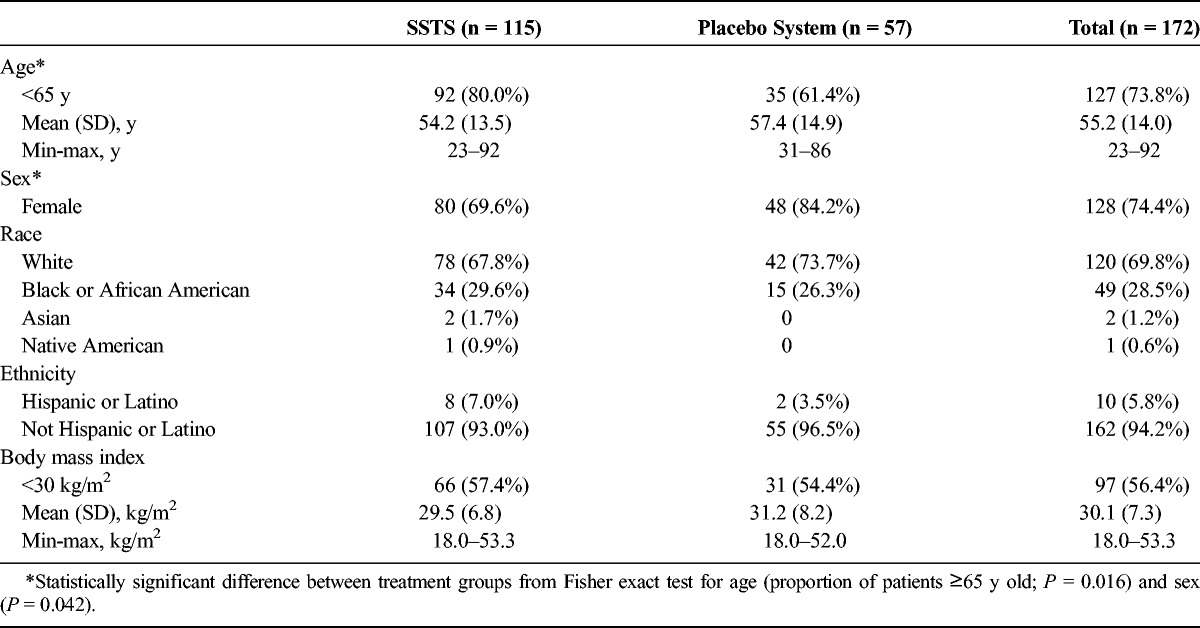
Of the 172 patients who received the study drug, 105 (61.0%) completed the 48-hour study period (Fig. 2), and 40 (23.2%) completed the 72-hour study period. One patient who was randomized to receive SSTS instead received a placebo system and therefore was included in the SSTS group for the analysis of efficacy data but was included in the placebo system group for the analysis of safety data. Demographics were similar for the 2 treatment groups, except for a significantly higher proportion of older patients (≥65 years) and female patients in the placebo system group (Table 1). Most of the surgeries were open lower abdominal/pelvic procedures (52%), followed by laparoscopic-assisted open procedures (36%) and open upper abdominal procedures (12%). While the protocol allowed spinal anesthesia, all patients received a general anesthetic in this study.
FIGURE 2.
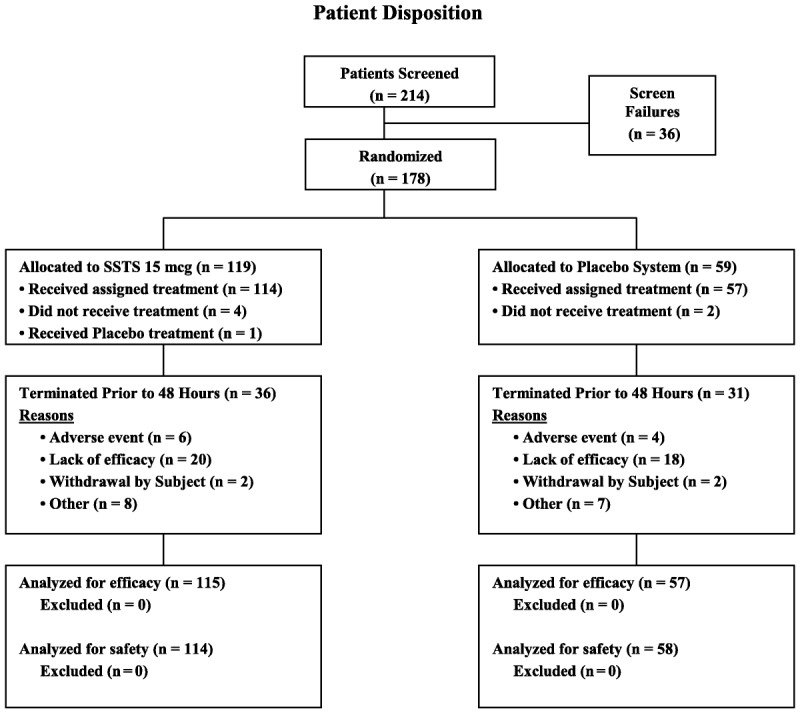
Patient disposition flow diagram. mcg indicates microgram.
TABLE 1.
Demographics and Baseline Characteristics: Intent-to-Treat Population
Efficacy Results
The primary end point, SPID48 score, was significantly higher in the SSTS group than in the placebo system group (least squares mean [SEM], 105.60 [10.14] vs 55.58 [13.11]; P = 0.001). Summed pain intensity difference and TOTPAR scores were also significantly higher in the SSTS group at all evaluation time points from 1 hour (SPID) and 2 hours (TOTPAR) until 72 hours. These summed scores were generated from the original pain intensity and pain relief scores at each time point, which are shown in Figures 3A and B, respectively. Analgesia resulting from use of IV morphine was not included in the summed pain scores (SPID or TOTPAR) because premorphine pain intensity scores were carried forward for 1 hour after morphine dosing for all these end points in order to be conservative in representing the analgesic effect of the active drug. Figures 3A and B, however, show the original pain intensity and pain relief values without any imputation for either IV morphine use or patient dropout; therefore, not surprisingly, the pain scores are similar after the first day for any patients remaining in the study.
FIGURE 3.
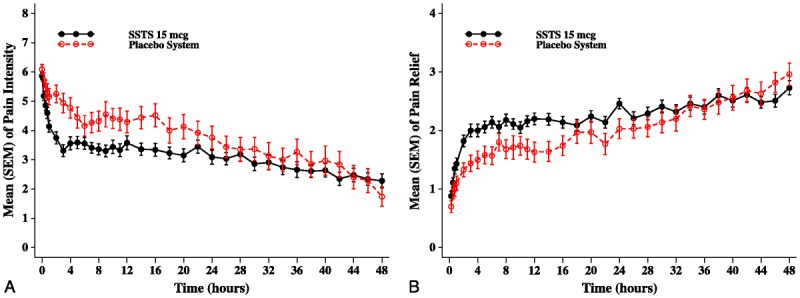
Mean (SEM) of (A) pain intensity scores and (B) pain relief scores by evaluation time point (intent-to-treat population).
To demonstrate onset of analgesia, the patient’s baseline pain intensity score must be taken into account as well as utilization of imputation to adjust for rescue analgesics. Therefore, PID to baseline scores by evaluation time point were evaluated following the initial dose of study drug, and the results from the first 4 hours are plotted in Figure 4. The SSTS group had significantly greater PID scores (ie, a greater drop in pain intensity compared with baseline) than in the placebo system group as early as 45 minutes after the first dose of study drug, and these differences were maintained for a majority of time points throughout the study. Following the initial dose of study drug and throughout the study, patients could dose every 20 minutes as needed; however, the average interdosing interval for the SSTS group throughout the 48-hour study was 100 minutes and for the placebo system group was 79 minutes (P < 0.05).
FIGURE 4.
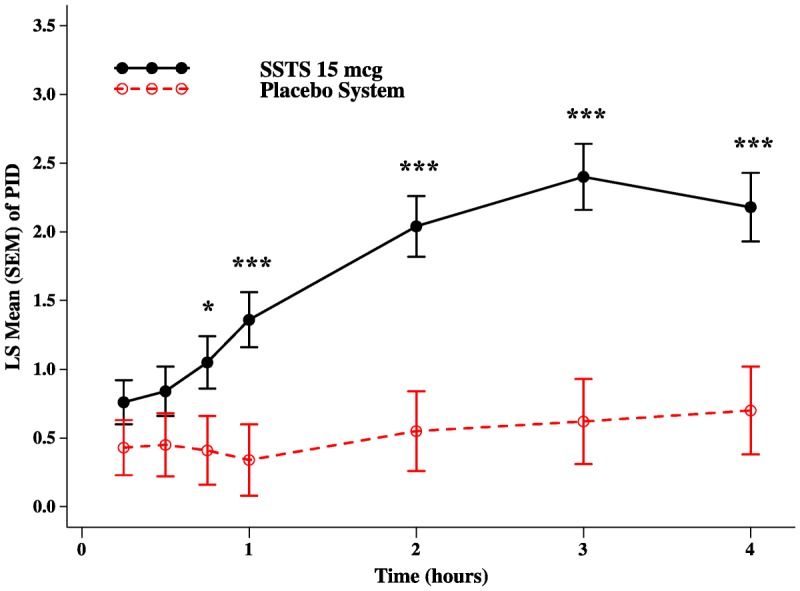
Least squares mean (SEM) of PID scores in the first 4 hours after dosing; *P < 0.05; ***P < 0.001.
A lower proportion of patients in the SSTS group prematurely discontinued the study prior to 48 hours because of inadequate analgesia (SSTS: 17.4%; placebo system: 31.6%; P = 0.035), and over this same time period, a lower proportion of SSTS patients required IV morphine as rescue for inadequate analgesia (SSTS: 33.0%; placebo system: 66.7%; P < 0.001). The placebo system group had earlier discontinuations because of inadequate analgesia (P = 0.022; Fig. 5A) and earlier use of IV morphine as rescue (P < 0.001) compared with the SSTS group (Fig. 5B). The mean cumulative number of doses of IV morphine used was statistically lower in the SSTS group than in the placebo system group for all time periods (Table 2), although neither group used a significant amount.
FIGURE 5.
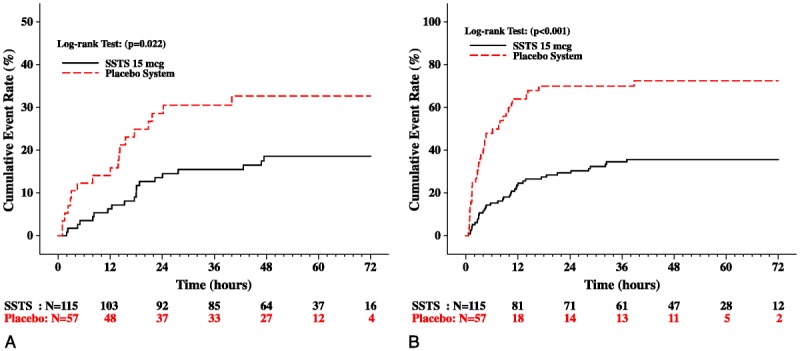
Kaplan-Meier cumulative event rates for (A) time to termination from the study due to inadequate analgesia and (B) time to take first rescue medication due to inadequate analgesia (intent-to-treat population). Numbers below x axis indicate the number of patients followed at each time point.
TABLE 2.
Intravenous Morphine Use for Inadequate Analgesia

In the SSTS group, more patients reported success (ie, responded good or excellent) on the PGA at 24 hours (69.6% vs 42.1%; P < 0.001), 48 hours (67.8% vs 45.6%; P = 0.005), and 72 hours (67.0% vs 45.6%; P = 0.007) and more health care professionals reported success on the HPGA at 24 hours (70.4% vs 43.9%; P < 0.001), 48 hours (69.6% vs 49.1%; P = 0.009), and 72 hours (69.6% vs 47.4%; P = 0.005) than in the placebo system group.
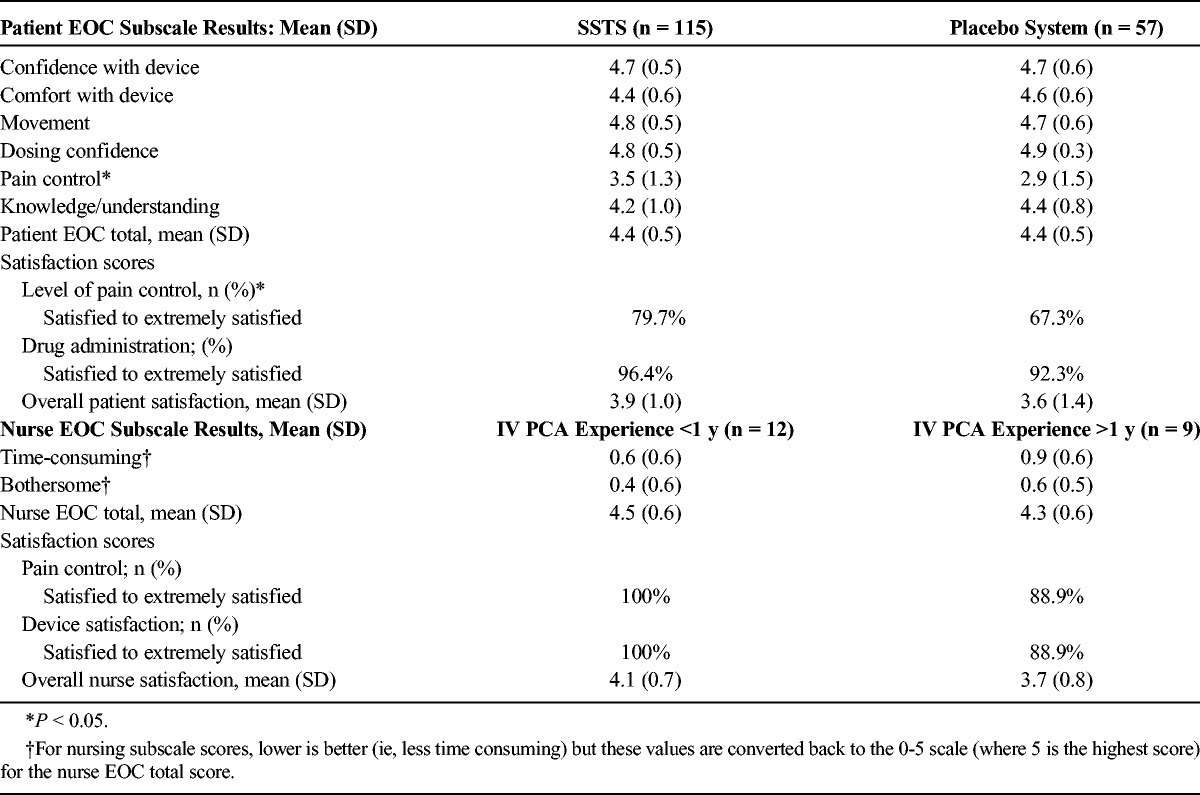
Patient EOC questionnaire results were similar in both treatment groups, except that patients in the SSTS group had better scores for questions related to pain control than patients in the placebo system group (Table 3). Nurses filled out 1 EOC questionnaire either after setting up the system in at least 10 patients or at the end of the study, whichever occurred first. The nurse EOC results were compared for nurses with less than 1 year of experience setting up IV PCA pumps versus nurses with more than 1 year of experience. Overall, both patients and nurses rated the ease of care of the system as greater than a 4 on the 0- to 5-point scale. There was no statistical difference between the 2 nursing groups for any EOC score.
TABLE 3.
Ease-of-Care Questionnaire Results
The average interdosing interval of 100 minutes for patients in the SSTS group resulted in sufentanil venous plasma levels of 71 and 69 pg/mL at 24 and 48 hours, respectively.
Safety
All adverse events in the SSTS group were mild or moderate in severity, whereas 1 adverse event in the placebo group was rated by the clinical investigator as severe (abdominal pain). Overall, 23.7% of patients in the SSTS group and 25.9% of patients in the placebo system group had 1 or more adverse events considered possibly or probably related to study drug by the investigator. There were no statistically or clinically meaningful differences between treatment groups for the proportion of patients with any possible or probably related adverse event (Table 4). Sedation does not appear as an adverse event in Table 4, as only 1 patient (0.9%) had this adverse event reported (in the SSTS group).
TABLE 4.
Possibly or Probably Related Adverse Events (>1% in Either Treatment Group)
One patient in the SSTS group had a treatment-emergent serious adverse event of moderate atrial fibrillation that was considered to be unrelated to study drug by the site investigator. Twelve patients (7 SSTS, 5 placebo) discontinued the study because of an adverse event, most commonly back pain (3 patients) and nausea and vomiting (2 patients each).
There were no statistically significant differences between treatment groups for mean changes from baseline to 48 hours or final evaluation for any laboratory variable. In addition, there were no statistically significant differences between treatment groups for mean changes from baseline in oxygen saturation. Average mean oxygen saturation values ranged from 96.72% to 98.10% for the SSTS-treated patients and 96.39% to 98.50% for placebo-treated patients at evaluation time points up through 72 hours.
DISCUSSION
This phase 3 study demonstrates the efficacy and safety of the SSTS in the treatment of postoperative pain in patients following open abdominal surgery compared with placebo. Both pain intensity and pain relief assessments throughout the study demonstrated consistently greater pain control for the SSTS group compared with the placebo system group. Patients could initiate study drug dosing only if they had a pain intensity score of 5 or higher on the 11-point NRS, thereby limiting the study to patients suffering from moderate to severe pain. The SSTS group averaged a PID score of greater than 1.3 by 1 hour after dosing, demonstrating a rapid onset of clinically significant analgesia.10 This rapid onset of action is possible because of the high lipophilicity of sufentanil (octanol:buffer partition coefficient [OBPC] of 1757:1) as well as a 20% nonionized fraction (at pH of 7.4).11 Lipophilic, nonionized drug molecules have rapid uptake from sublingual tissues into the plasma as well as rapid uptake from the plasma to the μ-opioid effector site in the central nervous system (CNS) (plasma:CNS equilibration half-life t½ke0 = 6.2 minutes for sufentanil).12 Fentanyl is less lipophilic (OBPC of 816:1) and has a lower nonionized fraction of 9%.11 Morphine is not lipophilic (OBPC of 1:1) and, therefore, even when delivered IV, has a t½ke0 of 2.8 hours, and the active metabolite morphine-6-glucuronide has an even more delayed equilibration (t½ke0 = 6.4 hours).11,13
Minimal use of rescue IV morphine over the entire 72-hour time period was required because of inadequate analgesia for the SSTS group (a total of 1.9 doses = 3.8 mg). The placebo group used statistically more IV morphine (8 mg); however, this amount is surprisingly low. This can partially be attributed to the earlier and higher dropout rate of placebo patients (32%), thereby giving them less time in the study with which to utilize the IV morphine rescue dosing. It could also be true that the 2-mg IV dose of morphine, given its aforementioned slow CNS equilibration, was not experienced by patients as sufficient analgesia or, in fact, resulted in adverse effects, such that the patient minimized requesting this drug.
This study had a relatively high placebo-response rate (46.6% completed 48 hours), which, considering the availability of rescue IV morphine, is fairly typical for placebo-controlled analgesia studies in general. It is possible that the novelty of the device attributed to this effect.
Approximately 17% of patients in the SSTS group dropped out of the study because of inadequate analgesia, significantly fewer than the placebo patients, and the dropouts occurred later in the SSTS group. Given the nature of a placebo-controlled study, where factors that will increase the placebo-response rate are purposely diminished (eg, no adjuvant analgesics or local anesthetic field blocks) and patients are unsure if they are receiving active study drug and may tend to drop out to guarantee access to analgesics, this rate of dropout from the active group is also neither surprising nor uncommon for large phase 3 placebo-controlled analgesic studies.
Patient and health care professional global assessments as well as the EOC questionnaire ratings all demonstrate that both patients and health care providers found the system to provide adequate analgesia with a user-friendly device. Regardless of whether patients were randomized to active or placebo, scores on ease of use of the system were high, with the exception of pain relief, which appropriately was lower in the placebo system group. In an open-label, randomized study of SSTS compared with IV PCA morphine in patients following either open abdominal surgery or major joint replacement surgery, patient and nurse EOC scores were higher for the SSTS compared with IV PCA for both the total EOC score as well as each subscale score.14
The safety and tolerability of the SSTS were measured using standard adverse event reporting and vital sign measurements. There were no differences for related adverse events or changes in vital signs between active and placebo groups. To reflect the real-world surgical population, the protocol did not limit enrollment by age or body mass index, resulting in more than a quarter of the patients ranging from 65 to 92 years of age and 44% of the population rated as obese (body mass index ≥30 kg/m2). The similar safety profile of SSTS compared with placebo is encouraging for the use of this product in postoperative patients who often present with multiple comorbidities. Furthermore, there is an inherent additional safety factor in a system that cannot have a prescribing or programming error as the result of a fixed-dose, fixed-lockout paradigm, as well as having a noninvasive route of administration. These phase 3 study data are in agreement with earlier phase 2 dose-finding studies, which demonstrated an adverse event profile similar to placebo with only pruritus statistically higher in the active versus placebo groups.7
The slightly higher number of enrolled patients 65 years or older and female patients in the placebo group could possibly have affected the adverse event profile in this group. The “as-needed” dosing of both the SSTS and the rescue IV morphine is specifically to allow tailoring of the drug dose for individual patient’s analgesic requirements, thereby adjusting for any demographic influence. However, we cannot fully dismiss that these demographic variables could have impacted the results of the study.
The SSTS contains a number of security features, including a patient-specific RFID technology to minimize “proxy” dosing, which is an advantage over traditional IV PCA, which can allow family members to dose the patient, resulting in adverse events. Other features include a security tether, a tamper-resistant drug cartridge to limit health care worker diversion, and an electronic tablet count on both the dispensing device and the cartridge RFID label, which can be reconciled to identify possible diversion. Although health care providers should be observant for patient diversion of medication, this issue in hospitalized patients is limited compared with the much larger problem of diversion of outpatient-prescribed opioids.
In summary, the SSTS is an investigational patient-administered opioid system for the management of moderate to severe acute pain in a hospital setting. The system is preprogrammed and noninvasive, overcoming some of the issues with IV PCA, and results from this study suggest it is an effective analgesic treatment for patients following major abdominal surgery and possesses an encouraging safety profile that would integrate well within a multimodal approach to acute pain management in the hospital setting.
ACKNOWLEDGMENTS
AcelRx Pharmaceuticals, the study sponsor, thanks the study subjects, PharmaNet/i3, a subsidiary of inVentiv Health Clinical, the research coordinators, and the investigators for their contribution.
Footnotes
Funding was received from AcelRx Pharmaceuticals, Redwood City, CA.
These data were previously presented in part as a poster at 2013 spring and fall meetings of the American Society of Regional Anesthesia and Pain Medicine, the 2013 American Society of Anesthesiologists (ASA) Annual Meeting, and the 2013 ASA Postgraduate Assembly Annual Meeting
H.S.M. received honoraria and research funding; F.G.R., T.J.G., and K.A.A. have received research funding; Y.C. is a paid consultant and M.A.E. and P.P.P. are employees of AcelRx.
REFERENCES
- 1. Ballantyne JC, Carr DB, Chalmers TC, Dear KBG, Angelillo IF, Mosteller F. Postoperative patient-controlled analgesia: meta-analyses of initial randomized control trials. J Clin Anesth. 1993; 5: 182– 193. [DOI] [PubMed] [Google Scholar]
- 2. Hudcova J, McNicol E, Quah C, Lau J, Carr DB. Patient-controlled opioid analgesia versus conventional opioid analgesia for postoperative pain. Cochrane Database Syst Rev. 2006; 4: CD003348. [DOI] [PubMed] [Google Scholar]
- 3. Webster J, Osborne S, Rickard C, Hall J. Clinically-indicated replacement versus routine replacement of peripheral venous catheters. Cochrane Database Syst Rev. 2010; 3: CD007798. [DOI] [PubMed] [Google Scholar]
- 4. Panchal SJ, Damaraju CV, Nelson WW, Hewitt DJ, Schein JR. System-related events and analgesic gaps during postoperative pain management with the fentanyl iontophoretic transdermal system and morphine intravenous patient-controlled analgesia. Anesth Analg. 2007; 105: 1437– 1441. [DOI] [PubMed] [Google Scholar]
- 5. Hicks RW, Sikirica V, Nelson W, Schein JR, Cousins DD. Medication errors involving patient-controlled analgesia. Am J Health Syst Pharm. 2008; 65: 429– 440. [DOI] [PubMed] [Google Scholar]
- 6. Meissner B, Nelson W, Hicks R, Sikirica V, Gagne J, Schein J. The rate and costs attributable to intravenous patient-controlled analgesia errors. Hosp Pharm. 2009; 44: 312– 324. [Google Scholar]
- 7. Minkowitz HS, Singla NK, Evashenk MA, et al. Pharmacokinetics of sublingual sufentanil tablets and efficacy and safety in the management of postoperative pain. Reg Anesth Pain Med. 2013; 38: 131– 139. [DOI] [PubMed] [Google Scholar]
- 8. Harding G, Vallow S, Leidy NK, et al. Ease of care with patient controlled analgesia systems: questionnaire development and validation. J Adv Nurs. 2007; 59: 530– 541. [DOI] [PubMed] [Google Scholar]
- 9. Harding G, Schein JR, Nelson WW, et al. Development and validation of a new instrument to evaluate the ease of use of patient-controlled analgesic modalities for postoperative patients. J Med Econ. 2010; 13: 42– 54. [DOI] [PubMed] [Google Scholar]
- 10. Bijur PE, Latimer CT, Gallagher EJ. Validation of a verbally administered numerical rating scale of acute pain for use in the emergency department. Acad Emerg Med. 2003; 10: 390– 392. [DOI] [PubMed] [Google Scholar]
- 11. Bernards CM. Clinical implications of physicochemical properties of opioids. In: Stein C. Opioids in Pain Control: Basic and Clinical Aspects. Cambridge, UK: Cambridge University Press; 1999. [Google Scholar]
- 12. Scott JC, Cooke JE, Stanski DR. Electroencephalographic quantitation of opioid effect: comparative pharmacodynamics of fentanyl and sufentanil. Anesthesiology. 1991; 74: 34– 42. [DOI] [PubMed] [Google Scholar]
- 13. Lotsch J, Skarke C, Schmidt H, Grosch S, Geisslinger G. The transfer half-life of morphine-6-glucuronide from plasma to effect site assessed by pupil size measurement in healthy volunteers. Anesthesiology. 2001; 95: 1329– 1338. [DOI] [PubMed] [Google Scholar]
- 14. Melson T, Boyer DL, Minkowitz H, Palmer PP, Royal MA. Sufentanil NanoTab PCA System: results from 2 ease of use studies [International Anesthesia Research Society abstract]. Anesth Analg. 2013; 116 (suppl): S258. [Google Scholar]