

Abstract
Purpose of review
Systemic lupus erythematosus (SLE) is characterized by autoantibodies directed against nuclear autoantigens normally concealed from immune recognition in healthy individuals. Here we summarize recently identified mechanisms of abnormal cell death leading to exposure and aberrant processing of nucleoprotein self antigens, and discuss their role in the SLE pathogenesis.
Recent findings
During the past few years, the unveiling of several new forms of cell death has expanded our understanding beyond the simple view of “apoptotic” versus “necrotic” cell death. SLE patients show abnormalities in cell death at several levels, including increased rates of apoptosis, necrosis, and autophagy, as well as reduced clearance of dying cells. These abnormalities lead to an increased autoantigen burden and also antigen modifications, such as nucleic acid oxidation that increase the inflammatory properties of self antigens. Recent investigations have highlighted the role of opsonins in determining the immunogenic versus tolerogenic characteristics of self antigens.
Summary
Dysregulation of different forms of programmed cell death contributes to increased exposure, availability, and immunogenic characteristic of intracellular self antigens, which all participate in development of lupus autoimmunity. As our understanding of abnormalities of cell death in SLE advances, potential therapeutic opportunities await human implementation.
Keywords: systemic lupus erythematosus, cell death, apoptosis, NETosis, complement
Introduction
It is becoming increasingly apparent that nucleoprotein complexes (NPC) containing either DNA or RNA play a dominant role in the pathogenesis of systemic lupus erythematosus (SLE). Evidence includes that both DNA complexes (including chromatin and extracellular proteins such as LL37), and RNA complexes (including SmRNP, Ro and La particles) are targeted by autoantibodies. These NPC particles are thought to be derived from dead and dying cells and contribute to inflammation by several routes. Dead cell-derived NPC may activate the innate immune system leading to inflammatory cytokine production, either directly or following the formation of antigen-antibody complexes (immune complexes or ICs). In addition, nucleoprotein particles that are ingested following abnormal cell death may be processed and presented by antigen presenting cells leading to the generation of autoreactive T cell responses. Expression of self antigens on the surface of apoptotic cells are thought to play a role in positive selection of low affinity autoreactive B cells, and antigens from dying cells in the germinal center may promote the formation of self antigen-containing ICs. NPC containing ICs provoke inflammation by activation of the complement cascade and by activating Fcg receptors (FcgR) on monocytes, macrophages, dendritic cells and neutrophils as well as intracellular Toll-like receptors (TLRs) when internalized. As the role of increased rates of apoptosis and decreased apoptotic cell clearance in SLE has been extensively reviewed before [1], we will focus our discussion on how other cell death abnormalities and dead-cell derived self antigens, in particular NPC, may contribute to the SLE pathogenesis.
Cell death pathways implicated in SLE
NETosis
Neutrophil extracellular traps (NETs) were first described in 2004 as an important defense mechanism in infection, mediating bacterial trapping and killing through the extrusion of nuclear material including DNA and histones in a web-like structure [2]. In SLE, low-density granulocytes (LDGs) extrude NETs spontaneous when isolated ex vivo, and in vitro studies show that IgG anti-RNP autoantibodies can mediate NETosis by type I IFN-primed neutrophils [3, 4]. The balance between apoptosis and NETosis is partly regulated by Akt, which functions downstream of the NADPH oxidase inhibiting apoptosis and promoting NETosis [5*]. Thus, Akt may be a potential therapeutic target to reduce NETosis in vivo [5*].
Oxidation, as well as PAD4-mediated citrullination of histones, is thought to be necessary for NETosis. Whereas Shlomchik et al [6] reported that MRL/lpr mice deficient in Nox2, and therefore impaired in NADPH-mediated oxidative pathway and NETosis, develop a worse lupus-like disease compared to wild type mice, NZM2328 mice treated with a PAD4 inhibitor had reduced immune complex deposition and renal inflammation [7**]. Whether differences in these results are related to the different mouse models tested, unexpected effects of genetic manipulation or “off target” effects of the chemical PAD4 inhibitor, remain to be determined. Another argument against an in vivo role for NETosis is that oxidation was shown not to be necessary for in vivo NET production in an IC murine model of inflammation [8]. If indeed, NETosis is involved in human SLE, another possible therapeutic target may be signal inhibitory receptor on leukocytes-1 (SIRL-1), which, upon ligation, inhibits both spontaneous and antibody-induced NETosis in neutrophils from SLE patients with low or moderate disease activity [9*]. Currently available therapies such as acetylsalicylic acid, but not dexamethasone, are able to reduce NETosis both in vitro and in vivo [10].
Reduced NET degradation in vitro is associated with a more severe clinical disease, including nephritis [11–13]. NETs are rendered resistant to nuclease digestion by autoantibodies [11, 13] and also by oxidation of DNA [14**]. Some investigators reported that macrophages clear NETs in a silent non-inflammatory manner [15*], while others demonstrated LL-37-mediated inflammasome activation following ingestion of NETs by macrophages [16]. Indeed, NETs could contain different molecules depending on the inducing stimulus [17] and such differential composition likely affects their inflammatory properties. In summary, NETs could be a potent source of modified autoantigens that promote inflammation in SLE, both by activation of the innate immune system as well as by serving as an autoantigen within IC. However their role in patients with SLE needs to be evaluated in greater detail before NETs can be clearly implicated in the pathogenesis of the disease.
Autophagy
Autophagy, or self-cannibalism, is an essential system to maintain intracellular homeostasis to ensure disposal of non-functional, damaged or unnecessary proteins and organelles. The process is regulated by the autophagy-related gene family, of which atg5 has been linked to development of SLE by genetic studies [18, 19]. DNA immune complexes (DNA-ICs) phagocytosed by plasmacytoid dendritic cells (pDCs) induce IFNa by activating TLR9, and this process requires a noncanonical autophagy pathway named LC3-associated phagocytosis (LAP). Deficiencies in this pathway (i.e. Atg7−/− cells) do not affect the phagocytosis of DNA-ICs, but decrease TLR9 engagement and subsequent IFNa production by murine pDCs [20]. While LC3 amplifies the response to intracellular DNA, another member of the autophagy pathway, Beclin-1, acts as an inhibitor instead: in particular, Beclin-1 prevents the activity of cGAS, a cytosolic DNA sensor, and promotes autophagy-mediated degradation of intracellular DNA, thus inhibiting type I IFN production [21]. In an SLE model, autophagy preceded lupus disease onset, and Atg7−/− B cells failed to efficiently develop into plasma cells [22*], suggesting that activation of B cell autophagy is important for terminal B cell development, and that selective autophagy inhibition could be considered as an approach to limit plasma cell differentiation. Of note, the same authors showed that SLE patients displayed increased B cell autophagy, which supported increased plasmablast development [22*]. Together, these studies demonstrate important roles of autophagy in the immunological response to self DNA and in the development of antibody-producing plasma cells, both key events in loss of self tolerance.
Necroptosis
Necroptosis, a form of caspase-independent cell death, was initially described as a defense mechanism against intracellular pathogens, but has been shown to occur under different pathological conditions including myocardial infarction, stroke, ischemia-reperfusion injury, pancreatitis, and inflammatory bowel disease [23]. Necroptosis is induced after triggering of death receptors, including TNFR1 and Fas, but surface TLRs and the cytoplasmic DNA sensor DAI have also been implicated in this process through regulated assembly of the “necrosome” [24]. Interestingly, type I IFNs were recently shown to induce necroptosis in mouse embryonic fibroblasts [25]. Further, murine macrophages lacking type I interferon receptor (ifnar−/−) were shown to be resistant to Salmonella-induced necroptosis [26], arguing for a role of type I IFNs in promoting necroptosis. Given the increased expression of type I IFNs in SLE patients, it will be of interest to investigate this pathway in SLE since therapeutic agents targeting components within the necrosome, including necrostatin-1 and necrosulfonamide, have shown encouraging results in preventing mortality in preclinical models for TNFa-induced shock [27].
MicroRNA-mediated regulation of cell death in SLE
MicroRNAs (miRNAs) are small, 19–25 nucleotide long, sequences of non-coding RNA able to regulate mRNA expression post-transcriptionally through targeted degradation of mRNA or by inhibiting translation. One well-studied cluster of miRNAs, the miR-17–92 family, exhibits anti-apoptotic functions through repressing Bim and PTEN [28] and was found to be decreased in SLE patients in two independent cohorts [29]. Several other miRNAs, including miR-29b and miR-29c target anti-apoptotic members of the Bcl-2 family. Hong and colleagues found that glucocorticoids increased the expression of miR-29b and miR-29c in plasmacytoid dendritic cells rendering them more susceptible to apoptosis [30]. However, in presence of a TLR9 agonist, glucocorticoids were unable to induce miR-29b and miR-29c and subsequent cell death. Thus, TLR activation-mediated down-regulation of miR-29b and miR-29c may be an efficient way of protecting cells from apoptosis and promoting their continued inflammatory response. T cells from SLE patients have been described to have increased levels of miR-29b, suggesting that those cells would be more prone to undergo apoptosis [31]. However, considering DNA and RNA-containing ICs as the main TLR ligands in SLE patients, it would be of interest to also study miR-29b and miR-29c expression in TLR7 or TLR9-expressing cells, to determine whether decreased miR-29b and miR-29c levels within those cells may contribute to increased longevity and chronic inflammation in SLE patients.
MiR-155 expression is increased in SLE as well as in lupus-prone mice and has been linked to several immune modulatory functions including autoantibody production and type I IFN regulation [32–34] and, recently, also to autophagy through the targeting the mTOR pathway [35]. The possible role of miR-155 in the increased autophagy seen in SLE patients is however not known [22]. Das and colleagues found that miR-21 expression was up-regulated in phagocytosing macrophages, and was essential to promote an anti-inflammatory response. Furthermore, overexpression of miR-21 reduced the LPS-induced release of pro-inflammatory cytokines from macrophages even in the absence of apoptotic cells [36]. Thus, miR-21 is suggested to be an important anti-inflammatory mediator during clearance of apoptotic cells, and, possibly, under other pathophysiological conditions. However, in lupus T cells, miR-21 expression is increased and highly related to disease activity and to B and T cell activation [37]. Furthermore, activation of both murine and human T cells was effectively reversed by miR-21 silencing, no longer supporting B cell maturation into plasma cells [37, 38]. These important differences in the biology of miR-21 in different cell types cautions against therapeutic agents such as antigomirs, which may then have untoward effects in patients.
Accumulation, modification and downstream consequences of dead cell-derived antigens
Accumulation of dying cells: defects in apoptotic cell receptors and serum opsonins
Clearance of apoptotic cells can occur via direct recognition by scavenger receptors on phagocytes, and with the aid of specific serum opsonins including MFG-E8, Gas-6, Protein-S and complement components such as C1q and C3b, that act as molecular bridges by binding to both AC and different endocytic receptors on phagocytes [1]. Given the central importance of clearing dead cells in an efficient and immunologically silent manner, knockout studies have indicated a high degree of redundancy and/or compensatory mechanisms of dead cell receptors and opsonins for apoptotic cells. Recent evidence suggests that efficient clearance of early apoptotic cells, before proceeding to secondary necrosis, relies on the induction of MerTK, the receptor for Gas-6 and Protein-S, in M2c polarized macrophages, resulting in anti-inflammatory cytokine production [39]. In MerTK knockout mice, apoptotic cells accumulate in germinal centers promoting aberrant B and T cell responses and development of antinuclear antibodies [40]. In SLE patients, soluble Mer ectodomain shedding was shown to correlate with increased disease activity and autoantibody levels [41], suggesting it may be employed as a disease biomarker.
Deficiency of MFG-E8, a bridging molecule binding phosphatidylserine on AC and av integrins on phagocytes, causes altered processing of dead cell-associated antigens, but is not sufficient to induce lupus in mice with non-autoimmune genetic background (C57BL/6) [42]. In contrast, MFG-E8−/− mice crossed to mice deficient in another apoptotic cell receptor, Tim-4 that binds directly to phosphatidylserine on AC, resulted in increased autoantibody production [43]. Whether these findings are explained by the numbers of apoptotic cells that fail to be engulfed or by specific signals generated by phagocytes receiving a cooperative signal remains to be determined. Relevant to human SLE, Lauber et al [44] showed that glucocorticoids enhance MFG-E8 expression in human SLE monocytes leading to increased clearance of apoptotic cells [44]. Thus, in addition to their general immunosuppressive effects, glucocorticoids may ameliorate disease also by enhancing the opsonization and swift processing of apoptotic cells as has been shown in other contexts [39, 45–47].
While SLE is a polygenic disease, over 90% of individuals carrying genetic or acquired deficiencies in the first component of the classical complement cascade, C1q, develop lupus, and C4 and C2 deficiencies are considered major susceptibility factors. Many mechanisms via which early complement components help preventing lupus autoimmunity have been suggested, including: 1) C1q binding to and clearance of apoptotic cells either directly [48–51] or via the induction of C3b deposition [52, 53], 2) C1q-mediated induction of the expression of serum opsonins such as Gas-6 and Protein-S and their receptor MerTK [54*], 3) C1q-driven suppression of IFNa-production by pDC either directly [55] or by mediating the binding of SLE IC to monocytes instead of pDC [56]; 4) C1q-mediated inhibition of inflammasome activation in human macrophages [57*], and 5) C3b/CR3-mediated anti-inflammatory signals [58, 59]; 5) Mac-1/CR3-mediated negative regulation of FcγRIIA-dependent neutrophil recruitment in response to in vivo transferred SLE IC [60]. Furthermore, similarly to what was previously shown for MFG-E8 [42], C3 activation products were recently reported to regulate the processing and presentation of dead cell-derived antigens by murine dendritic cells [61*], further expanding the role of complement in the control of the immune responses to self.
Two newly identified C1q receptors, SCARF1 [62*] and LAIR-1[63*], have been shown to mediate lupus protective functions. SCARF1 is highly expressed on endothelial cells, B-1 cells, and CD8a+ DC, the latter known to be very important in the uptake, presentation and tolerance to dead cell-derived antigens. Strikingly, the single genetic ablation of SCARF1 was shown to lead to impaired apoptotic cell uptake and accumulation, immune cell activation, and a lupus-like disorder in mice, characterized by antinuclear autoantibodies and nephritis[62*]. LAIR-1 is expressed on both myeloid and lymphoid cells, but a reduction in expression on SLE B cells [64] and pDCs [65] has been reported. C1q binding to LAIR-1 delivers an ITIM-mediated inhibitory signal, resulting in the blockade of dendritic cell differentiation from monocytes, and inhibition of IFNa production by pDC [63*]. Interestingly, both phenomena were reversed by the addition of LAIR-2, a soluble LAIR-1 inhibitor, suggesting that therapeutic blockade of LAIR-2 and/or agonistic targeting of LAIR-1 may represent promising strategies to decrease DC differentiation and IFNa production in SLE. The new studies are consistent with a protective role of complement in preventing lupus autoimmunity and offer additional therapeutic approaches to enhance removal of apoptotic cells and/or simulate their immune suppressive effects.
Modified autoantigens
Apoptotic and secondary necrotic cells are a source of modified autoantigens with increased immunogenic properties. Oxidative stress and other posttranslational modifications may render autoantigens immunogenic in SLE. As mentioned above, oxidation of 8-hydroxyguanosine (8-OHG) renders DNA resistant to degradation by the endonuclease, TREX1, and 8-OHG DNA is prevalent in NETs and in UV-exposed skin lesions of lupus patients [14]. Further, mice lacking the MFG-E8 were shown to have an increased IgM response specific for the 4-oxo-2-nonenal (ONE) protein modification [66], a highly reactive aldehyde resulting from fatty acid peroxidation, reacting with late apoptotic, secondary necrotic cells. Interestingly, an increase IgG response to ONE-modified proteins crossreacting with DNA was also described in Fas-deficient MRL-lpr mice [67], suggesting that the lipid peroxidation and, in particular the ONE modification, may be of particular pathophysiological significance in lupus, though the relevance of the ONE modification in human disease has yet to be determined. On the other hand, a recent study demonstrated the beneficial effects of lipid oxidation, and in particular generation of oxidized phosphatidylethanolamine (OxPE), by the enzyme, 12/15 lipoxigenase (12/15 LO). Genetic deficiency of 12/15-LO during inflammation caused aberrant apoptotic cell clearance and enhanced uptake by inflammatory monocytes, resulting in T cell activation in response to apoptotic cell-derived self antigens, autoantibody production, and glomerulonephritis [68**]. 12/15 LO-mediated lipid oxidation was shown to promote clearance of apoptotic cells by tolerogenic resident macrophages, blocking uptake by newly recruited inflammatory monocytes and macrophages [68**]. The underlying mechanism is not fully understood, but seems to partly depend on the OxPE-dependent sequestration of soluble MFG-E8 by alternatively activated resident macrophages. These data suggest that, despite many reports of the noxious consequences of oxidative changes in SLE, care should be taken in designing therapies that broadly target oxidative events in lupus.
Cleanup of inflammatory mediators
Contrary to apoptotic cell death, primary and secondary necrosis, as well as NETosis, leads to spillage of danger signals including DNA and RNA-containing NPC that interact with TLRs and other sensors leading to immune cell activation and loss of tolerance. Such danger molecules usually require carrier macromolecules to enter cells, whether these are autoantibodies, peptides such as LL37 released by NETosis or even amyloid-like aggregates [3, 69]. To determine whether degradation of the potentially inflammatory nucleic acid DAMP can prevent or resolve lupus-like disease, studies in our laboratory focused on lupus-prone mice that were hyperresponsive to RNA through overexpression of TLR7. Sun et al [70] showed that mice that expressed high levels of RNase were significantly protected from disease as determined by increased survival, decreased IC deposition in the kidneys, and dampened T/B cell expansion and activation. Furthermore, expansion of a unique subset of T1 B cells that were responsible in part for autoantibody production was reduced in RNAse transgenic mice [71].
Conclusions
Cell death plays a prominent role in the SLE pathogenesis by providing 1) increased availability, 2) pathogenic modifications, and 3) aberrant processing and presentation of self antigens, in particular NPC (Figure 1). Several potential therapies have been proposed, including PAD4 targeting for NETosis inhibition, degradation of extracellular RNA by circulating RNase, and modulation of the LAIR-1/LAIR-2 axis to inhibit IFNa production. Future studies will determine which of these strategies targeting different aspects and/or consequences of aberrant cell death pathways will be useful to reduce disease activity and, possibly, prevent autoimmunity in SLE patients.
Figure 1. An overview of the aberrant cell death and processing of self antigens in the pathogenesis of systemic lupus erythematosus (SLE) and possible therapeutic implications.
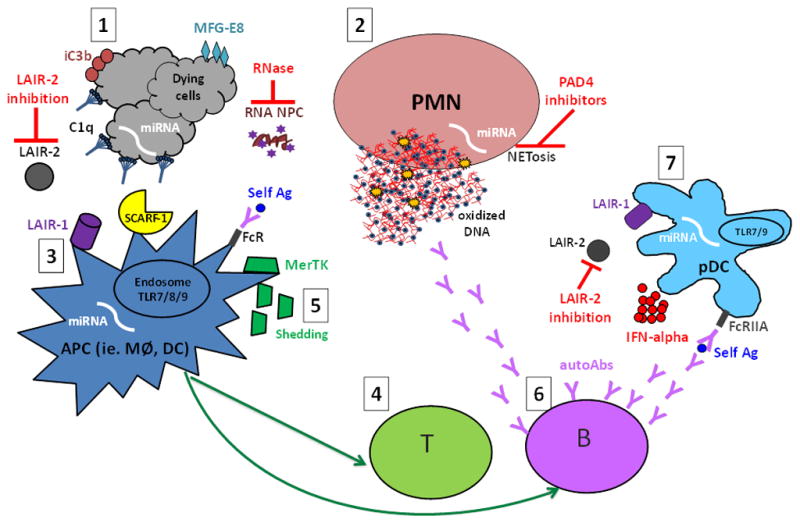
1. Aberrant cell death, i.e. increased or unwanted cell death, including NETosis and release of neutrophil (PMN) extracellular traps (NETs) (2), may provide increased availability and/or modified self antigens (self Ag), such as 8-OHG-carrying oxidized DNA- and RNA -containing nucleoprotein complexes (NPCs). Opsonins, in particular the complement components C1q and iC3b as well as MFG-E8, recognize dying cells or cell remnants and mark them for silent processing through interaction with, among others, the recently identified C1q receptors (LAIR-1 and SCARF-1 (3). In the absence of MFG-E8, self antigens are preferentially targeted for antigen presentation to T cells (4) instead of degradation. Further, MerTK-mediated swift uptake of early apoptotic cells (AC) was shown to be important to mediate the immunosuppressive effects of AC, while extracellular MerTK shedding seems to correlate with disease activity and has been proposed as a novel lupus biomarker (5). In genetically predisposed individuals these aberrations could lead to activation of autoreactive T and B cells and development of lupus autoimmunity, characterized by the production of autoantibodies (autoAbs) targeting NPCs (6). NPC-containing immune complexes are efficiently recognized by plasmacytoid dendritic cells (pDCs) and other antigen presenting cells (APC) via FcR-mediated uptake, and give rise to IFN-alpha production through TLR7/9 interactions in the endosomal compartment, a process inhibited by the binding of C1q to LAIR-1 (7). IFN-alpha has been reported to have many different adjuvant functions including up-regulation of the antigen presentation capacity of APC and increased production of class-switched antibodies by plasma cells, thus resulting in a vicious circle of increased autoantibody-self antigen interactions. Many novel therapeutic targets have been proposed, such as modulation of the LAIR-1/LAIR-2 axes (LAIR-2 being a soluble LAIR-1 inhibitor), RNase-mediated degradation of circulating RNA-containing complexes, and NETosis inhibition via PAD4 targeting. Several microRNAs (miRNAs) have been implicated in lupus pathogenesis by participating in the regulation of apoptosis, NETosis, antigen presentation, and production of IFN-alpha, and could theoretically be targeted via antigomirs. Further studies are needed to elucidate which of those may represent potential targets in human SLE.
Key points.
Apart from apoptosis, several forms of cell death such as necrosis, autophagy, NETosis and necroptosis, have been implicated in immunologic aberrations related to SLE.
Abnormal cell death can lead to increased availability, pathogenic modifications, immunogenic processing and/or presentation of self antigens.
Opsonins, including MFG-E8, C3 and C1q, have a key role in mediating the clearance of dying cells and in determining the immunogenic versus tolerogenic properties of self antigens.
MicroRNAs participate on several levels to contribute to cell death regulation in SLE.
Many new potential therapeutic targets have been proposed that may inhibit inflammation or reactivity to self that may be applicable to treatment of SLE.
Acknowledgments
This study was supported by NIH 1 F32 AR065837-01 (to LC), the Wenner-Gren Foundation (to CL), and NIH R01 NS 065933 (to KBE).
We thank Dr. Alice Wiedeman, University of Washington, for review of the manuscript
Abbreviations
- AC
apoptotic cell
- DC
dendritic cell
- FcgR
Fc gamma receptor
- IC
immune complex
- IFN
interferon
- IFNa
interferon alpha
- miR
microRNA
- miRNA
microRNA
- NET
neutrophil extracellular trap
- NPC
nucleoprotein complexes
- pDC
plasmacytoid dendritic cell
- SLE
systemic lupus erythematosus
- TLR
Toll-like receptor
Footnotes
Conflict of interest:
The authors declare no conflict of interest
References
- 1.Elkon KB. Cell Survival and death in rheumatic diseases. In: Firestein G, Budd R, Gabriel SSR, Harris E, Sledge C, et al., editors. Kelley’s Textbook of Rheumatology. 9. Philadelphia: Elsevier Publishing; 2012. pp. 382–99. [Google Scholar]
- 2.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Romo GS, Caielli S, Vega B, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3(73):73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villanueva E, Yalavarthi S, Berthier CC, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187(1):538–52. doi: 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5*.Douda DN, Yip L, Khan MA, et al. Akt is essential to induce NADPH-dependent NETosis and to switch the neutrophil death to apoptosis. Blood. 2014;123(4):597–600. doi: 10.1182/blood-2013-09-526707. This is a novel finding of Akt being involved in regulation of the apoptosis-NETosis axis and may have therapeutic implications to promote cell death by apoptosis instead of NETosis. [DOI] [PubMed] [Google Scholar]
- 6.Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med. 2012;4(157):157ra41. doi: 10.1126/scitranslmed.3004801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7**.Knight JS, Zhao W, Luo W, et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest. 2013;123(7):2981–93. doi: 10.1172/JCI67390. This study demonstrates reduction of disease severity and autoantibody levels in lupus prone mice treated with the PAD inhibitor Cl-amidine. This implies an important role of histone citrullination and NETosis in development of murine SLE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen K, Nishi H, Travers R, et al. Endocytosis of soluble immune complexes leads to their clearance by FcgammaRIIIB but induces neutrophil extracellular traps via FcgammaRIIA in vivo. Blood. 2012;120(22):4421–31. doi: 10.1182/blood-2011-12-401133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9*.Van Avondt K, Fritsch-Stork R, Derksen RH, Meyaard L. Ligation of signal inhibitory receptor on leukocytes-1 suppresses the release of neutrophil extracellular traps in systemic lupus erythematosus. PLoS One. 2013;8(10):e78459. doi: 10.1371/journal.pone.0078459. This investigation is the first to pinpoint SIRL-1 as an effective inhibitory target of both induced and spontaneous release of NETs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lapponi MJ, Carestia A, Landoni VI, et al. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J Pharmacol Exp Ther. 2013;345(3):430–7. doi: 10.1124/jpet.112.202879. [DOI] [PubMed] [Google Scholar]
- 11.Hakkim A, Furnrohr BG, Amann K, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. 2010;107(21):9813–8. doi: 10.1073/pnas.0909927107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leffler J, Gullstrand B, Jonsen A, et al. Degradation of neutrophil extracellular traps co-varies with disease activity in patients with systemic lupus erythematosus. Arthritis Res Ther. 2013;15(4):R84. doi: 10.1186/ar4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leffler J, Martin M, Gullstrand B, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. 2012;188(7):3522–31. doi: 10.4049/jimmunol.1102404. [DOI] [PubMed] [Google Scholar]
- 14**.Gehrke N, Mertens C, Zillinger T, et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity. 2013;39(3):482–95. doi: 10.1016/j.immuni.2013.08.004. This study reports that UV-irradiation and NETosis induction result in oxidative modifications of DNA, rendering it resistant to degradation by the intracellular nuclease TREX1. Oxidized DNA, in turn, was shown to promote type I IFN production and lupus skin lesions in both in human and mice. [DOI] [PubMed] [Google Scholar]
- 15*.Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol. 2013;191(5):2647–56. doi: 10.4049/jimmunol.1300436. This study demonstrates that NETs are removed by macrophages in a C1q- and DNase I-dependent silent manner. This is important since deficiencies in C1q, DNase I and macrophage clearance are all seen in SLE patients and may thus contribute to inadequate removal or response to NETs. [DOI] [PubMed] [Google Scholar]
- 16.Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol. 2013;190(3):1217–26. doi: 10.4049/jimmunol.1202388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013;5(178):178ra40. doi: 10.1126/scitranslmed.3005580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez P, Alonso-Perez E, Rodriguez-Carrio J, Suarez A. Influence of Atg5 mutation in SLE depends on functional IL-10 genotype. PLoS One. 2013;8(10):e78756. doi: 10.1371/journal.pone.0078756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou XJ, Lu XL, Lv JC, et al. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann Rheum Dis. 2011;70(7):1330–7. doi: 10.1136/ard.2010.140111. [DOI] [PubMed] [Google Scholar]
- 20.Henault J, Martinez J, Riggs JM, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37(6):986–97. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang Q, Seo GJ, Choi YJ, et al. Crosstalk between the cGAS DNA Sensor and Beclin-1 Autophagy Protein Shapes Innate Antimicrobial Immune Responses. Cell host & microbe. 2014;15(2):228–38. doi: 10.1016/j.chom.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22*.Clarke AJ, Ellinghaus U, Cortini A, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. 2014 doi: 10.1136/annrheumdis-2013-204343. This study highlights the important role of autophagy in development of plasmablasts and autoimmunity in human and mice. Importantly, in mice, autophagy preceeded development of autoimmunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Linkermann A, Green DR. Necroptosis. The New England journal of medicine. 2014;370(5):455–65. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy JM, Czabotar PE, Hildebrand JM, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39(3):443–53. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 25.Thapa RJ, Nogusa S, Chen P, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013;110(33):E3109–18. doi: 10.1073/pnas.1301218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson N, McComb S, Mulligan R, et al. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nature immunology. 2012;13(10):954–62. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takahashi N, Duprez L, Grootjans S, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell death & disease. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013;20(12):1603–14. doi: 10.1038/cdd.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlsen AL, Schetter AJ, Nielsen CT, et al. Circulating microRNA expression profiles associated with systemic lupus erythematosus. Arthritis Rheum. 2013;65(5):1324–34. doi: 10.1002/art.37890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong Y, Wu J, Zhao J, et al. miR-29b and miR-29c are involved in Toll-like receptor control of glucocorticoid-induced apoptosis in human plasmacytoid dendritic cells. PLoS One. 2013;8(7):e69926. doi: 10.1371/journal.pone.0069926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin H, Zhu X, Liang J, et al. MicroRNA-29b contributes to DNA hypomethylation of CD4+ T cells in systemic lupus erythematosus by indirectly targeting DNA methyltransferase 1. J Dermatol Sci. 2013;69(1):61–7. doi: 10.1016/j.jdermsci.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 32.Thai TH, Patterson HC, Pham DH, et al. Deletion of microRNA-155 reduces autoantibody responses and alleviates lupus-like disease in the Fas(lpr) mouse. Proc Natl Acad Sci U S A. 2013;110(50):20194–9. doi: 10.1073/pnas.1317632110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wen Z, Xu L, Chen X, et al. Autoantibody induction by DNA-containing immune complexes requires HMGB1 with the TLR2/microRNA-155 pathway. J Immunol. 2013;190(11):5411–22. doi: 10.4049/jimmunol.1203301. [DOI] [PubMed] [Google Scholar]
- 34.Zhou H, Huang X, Cui H, et al. miR-155 and its star-form partner miR-155* cooperatively regulate type I interferon production by human plasmacytoid dendritic cells. Blood. 2010;116(26):5885–94. doi: 10.1182/blood-2010-04-280156. [DOI] [PubMed] [Google Scholar]
- 35.Wan G, Xie W, Liu Z, et al. Hypoxia-induced MIR155 is a potent autophagy inducer by targeting multiple players in the MTOR pathway. Autophagy. 2014;10(1):70–9. doi: 10.4161/auto.26534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Das A, Ganesh K, Khanna S, et al. Engulfment of Apoptotic Cells by Macrophages: A Role of MicroRNA-21 in the Resolution of Wound Inflammation. J Immunol. 2014;192(3):1120–9. doi: 10.4049/jimmunol.1300613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stagakis E, Bertsias G, Verginis P, et al. Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: miR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann Rheum Dis. 2011;70(8):1496–506. doi: 10.1136/ard.2010.139857. [DOI] [PubMed] [Google Scholar]
- 38.Garchow BG, Bartulos Encinas O, Leung YT, et al. Silencing of microRNA-21 in vivo ameliorates autoimmune splenomegaly in lupus mice. EMBO Mol Med. 2011;3(10):605–15. doi: 10.1002/emmm.201100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zizzo G, Hilliard BA, Monestier M, Cohen PL. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol. 2012;189(7):3508–20. doi: 10.4049/jimmunol.1200662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khan TN, Wong EB, Soni C, Rahman ZS. Prolonged apoptotic cell accumulation in germinal centers of Mer-deficient mice causes elevated B cell and CD4+ Th cell responses leading to autoantibody production. J Immunol. 2013;190(4):1433–46. doi: 10.4049/jimmunol.1200824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zizzo G, Guerrieri J, Dittman LM, et al. Circulating levels of soluble MER in lupus reflect M2c activation of monocytes/macrophages, autoantibody specificities and disease activity. Arthritis Res Ther. 2013;15(6):R212. doi: 10.1186/ar4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng Y, Elkon KB. Autoimmunity in MFG-E8-deficient mice is associated with altered trafficking and enhanced cross-presentation of apoptotic cell antigens. J Clin Invest. 2011;121(6):2221–41. doi: 10.1172/JCI43254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyanishi M, Segawa K, Nagata S. Synergistic effect of Tim4 and MFG-E8 null mutations on the development of autoimmunity. International immunology. 2012;24(9):551–9. doi: 10.1093/intimm/dxs064. [DOI] [PubMed] [Google Scholar]
- 44.Lauber K, Keppeler H, Munoz LE, et al. Milk fat globule-EGF factor 8 mediates the enhancement of apoptotic cell clearance by glucocorticoids. Cell Death Differ. 2013;20(9):1230–40. doi: 10.1038/cdd.2013.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McColl A, Bournazos S, Franz S, et al. Glucocorticoids induce protein S-dependent phagocytosis of apoptotic neutrophils by human macrophages. J Immunol. 2009;183(3):2167–75. doi: 10.4049/jimmunol.0803503. [DOI] [PubMed] [Google Scholar]
- 46.Giles KM, Ross K, Rossi AG, et al. Glucocorticoid augmentation of macrophage capacity for phagocytosis of apoptotic cells is associated with reduced p130Cas expression, loss of paxillin/pyk2 phosphorylation, and high levels of active Rac. J Immunol. 2001;167(2):976–86. doi: 10.4049/jimmunol.167.2.976. [DOI] [PubMed] [Google Scholar]
- 47.Scannell M, Flanagan MB, deStefani A, et al. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J Immunol. 2007;178(7):4595–605. doi: 10.4049/jimmunol.178.7.4595. [DOI] [PubMed] [Google Scholar]
- 48.Verneret M, Tacnet-Delorme P, Osman R, et al. Relative Contribution of C1q and Apoptotic Cell-Surface Calreticulin to Macrophage Phagocytosis. Journal of innate immunity. 2014 doi: 10.1159/000358834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fraser DA, Laust AK, Nelson EL, Tenner AJ. C1q differentially modulates phagocytosis and cytokine responses during ingestion of apoptotic cells by human monocytes, macrophages, and dendritic cells. J Immunol. 2009;183(10):6175–85. doi: 10.4049/jimmunol.0902232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ogden CA, deCathelineau A, Hoffmann PR, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. The Journal of experimental medicine. 2001;194(6):781–95. doi: 10.1084/jem.194.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paidassi H, Tacnet-Delorme P, Garlatti V, et al. C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J Immunol. 2008;180(4):2329–38. doi: 10.4049/jimmunol.180.4.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. The Journal of experimental medicine. 1998;188(12):2313–20. doi: 10.1084/jem.188.12.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quartier P, Potter PK, Ehrenstein MR, et al. Predominant role of IgM-dependent activation of the classical pathway in the clearance of dying cells by murine bone marrow-derived macrophages in vitro. Eur J Immunol. 2005;35(1):252–60. doi: 10.1002/eji.200425497. [DOI] [PubMed] [Google Scholar]
- 54*.Galvan MD, Foreman DB, Zeng E, et al. Complement component C1q regulates macrophage expression of Mer tyrosine kinase to promote clearance of apoptotic cells. J Immunol. 2012;188(8):3716–23. doi: 10.4049/jimmunol.1102920. This paper describes downstream events following C1q engagement by macrophages engulfing apoptotic cells (AC). The increase in AC phagocytosis induced by C1q was shown to be dependent on the expression of Mer and incubation of macrophages with C1q induced MerTK and Gas-6 expression on macrophages at both the mRNA and the protein levels. This suggests that C1q engagement promotes an efferocytic phenotype on the responding macrophages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lood C, Gullstrand B, Truedsson L, et al. C1q inhibits immune complex-induced interferon-alpha production in plasmacytoid dendritic cells: a novel link between C1q deficiency and systemic lupus erythematosus pathogenesis. Arthritis Rheum. 2009;60(10):3081–90. doi: 10.1002/art.24852. [DOI] [PubMed] [Google Scholar]
- 56.Santer DM, Hall BE, George TC, et al. C1q deficiency leads to the defective suppression of IFN-alpha in response to nucleoprotein containing immune complexes. J Immunol. 2010;185(8):4738–49. doi: 10.4049/jimmunol.1001731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57*.Benoit ME, Clarke EV, Morgado P, et al. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol. 2012;188(11):5682–93. doi: 10.4049/jimmunol.1103760. This report describes different mechanisms through which C1q may act as a modulator of macrophage inflammation in a fully human autologous system. C1q binding to autologous apoptotic lymphocytes induced decreased expression of genes involved in the JAK/STAT signaling pathway and was for the first time shown to modulate inflammasome activation in human macrophages by inhibiting caspase-1-dependent cleavage of IL-1beta. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amarilyo G, Verbovetski I, Atallah M, et al. iC3b-opsonized apoptotic cells mediate a distinct anti-inflammatory response and transcriptional NF-kappaB-dependent blockade. Eur J Immunol. 2010;40(3):699–709. doi: 10.1002/eji.200838951. [DOI] [PubMed] [Google Scholar]
- 59.Reed JH, Jain M, Lee K, et al. Complement receptor 3 influences toll-like receptor 7/8-dependent inflammation: implications for autoimmune diseases characterized by antibody reactivity to ribonucleoproteins. The Journal of biological chemistry. 2013;288(13):9077–83. doi: 10.1074/jbc.M112.403303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosetti F, Tsuboi N, Chen K, et al. Human lupus serum induces neutrophil-mediated organ damage in mice that is enabled by Mac-1 deficiency. J Immunol. 2012;189(7):3714–23. doi: 10.4049/jimmunol.1201594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61*.Baudino L, Sardini A, Ruseva MM, et al. C3 opsonization regulates endocytic handling of apoptotic cells resulting in enhanced T-cell responses to cargo-derived antigens. Proc Natl Acad Sci U S A. 2014;111(4):1503–8. doi: 10.1073/pnas.1316877111. Murine C3-deficient dendritic cells were characterized by altered intracellular processing of dead cell cargo without having any defect in dead cell pahgocytosis. In particular, C3−/− DCs showed accelarated processing of phagocytosed dying cells and decreased presentation of dead cell-derived antigent on the MHC-II pathway, leading to decreased CD4 T cell responses. This is original in linking complement C3 to the regulation of processing and presentation of dying cells, with implication for the development of lupus autoimmunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62*.Ramirez-Ortiz ZG, Pendergraft WF, 3rd, Prasad A, et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nature immunology. 2013;14(9):917–26. doi: 10.1038/ni.2670. This manuscript reports how ablation of the gene encoding the scavenger receptor, SCARF1, causes apoptotic cell accumulation and a severe lupus like disorder in non-autoimmune prone C57BL/6 mice. SCARF1 was found to be highly expressed on endothelial cells and on CD8+ DC, and to mediate clearance of apoptotic cells by binding to the complement component C1q. Of note, monogenic lupus is an extraordinarily rare event, underlying the importance of this receptor for the prevention of autoimmunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63*.Son M, Santiago-Schwarz F, Al-Abed Y, Diamond B. C1q limits dendritic cell differentiation and activation by engaging LAIR-1. Proc Natl Acad Sci U S A. 2012;109(46):E3160–7. doi: 10.1073/pnas.1212753109. This paper identifies the inhibitor receptor LAIR-1 as a novel C1q receptor. LAIR-1 engagement by C1q triggers an ITIM-mediated signaling pathway leading to inhibition of IFN-a production by pDC, and preventing DC differentiation from monocytes. Interestingly, C1q was also shown to bind to LAIR-2, a soluble inhibitor of LAIR-1, this latter binding leading to reversal of C1q-mediated IFN-a blockade and DC differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Colombo BM, Canevali P, Magnani O, et al. Defective expression and function of the leukocyte associated Ig-like receptor 1 in B lymphocytes from systemic lupus erythematosus patients. PLoS One. 2012;7(2):e31903. doi: 10.1371/journal.pone.0031903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kanakoudi-Tsakalidou F, Farmaki E, Tzimouli V, et al. Simultaneous changes in serum HMGB1 and IFN-alpha levels and in LAIR-1 expression on plasmatoid dendritic cells of patients with juvenile SLE. New therapeutic options? Lupus. 2014;23(3):305–12. doi: 10.1177/0961203313519157. [DOI] [PubMed] [Google Scholar]
- 66.Chikazawa M, Otaki N, Shibata T, et al. An apoptosis-associated mammary protein deficiency leads to enhanced production of IgM antibodies against multiple damage-associated molecules. PLoS One. 2013;8(7):e68468. doi: 10.1371/journal.pone.0068468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Otaki N, Chikazawa M, Nagae R, et al. Identification of a lipid peroxidation product as the source of oxidation-specific epitopes recognized by anti-DNA autoantibodies. The Journal of biological chemistry. 2010;285(44):33834–42. doi: 10.1074/jbc.M110.165175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68**.Uderhardt S, Herrmann M, Oskolkova OV, et al. 12/15-lipoxygenase orchestrates the clearance of apoptotic cells and maintains immunologic tolerance. Immunity. 2012;36(5):834–46. doi: 10.1016/j.immuni.2012.03.010. This study describes the beneficial consequences of lipid oxidation by the enzyme 12/15-lipoxygenase (12/15-LO). 12/15-LO expression resulted in the surface exposure of lipid oxidation products on alternatively activated resident macrophages and increased apoptotic cell (AC) uptake, thus diverting AC uptake from inflammatory macrophages. Such protective sorting was at least in part mediated via the sequestration of soluble MFG-E8 by resident macrophages. Genetic ablation of 12/15-LO was reported to cause aberrant AC uptake by inflammatory monocytes, presentation of dead cell-derived antigens, and a lupus-like disease in mice. [DOI] [PubMed] [Google Scholar]
- 69.Di Domizio J, Dorta-Estremera S, Gagea M, et al. Nucleic acid-containing amyloid fibrils potently induce type I interferon and stimulate systemic autoimmunity. Proc Natl Acad Sci U S A. 2012;109(36):14550–5. doi: 10.1073/pnas.1206923109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun X, Wiedeman A, Agrawal N, et al. Increased ribonuclease expression reduces inflammation and prolongs survival in TLR7 transgenic mice. J Immunol. 2013;190(6):2536–43. doi: 10.4049/jimmunol.1202689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Giltiay NV, Chappell CP, Sun X, et al. Overexpression of TLR7 promotes cell-intrinsic expansion and autoantibody production by transitional T1 B cells. The Journal of experimental medicine. 2013;210(12):2773–89. doi: 10.1084/jem.20122798. [DOI] [PMC free article] [PubMed] [Google Scholar]