

Abstract
Aurintricarboxylic acid (ATA) is a potent inhibitor of many enzymes needed for cell and virus replication, such as polymerases, helicases, nucleases, and topoisomerases. This study examines how ATA interacts with the helicase encoded by the hepatitis C virus (HCV) to reveal that ATA interferes with both nucleic acid and ATP binding to the enzyme. We show that ATA directly binds HCV helicase to prevent the enzyme from interacting with nucleic acids and to modulate the affinity of HCV helicase for ATP, the fuel for helicase action. Amino acid substitutions in the helicase DNA binding cleft or its ATP binding site alter the ability of ATA to disrupt helicase-DNA interactions. These data, along with molecular modeling results, support the notion that an ATA polymer binds between Arg467 and Glu493 to prevent the helicase from binding either ATP or nucleic acids. We also characterize how ATA affects the kinetics of helicase-catalyzed ATP hydrolysis, and thermodynamic parameters describing the direct interaction between HCV helicase and ATA using microcalorimetry. The thermodynamics of ATA binding to HCV helicase reveal that ATA binding does not mimic nucleic acid binding in that ATA binding is driven by a smaller enthalpy change and an increase in entropy.
Keywords: ATPase, RNA-binding protein, calorimetry, nonstructural protein 3 (NS3)
INTRODUCTION
The red dye aurintricarboxylic acid (ATA, PubChem1 CID 2259) is a triphenylmethane that inhibits most enzymes that synthesize, degrade or manipulate DNA and RNA.2, 3 The molecular basis for the sensitivity of nucleic acid enzymes to submicromolar ATA concentrations likely results from the ability of ATA to form polymers in solution that resemble nucleic acids (Fig. 1).4, 5 ATA polymers bind RNA6 and also bind to nucleic acid binding clefts on nucleases and similar proteins.3, 4, 7–10
Figure 1.

Aurintricarboxylic Acid (ATA). Chemical structure of an ATA monomer and an idealized ATA polymer formed in aqueous solution.5
ATA is part of Sigma’s Library of Pharmacologically Active Compounds (LOPAC), and consequently ATA has been observed to inhibit a wide variety of proteins. Our interest in ATA, stems from our recent discovery that ATA inhibits the helicase encoded by the hepatitis C virus (HCV),11 but the list of ATA targets includes over 100 listed in PubChem Bioassay.12 This list includes other viral enzymes,13 Bloom’s syndrome helicase (BLM) (Pubchem AID 2364), and the Human RECQ1 protein (AID 2549). ATA also inhibits other ATP-fueled proteins like topoisomerase II,14 protein kinases in the JAK/STAT pathway, and other kinases needed in metabolism.15–18
HCV infects almost 150 million people today causing liver cancer, cirrhosis, and liver failure. HCV is a single stranded, positive sense RNA virus with a genome encoding a single multifunctional polyprotein. Host and viral proteases cleave the HCV polyprotein into 10 structural and nonstructural proteins. Besides inhibiting HCV helicase, ATA also inhibits the HCV RNA dependent RNA polymerase, which is formed by nonstructural protein 5B (NS5B).10 The HCV helicase is part of the multifunctional nonstructural protein 3 (NS3), which is also a protease when NS3 binds its essential protease cofactor, nonstructural protein 4A (NS4A). HCV helicase unwinds duplex RNA and DNA, even though only duplex RNA intermediates are known to be involved in HCV replication.19 Two recently approved antiviral drugs (boceprevir20 and telaprevir21) inhibit the protease function of the NS3/NS4A complex, but relatively few potent NS3 helicase inhibitors have been reported, and no HCV helicase inhibitors have entered clinical trials.22 HCV and related viruses encode the only proteins in which protease and helicase active sites are covalently linked in the same polypeptide. The NS3 N-terminal domain is the protease and the NS3 C-terminal domain is the helicase. Each NS3 domain influences the activity of the other,23, 24 but it is still not clear exactly how the NS3 helicase and protease are coordinated during HCV replication. Only a few compounds are known that inhibit both the NS3/NS4A protease and helicase functions, and ATA is not one of these.25
Because HCV helicase is a motor protein fueled by ATP hydrolysis, helicase inhibitors can act either by preventing ATP binding, ATP hydrolysis, RNA binding, the ability of NS3 to move along nucleic acids, or the ability of NS3 to separate a duplex. Mukherjee et al. showed that ATA causes NS3 to dissociate from single stranded DNA, an effect that might be due to ATA binding to the NS3 nucleic acid binding site, to an allosteric site, or both.11 The helicase portion of NS3 (i.e. NS3h) has three domains, two of which resemble conserved motor domains shared by all helicases and related proteins. ATP binds between the motor domains, and one strand of RNA (or DNA) binds the cleft that separates the motor domains from a novel helical domain not seen in other helicase structures.26 ATP binding between the NS3h motor domains causes the ATP binding cleft to close so that NS3h binds RNA more weakly and can slide toward the 5′ end of RNA like a Brownian motor.27 Because compounds that mimic ATP, like non-hydrolyzable nucleotide analogs, also cause NS3h to release DNA,28, 29 it is possible that ATA might cause NS3h to release DNA by binding to the ATP-binding site rather than the RNA-binding site. Such a hypothesis that ATA binds NS3h in place of ATP is supported by the observation that a similar triphenylmethane dye called blue HT inhibits HCV helicase by binding in place of ATP (PDB file 2ZJO).30 The goal of this study is therefore to determine if ATA interacts with HCV helicase like DNA, or if ATA interacts with NS3h at the ATP binding site, like blue HT.
Here we present evidence that ATA directly binds NS3h, and that ATA influences the binding of both nucleic acid and ATP to NS3h. These data can be interpreted as further evidence of communication between the ATP and nucleic acid binding sites on NS3h, or as evidence that ATA inhibits HCV helicase by interacting with both the ATP and nucleic acid binding sites. We performed assays monitoring NS3-catalyzed ATP hydrolysis in the presence and absence of ATA, and various concentrations of DNA, along with direct binding assays with ATA, wildtype NS3h, and NS3h harboring amino acid substitutions known to affect either ATP binding or DNA binding to NS3.
EXPERIMENTAL PROCEDURES
Materials
ATA (catalog #A1895, lot #051M0200V) was from Sigma (St. Louis, MO). The truncated C-terminally His-tagged NS3 proteins lacking the protease domain (NS3h) were purified as described before: NS3h (wildtype),31 NS3h_D290N, NS3h_E291Q,32 NS3h_H369K, and NS3h_E493Q.33 NS3h R467E was generated for this study using the Quik-Change site directed mutagenesis kit (Agilent technologies) to alter the p24NS3h_1b(con1) plasmid using the oligonucleotides 5′-GCG GCG AGG CAG GAC TGG TGA GGG CAG GAT GGG CAT TTA C -3′ and 5′-GTA AAT GCC CAT CCT GCC CTC ACC AGT CCT GCCTCG CCG C-3′. The R467E protein was expressed and purified as described for the wildtype enzyme.31 Synthetic oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA).
Electrophoretic mobility shift assay
Binding assays containing 25 mM MOPS, pH 7.5, 1.25 mM MgCl2 10 nM Cy5-dT15 (5′-/5Cy5/-TTT TTT TTT TTT TTT-3′) and 30 nM NS3h were incubated for 5 min at room temperature. Following addition of indicated concentrations of ATA, the binding reactions were incubated another 5 min at 23 °C. A 15% polyacrylamide Tris Borate EDTA (TBE) gel was pre-run at 4 °C for 30 min at 100 V. Ten microliters of each sample was loaded onto the gel. The gel was run 5 min at 200V to allow samples to enter the gel, then 60 min at 100 V at 4°C. The gel was scanned on a BioRad Molecular Imager FX Phosphorimager.
Fluorescence polarization (FP)-based DNA binding assay
Binding assays were performed as described by Mukherjee et al.11 in low volume 384-well microplates (Greiner Bio-One (Kremsmünster, Austria, catalog #784076–25). Reactions were assembled in 19 μl, then 1 μl of DMSO or ATA dissolved in DMSO was added, such that the final concentrations in each assay was 5 nM Cy5-dT15, 25 mM MOPS, pH 7.5, 1.25 mM MgCl2, 0.0025 mg/ml BSA, 0.005% (v/v) Tween 20, 0.025 mM DTT and 5% DMSO (v/v), and indicated amounts of NS3h or ATA. Polarization was monitored with a TECAN Infinite M1000 PRO multi-mode microplate reader by exciting at 635 nm (5 nm bandwidth) and measuring total fluorescence intensity, parallel and perpendicular polarized light at 667 nm (20 nm bandwidth). G-factors were calculated from wells with Cy5-dT15 alone.
To calculate the concentration of NS3h needed to bind 50% of the oligonucleotide (EC50), and to calculate the amount of ATA needed to displace 50% of the protein (IC50), polarization values were fitted to standard dose response equations using GraphPad Prism (v. 6). The Ki of ATA was calculated for each compound using the Cheng-Prusoff relationship,34 i.e. equation 1, by assuming ATA competes with the DNA oligonucleotide to bind NS3h.
| (1) |
In equation 1, IC50 is the concentration of ATA that reduces polarization by 50%, EC50 is the concentration of NS3h needed to increase polarization by 50%, and [Protein] is the NS3h concentration used to measure IC50.
ATP hydrolysis (ATPase) assays
NS3h-catalyzed ATP hydrolysis was monitored using two different colorimetric assays that monitor inorganic phosphate released from ATP. Assay 1, which has a larger dynamic range but is less sensitive, was used when ATP concentrations were greater than 1 mM (Fig. 4) and assay 2 was used when ATA concentrations were less than 1 mM (Fig. 5). ATA did not interfere with either assay, as evidenced by the fact that up to 100 μM ATA did not affect the absorbance of inorganic phosphate controls in either assay.
Figure 4.
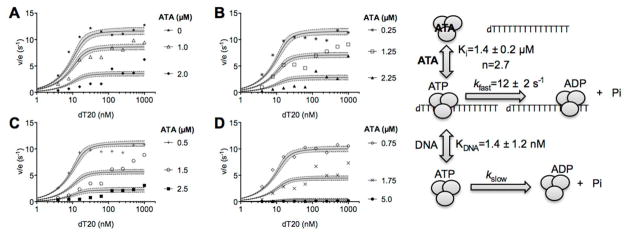
Effect of ATA on the ability of DNA to stimulate NS3h-catalyzed ATP hydrolysis. Specific activity of NS3h (mol ATP hydrolyzed/s/mol NS3h) in assays with 1 mM ATP and indicated concentrations of ATA and the oligonucleotide dT20. Data shown in panels A–D are fitted to Eqs. 2–4 by nonlinear regression, with indicated parameters. Uncertainties are 95% confidence intervals (dotted lines, grey shading) from the non-linear regression.
Figure 5.
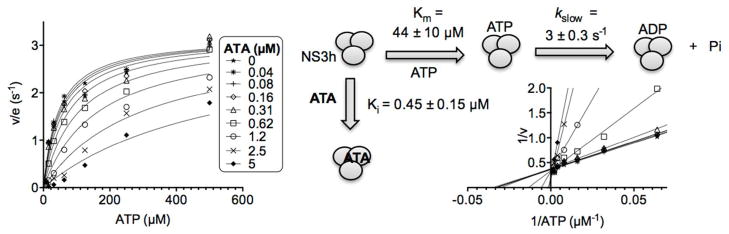
Effect of ATA on helicase catalyzed ATP hydrolysis in the absence of nucleic acids. Specific activities observed at indicated ATP and ATA concentrations in the absence of DNA. Data are fitted to Eq. 5 (methods) by non-linear regression with indicated parameters. Uncertainties are 95% confidence intervals from the non-linear regression. (Inset) Data on a Lineweaver-Burk plot with lines fitted by linear regression.
Assay 1
Assays were assembled in 27 μl in clear 96-well microtiter plates (Corning Inc, Catalog #9017) and initiated by adding 3 μl of ATP such that the final reactions contained 15 nM NS3h, 1 mM ATP, 25 mM MOPS, pH 6.5, 1.25 mM MgCl2, 5% DMSO, 50 μg/mL BSA, and 0.01% Tween 20 and indicated concentrations of ATA and oligonucleotide dT20 (5′-TTT TTT TTT TTT TTT TTT TT). After 15 minutes at 23°C, reactions were terminated by adding 200 μL of a solution containing 0.034% (w/v) malachite green, 1 N HCl, 1% ammonium molybdate, and 0.025% Tween 20, followed within 10 seconds by addition of 25 μL of 35% (w/v) sodium citrate. After 20 minutes at 23°C, an absorbance at 630 nm was read in a Varioskan multimodal plate reader (ThermoFisher, Inc.). Phosphate released was determined from a standard curve after subtracting A630 values obtained in a reaction lacking NS3h.
Concentration of ATP cleaved in 15 minutes (v) and total enzyme concentration (Et) were used to calculate specific activities (v/Et), which were globally fit to equations 2–4 using GraphPad Prism (v. 6.0).
| (2) |
| (3) |
| (4) |
In Equation 2–4, Et is NS3h concentration, Dt is dT20 concentration, I is ATA concentration, Ki is the apparent dissociation constant for NS3h and ATA, n is the Hill coefficient, ED is the NS3h-DNA complex, EI is the NS3h-ATA complex, and kfast is the turnover rate of DNA-stimulated NS3h-catalyzed ATP hydrolysis.
Assay 2
Assays were assembled in 45 μl in clear 96-well microtiter plates (Corning Inc, Catalog #9017) and initiated by adding 5 μl of a 10x solution containing ATP and MgCl2 at equimolar concentrations such that the final concentrations were 15 nM NS3h, 25 mM MOPS, pH 6.5, 1.25 mM (excess) MgCl2, 5% DMSO, 50 μg/mL BSA, 0.01% Tween 20, and indicated concentrations of MgATP2− and ATA. After incubation for 30 minutes at 37°C, reactions were terminated by adding 100 μL of BioMol Green™ reagent (Enzo Life Sciences). After 60 minutes at 23 °C, an absorbance at 620 nm was read. The amount of phosphate released was determined from a standard curve after subtracting A620 values obtained in a reaction lacking NS3h.
Concentration of ATP cleaved in 30 minutes (v) and total enzyme concentration (Et) were used to calculate specific activities (v/Et), which were globally fit to equation 5 using GraphPad Prism (v. 6.0).
| (5) |
In Equation 4, Et is NS3h concentration, I is ATA concentration, Ki is the apparent dissociation constant for NS3h and ATA, and kslow is the turnover rate of NS3h-catalyzed ATP hydrolysis in the absence of DNA.
Differential Scanning Calorimetry (DSC)
Experiments were performed in a Nano-DSC (TA Instruments). Wildtype NS3h was diluted to 10 μM in a solution containing 25 mM MOPS, pH 7.0, 1.25 mM MgCl2, 5% DMSO, 50 μg/mL BSA, and 0.01% Tween 20. In experiments containing DNA, dT20 was also at 10 μM. ATA was added at the concentrations indicated ranging from 10 μM to 5 mM. Heating was performed in the ranges 20–100°C, at a rate of 1°C/min. In each experiment, a duplicate heating was conducted on the same sample to serve as a baseline for data analysis. This second heat of the sample could be used because the components of the buffer were thermally stable and the protein did not refold when heated to 100 °C. The second heat scan also worked well as the baseline because of the similar thermal history of the sample and reference scan. Data was analyzed with NanoAnalyze™ software using the two-state scaled model (version 2.3.6, TA Instruments, New Castle, DE)
Binding by Isothermal Titration Calorimetry (ITC)
Binding of ATA to NS3h was monitored using a Nano-ITC (TA Instruments, New Castle, DE). NS3h was dialyzed overnight into a solution containing 25 mM MOPS, pH 7.0, 1.25 mM MgCl2, and 0.01% Tween 20. NS3h concentrations were determined after dialysis by absorbance at 280 nm using a molar extinction coefficient (e280) of 52,360 M−1cm−1. Enzyme was diluted in dialysis buffer to a final concentration of 20 μM for titrations with ATA and 15 μM for titrations with dT20. ATA was dissolved in the same dialysis buffer to 250 μM (or dT20 at 100 μM), and added to 174 μl of dialyzed NS3h in 16 incremental 3 μl injections at 300 second intervals at 5 °C. Data from the first injection was excluded, due to pre-equilibration mixing between the contents of cell and syringe at the syringe tip. Peak areas were integrated, normalized, and then fitted using the independent sites model by non-linear regression using NanoAnalyze™ (version 2.3.6, TA Instruments, New Castle, DE).
Molecular Modeling
The ATA heptamer (Fig. 1) was docked into PDB file 2ZJO 30 that was stripped of its ligand blue HT. Using UCSF Chimera 1.6.2,35 water molecules and counterions were removed from the PDB file, histidine protonation states were calculated, and incomplete side chains were automatically filled. A 3D conformation of the ATA heptamer from Fig. 1 was generated using Open Babel GUI, was saved as a PDB file, and positioned using a rigid body orienting code in UCSF DOCK 6.5.36 During modeling, the ligand-binding site of the protein was constrained to be the largest cluster of spheres surrounding the molecular surface of the protein as generated by UCSF DOCKS’s sphgen module. No additional constraints were made on the resolution of the binding site.
RESULTS
ATA Displaces DNA from NS3h. Mukherjee et al.11 recently reported that ATA is an HCV helicase inhibitor by using a polarization-based assay that monitors the ability of NS3h to bind a Cy5-labeled DNA oligonucleotide. They found that ATA decreases the polarization of NS3h to cause a polarization shift of a Cy5 labeled oligonucleotide with an IC50 value of 1.4 μM. They also found that ATA inhibited the ability of HCV helicase to unwind DNA and RNA with IC50 values of 0.6 ± 0.1 μM and 0.4 ± 0.2 μM, respectively.
The displacement of DNA from NS3h might result either from ATA interacting with the oligonucleotide or NS3h. If ATA binds to DNA it might cause a shift in the mobility of a DNA oligonucleotide in a gel, as is seen when an oligonucleotide binds its complement. To test this hypothesis, we analyzed the effect of ATA on the electrophoretic mobility of an NS3h-oligonucleotide complex using a gel-shift assay. When increasing concentrations of ATA (lanes 3–12, Fig. 2A) were applied to a solution containing NS3h (30 nM) and a Cy5-labeled oligonucleotide (Cy5-dT15, 10 nM) (lane 2, Fig. 2A), ATA caused the nucleotide to migrate more rapidly. At saturating ATA concentrations, the labeled oligonucleotide migrated at the same location where the oligonucleotide migrated in the absence of NS3h (lane 1, Fig. 2A). The concentration of ATA needed to shift 50% of the oligonucleotide to the position where free DNA migrates in the gel was 4 μM (lane 7, Fig. 2A), but lower concentrations of ATA caused the oligonucleotide to migrate slightly faster than it does in the absence of ATA. The lower concentration was more comparable to the concentration of ATA needed to decrease Cy5-dT15 polarization by 50% (circles, Fig. 2B). One explanation might be due to the fact that NS3h forms oligomers on DNA. The middle band in lane 2 (arrow, Fig. 2A) might represent an NS3h monomer bound to DNA and the more intense upper band might represent an NS3h oligomer bound to DNA. High concentrations of ATA did not appear to alter the mobility of Cy5-dT15 either in the presence of NS3h (lanes 3–13) or in the absence of NS3h (lanes 14–16).
Figure 2.
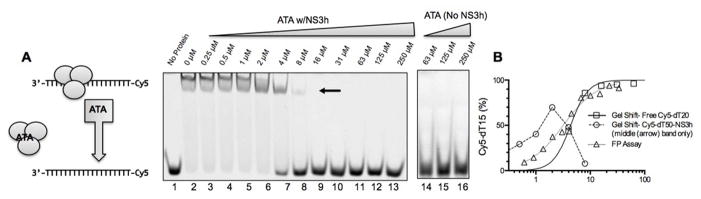
ATA displaces DNA from the HCV helicase. (A) Effect of ATA on the electrophoretic mobility of an oligonucleotide (Cy5-dT15) in the presence and absence of NS3h. The diagram on the left shows the likely band identity. Samples containing Cy5-dT15 alone (lane 1), or Cy5-dT15, NS3h and indicated concentrations of ATA were separated on a non-denaturing 15% polyacrylamide gel. The gel on the right (lanes 14–16) shows the electrophoretic mobility of Cy5-dT15 in the absence of NS3h, but in the presence of the highest concentrations of ATA analyzed. (B) Comparison of the effect of ATA on Cy5-dT15-NS3h florescence polarization as monitored with a FP-based DNA binding assay (triangles),11 with the amount of free Cy5-dT15 present in the lowest band in the gel shift assay (squares). The relative amount of the middle band (arrow) NS3h-shifted complex is also shown (circles).
Amino acid substitutions in both the NS3h ATP and DNA binding sites alter relative sensitivity of NS3h to ATA
To better localize the site where ATA interacts with NS3h, we examined the interaction of ATA with NS3h harboring amino acid substitutions that were previously demonstrated to disrupt the interaction of either ATP or DNA with NS3h (Fig. 3A). Three substitutions were in the ATP binding cleft, and two were in the DNA binding cleft. Asp290 binds the magnesium ion needed to bridge ATP to NS3h,29 and a D290N substitution causes magnesium to bind NS3h 500-times more weakly.32 Glu291 is the catalytic base needed to activate the water that attacks the gamma phosphate of ATP upon hydrolysis,29 and an E291Q mutation leads to a protein that cleaves ATP about 1,000 times more slowly.32 Arg467 acts as an “arginine finger” to stabilize the transition state. The R467E protein generated here retains the ability to bind DNA (Fig. 3B), but does not hydrolyze ATP or unwind DNA substrates (data not shown). Glu493 and His369 both contact DNA when it is bound to NS3h,26, 29 and both E493Q and H369K substitutions cause NS3h to bind DNA less tightly under optimal conditions.33
Figure 3.
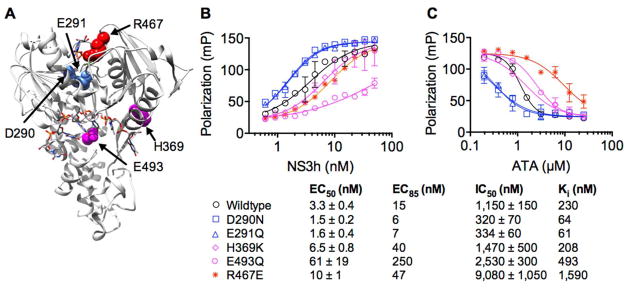
Effect of amino acid substitutions in the NS3h ATP and DNA binding clefts on the interaction of ATA with NS3h. (A) Locations of amino acids of interest mapped on PDB 3KQL,29 where DNA and an ATP analog are also shown as sticks. D290 and E291 (blue) line one side of ATP binding cleft of domain 1, R467 (red) on domain 2 completes the ATP-binding site, and H369 and E493 (magenta) line the DNA binding cleft. (B) Cy5-dT15 titrations with wildtype NS3 or its site-directed mutants. Points are averages of 2 replicates with error bars showing standard deviations. All assays were performed with 5 nM Cy5-dT15. Data are fitted to a four parameter dose-response equation with the indicated EC50 values, which approximate the dissociation constant describing the affinity of NS3h for DNA. (C) Ability of ATA to displace NS3h and each of its site-directed mutants from Cy5-dT15. Displacement assays were performed using the concentration of each NS3h protein that yielded 85% of the maximum observed change in polarization (EC85), which was calculated from the curve fits using Graphpad Prism. Data are fitted to standard dose-response equations with the IC50 values shown. Ki values were calculated for the concentration of DNA in each assay (shown) using Eq. 1 (methods). Errors are 95% confidence intervals of the curve fits.
We used the above FP-based DNA binding assay11 to investigate the ability of ATA to displace DNA bound to each of the above proteins (Fig. 3B). The FP-based binding assay confirmed earlier observations that the substitutions in the DNA binding site cause NS3h to bind DNA more weakly, and that the substitutions in the ATP binding site result in tighter DNA binding (Fig. 3B).33, 37 Less ATA was needed to displace DNA bound to D290N or E291Q than was needed to displace DNA from wildtype NS3, and more ATA was needed to displace DNA from H369K, E493Q, and R467E (Fig. 3C). Since the displacement assays were all performed at different NS3h concentrations (i.e. the EC85 values from Fig. 3B), data were normalized using the Cheng-Prusoff relationship (Eq. 1) to calculate a relative Ki for ATA with each protein.34 The resulting Ki’s were similar for both wildtype and the H369K protein, but the Ki for ATA was notably lower for the D290N and E291Q proteins, and higher for both E493Q and R467E.
ATA affects the affinity of NS3h for DNA
If a compound binds NS3h in place of nucleic acids, it should also prevent RNA or DNA from stimulating NS3h-catalyzed ATP hydrolysis. NS3h hydrolyzes ATP both in the presence and absence of DNA (or RNA) but nucleic acids stimulate rates of NS3h-catalyzed ATP hydrolysis by 10–100 fold (Fig. 4) depending on the nucleic acid sequence and base composition. Polypyrimidines, such as the oligonucleotide dT20 (5′ TTT TTT TTT TTT TTT TTT TT-3′) are some of the most efficient stimulators of NS3h-catalyzed ATP hydrolysis.38, 39 If a compound only binds NS3h in place of DNA, then inhibition of NS3h-catalyzed ATP hydrolysis should be observed at low levels of dT20, but high levels of dT20 should be able to compete with the inhibitor to still activate ATP hydrolysis. To test if ATA binds only the NS3h DNA binding site, we therefore examined initial rates of NS3h-catalyzed ATP hydrolysis at various concentrations of dT20 and ATA (Fig. 4). Data did not fit a model that assumes that dT20 and ATA compete for the same binding site on NS3h, as has been previously observed with NS3h inhibitors that simply mimic DNA.31 Instead, data fit a model that assumes ATA binds NS3h to produce a complex incapable of hydrolyzing ATP even when excess DNA is present (Eq. 2–4, methods). The model (Fig. 4) assumes that dT20 binds NS3h with a dissociation constant (KDNA) of 1.4 ± 1.2 nM, that ATA binds NS3h with a dissociation constant (Ki) of 1.4 ± 0.2 μM, and that in the absence of ATA, NS3h hydrolyzes ATP at a turnover rate (Kfast) of 11 ± 2 s−1 (uncertainties represent the 95% confidence intervals of the non- linear regression).
ATA affects the affinity of NS3h for ATP
Since all the above ATPase assays were performed with 1 mM ATP, a concentration that is over 10-times the Km of ATP in this reaction,32 they do not reveal whether or not ATA might influence the ability of NS3h to bind ATP. To address this issue and examine how ATA affects the ability of NS3h to hydrolyze ATP in the absence of nucleic acids, we used a more sensitive assay (assay 2, methods) to monitor the effect of ATA on NS3h-catalyzed ATP hydrolysis at lower ATP concentrations. ATA inhibited NS3h-catalyzed ATP hydrolysis even in the absence of DNA, and this inhibition was more pronounced at low ATP concentrations than at higher ATP concentrations (Fig. 5). Data fit a model that assumes that ATA acts as a competitive inhibitor (Eq. 4, methods), with a Ki (ATA) of 0.45 ± 0.15 μM, a Km (ATP) of 44 ± 10 μM, and a turnover rate (kslow) of 3 ± 0.3 s−1 (Fig. 5). Competitive inhibition was also clear when reciprocal data were fit by linear regression on a Lineweaver-Burk plot (Fig. 5 inset).
ATA binds NS3h in the absence of DNA or ATP
The above data suggest that ATA binds NS3h with a dissociation constant in the low micromolar range, but they do not directly show an ATA-NS3h interaction. Therefore, we next examined the direct interaction of ATA with NS3h in the absence of any helicase substrates using microcalorimetry.
Differential scanning calorimetry (DSC) was first used to examine the ATA-NS3h interaction. The goal of the DSC experiments were to understand if ATA forms a stable complex with NS3h. ATA increased the unfolding temperature of NS3h (compare Fig. 6A and Fig. 6C), although not to the same extent as a natural NS3h ligand like the oligonucleotide dT20 (compare Fig. 6A and Fig. 6B). In the presence of DNA, the unfolding temperature of NS3h increased by 10 °C, and this change was accompanied by a 300 kJ/mol increase in the enthalpy of unfolding (ΔΔH) (Fig. 6B). When ATA was added to NS3h, the unfolding temperature of NS3h increased by 8 °C, but ATA caused a decrease in the enthalpy of unfolding for NS3h (ΔΔH= −218 kJ/mol, Fig 6B). DSC data obtained from unfolding the NS3-dT20 complex fit a scaled van’t Hoff two-state model, which assumes that only one species melts in a single event (Fig. 6B). However, data obtained with the NS3h-ATA complex fit a model that assumes two species are present and that both undergo a two-state transition (Fig. 6C). The first species had the same Tm as free NS3h (red, line Fig. 6C), suggesting that not all NS3h was bound to ATA under these conditions. When ATA was added to a pre-assembled NS3h-dT20 complex (10 μM each), ATA lowered the magnitude and Tm of the observed melting event, lowering the observed melting temperature in a concentration dependent manner, most likely by replacing bound DNA (Fig. 6D). The Tm decreased 11 ± 3 °C and about 500 μM ATA was needed to decrease the Tm by 50% of this maximum change (Fig. 6E).
Figure 6.
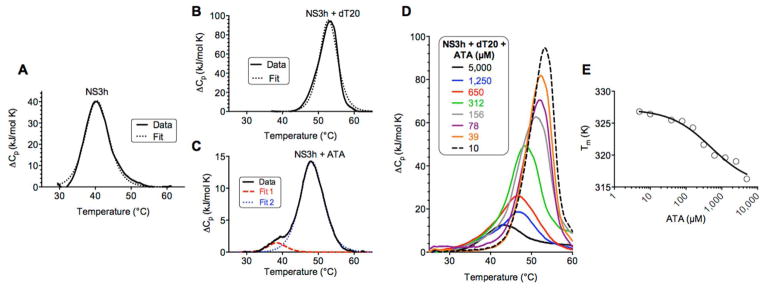
Interaction of NS3h, DNA and ATA as monitored by DSC. (A) NS3h (10 μM) was heated from 20–100 °C at a rate of 1 °C/min. Baseline corrected data were fitted to a scaled van’t Hoff two-state model (dotted line). (B) NS3h (10 μM) and dT20 (10 μM) were heated and analyzed as described in (A). (C) NS3h (10 μM) and 300 μM ATA were heated as described in (A) and deconvoluted data fitted to two scaled van’t Hoff two-state models (dotted lines). (D) NS3h (10 μM) and dT20 (10 μM) with indicated concentrations of ATA were heated as described in (A). Only baseline corrected raw data are shown in (D). (E) Melting temperatures observed at each ATA concentration for data shown in (D) fitted to a dose response equation.
Isothermal titration calorimetry (ITC) also revealed that ATA interacts with NS3h in the absence of DNA or ATP (Fig. 7). For comparison, we first performed a similar ITC experiment with NS3h and the oligonucleotide dT20 (Fig. 7A). ATA titrations generated notably smaller heats (Fig. 7B, C, D), but areas of these heats still fitted a standard n-site (non-cooperative) binding model. For these experiments, 20 μM NS3h was titrated with aliquots of a 250 μM ATA solution. Based on the dissociation constant determined, the “c value,” i.e., the product of the receptor concentration and the binding constant for each titration with ATA was ~18, which is well within the accepted range for ITC experiments.40 Determining the end point of the titration with ATA was challenging because even through both ATA and NS3h were in exactly the same buffer, there was still a clear heat of dilution remaining after NS3h was saturated with ATA, which was about twice that observed with DNA. Nevertheless, we were confident that end points were reached because similar heats were obtained in two additional titrations performed under the same conditions (Fig 7C, D), and because the initial heats observed in each titration were almost 20 kJ greater than the baseline. Thus, for model fitting, baseline heats were adjusted to zero based on the average of the heats of the last three injections.
Figure 7.
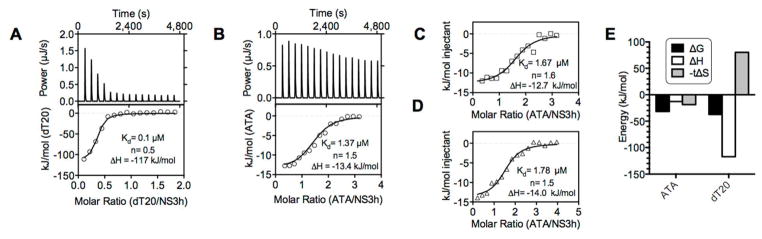
Interaction of NS3h with DNA, or ATA, as monitored by ITC. (A) NS3h (15 μM) was titrated with dT20 (100 μM) at 25°C, and heat released monitored with time (top panel). Resulting heats of injection were fitted by non-linear regression with indicated parameters (lower panel). (B) NS3h (20 μM) was titrated with ATA (250 μM) at 5°C, and heat released monitored with time (top panel). Note: In (A) and (B) data are plotted with exothermic events in the positive direction and the first data points removed for curve fits. (C), (D) Repeat titrations of NS3h with ATA under the same conditions as (B). (E) Comparison of thermodynamic parameters (adjusted to 25°C) describing the interaction of ATA or dT20 with NS3h.
NS3h bound dT20 more tightly than ATA with two NS3h protomers assembling on the oligonucleotide. More heat was released upon DNA binding to NS3h, however, and DNA binding was accompanied by a decrease in entropy (Fig. 7E). ATA bound NS3h differently, with 1.5 ATA monomers binding per NS3h with a dissociation constant of 1.3 ± 0.2 μM, and a ΔH of −13.4 ± 0.6 kJ/mol (Fig. 5B). The Kd determined using ITC was remarkably similar to most of the IC50’s and Ki’s determined above. A Kd of 1.3 μM corresponds to a ΔG of −30.9 ± 0.3 kJ/mol (at 5 °C), and considering that the change in enthalpy upon binding was only −13.4 kJ/mol, binding of ATA to NS3h appeared to be mainly driven by a positive change in entropy (ΔS= 63 J/mol K; at 5°C, −T ΔS= −17.5 kJ/mol).
DISCUSSION
Some of the data presented above could be interpreted to support the hypothesis that ATA binds NS3 helicase at the site normally occupied by RNA (or DNA) as has been observed with a variety of other nucleic acid enzymes.2, 7 However, several key observations do not fit this simple explanation. First, NS3h harboring amino acid substitutions in the ATP binding alter the ability of ATA to displace helicase-bound DNA (Fig. 3). Second, ATA modulates ATP binding in the absence of DNA (Fig. 4). Third, ATA modulates the ability of DNA to stimulate ATP hydrolysis in a cooperative manner, rather than as a linear competitive inhibitor of activation (Fig. 5).28, 32, 41 Last, the thermodynamic parameters describing the interaction of ATA do not resemble those describing the interaction of DNA with NS3h (Figs. 6 & 7).
To explain all our observations, we envision a model where an ATA polymer interacts with both the NS3h RNA-binding site, and the NS3h ATP-binding site, so that ATA causes a conformational change that prevents the protein from interacting with either ATP or nucleic acids (Fig. 8). If ATA binds in place of ATP, it could cause an abnormal rotation of the second motor domain (i.e. domain 2), as has been observed to occur when the similar triphenylmethane blue HT (aka methyl blue, CID 455837) binds NS3h.10 Domain 2 of NS3h is lined with positively charged amino acids, one of which is Arg467,42, 43 which acts as an arginine finger during ATP hydrolysis.29 Based on our site-directed mutagenesis data, we propose that ATA polymers bind near Arg467 in the second NS3h motor domain and Glu493 in the DNA binding cleft so that ATA might simultaneously interfere with the ability of NS3h to bind ATP and RNA (or DNA).
Figure 8.
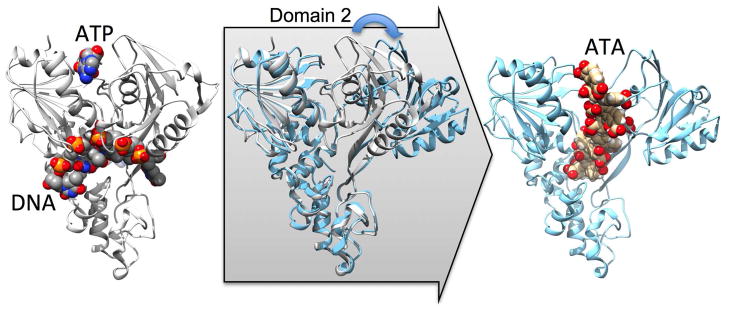
Molecular model depicting how ATA might inhibit HCV helicase. The left structure shows a ternary complex of NS3h, DNA, and an ATP analog (PDB file 3KQL29) with the protein depicted with ribbons and the ligands as space-filled models. The far right structure shows an ATA heptamer positioned in PDB file 2ZJO30 using UCSF Dock 6.5.36 The middle panel shows how NS3h domain 2 might rotate to accommodate ATA.
To test this hypothesis we used UCSF Dock 6.536 to examine the interaction of an ATA heptamer with PDB file 2ZJO30 after the blue HT ligand was removed. Remarkably the ATA polymer docked as predicted with one end of ATA within 3 Å of Glu493 in the DNA binding cleft, and the other end within 3 Å of Arg467 (Fig. 8 right panel). A comparison of the docked ATA model with PDB file 2ZJO29 (Fig. 8 left panel), reveals the conformational change that we propose releases DNA from the enzyme (Fig. 8, middle). Of note, altering either R467 or E493 causes the protein to be less responsive to ATA (Fig. 3). The observation that proteins harboring substitutions on the domain 1 side of the ATP-binding site are more responsive to ATA (Fig. 3) might be because such mutations cause the protein to assume an open conformation more frequently.
Our data lend less support to the ideas that ATA inhibits helicases because ATA binds nucleic acid substrates, or because ATA sequesters the metal ion cofactors needed for helicase action. The interaction of ATA with aluminum is a standard assay used to detect metal ions in water, and others have shown that ATA binds RNA.6 However, ATA does not shift the electrophoretic mobility of our DNA oligonucleotides (Fig. 2), and metal ion chelation seems unlikely because all assays were performed with a vast excess of divalent metal cofactors. One other explanation for the ability of ATA to inhibit NS3 helicase action is that ATA might disrupt the formation of NS3h oligomers that are needed to efficiently unwind DNA and RNA.44, 45 Monitoring how a small molecule disrupts NS3h oligomers is challenging. However, if the multiple bands seen on native gels with NS3h and DNA (Fig. 2) represent different oligomeric forms of NS3h, low levels of ATA appear to disrupt the NS3h oligomer, and this disruption might contribute to the potent ATA inhibition seen in helicase assays.
All studies here were performed with recombinant NS3 protein that was truncated to remove its protease domain (aka NS3h). Some studies have been repeated with full-length NS3 both in the presence and absence of NS4A, and we have not noted any differences in the sensitivity of these more biologically authentic helicase constructs to ATA. Likewise, we have no evidence that ATA also inhibits the protease function of NS3. Nevertheless, the NS3 protease domain, NS4A or other HCV or host proteins might influence how ATA interacts with NS3 in the cell.
On a final note, results here might inspire the development of more specific ATA derivatives as possible HCV antiviral agents. However, synthesis of more active analogs will likely be challenging because without a polymeric tail, triphenylmethanes, like blue HT and its optimized derivatives, are all relatively poor NS3h inhibitors, with IC50 values between 10 μM and 40 μM.10, 46 It might be worthwhile to start such a study by first examining the effect on HCV of the collection of ATA derivatives, which were previously synthesized to inhibit the reverse transcriptase encoded by the human immunodeficiency virus (HIV).5, 47 ATA derivatives might be particularly effective HCV antivirals since ATA inhibits both NS3h and the HCV NS5B RNA-dependent RNA polymerase.10
Acknowledgments
We thank Jean Ndjomou, Rajesh Kolli, and Collette Quinn for helpful advice and technical assistance with this study.
ABBREVIATIONS USED
- ATA
Aurintricarboxylic Acid
- HCV
hepatitis C virus
- NS3h
nonstructural protein 3 helicase domain
Footnotes
This work was supported by National Institutes of Health Grant AI088001 and a Research Growth Initiative Award from the UWM Research Foundation.
References
- 1.Bolton EE, Wang Y, Thiessen PA, Bryant SH. PubChem: integrated platform of small molecules and biological activities. Annual Rep Comp Chem. 2008;4:217–241. [Google Scholar]
- 2.Blumenthal T, Landers TA. The inhibition of nucleic acid-binding proteins by aurintricarboxylic acid. Biochem Biophys Res Commun. 1973;55:680–688. doi: 10.1016/0006-291x(73)91198-4. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh U, Giri K, Bhattacharyya NP. Interaction of aurintricarboxylic acid (ATA) with four nucleic acid binding proteins DNase I, RNase A, reverse transcriptase and Taq polymerase. Spectrochim Acta A Mol Biomol Spectrosc. 2009;74:1145–1151. doi: 10.1016/j.saa.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzalez RG, Blackburn BJ, Schleich T. Fractionation and structural elucidation of the active components of aurintricarboxylic acid, a potent inhibitor of protein nucleic acid interactions. Biochim Biophys Acta. 1979;562:534–545. doi: 10.1016/0005-2787(79)90116-3. [DOI] [PubMed] [Google Scholar]
- 5.Cushman M, Wang P, Schols D, De Clercq E. Structure investigation and anti-HIV activities of high-molecular weight ATA polymers. J Org Chem. 1992;57:7241–7248. [Google Scholar]
- 6.Skidmore AF, Beebee TJ. Characterization and use of the potent ribonuclease inhibitor aurintricarboxylic acid for the isolation of RNA from animal tissues. Biochem J. 1989;263:73–80. doi: 10.1042/bj2630073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez RG, Haxo RS, Schleich T. Mechanism of action of polymeric aurintricarboxylic acid, a potent inhibitor of protein--nucleic acid interactions. Biochemistry. 1980;19:4299–4303. doi: 10.1021/bi00559a023. [DOI] [PubMed] [Google Scholar]
- 8.Tan GS, Chiu CH, Garchow BG, Metzler D, Diamond SL, Kiriakidou M. Small Molecule Inhibition of RISC Loading. ACS Chem Biol. 2012;7:403–410. doi: 10.1021/cb200253h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorjsuren D, Kim D, Maloney DJ, Wilson DM, Simeonov A. Complementary non-radioactive assays for investigation of human flap endonuclease 1 activity. Nucleic Acids Res. 2011;39:e11. doi: 10.1093/nar/gkq1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y, Bopda-Waffo A, Basu A, Krishnan R, Silberstein E, Taylor DR, Talele TT, Arora P, Kaushik-Basu N. Characterization of aurintricarboxylic acid as a potent hepatitis C virus replicase inhibitor. Antivir Chem Chemother. 2009;20:19–36. doi: 10.3851/IMP1286. [DOI] [PubMed] [Google Scholar]
- 11.Mukherjee S, Hanson AM, Shadrick WR, Ndjomou J, Sweeney NL, Hernandez JJ, Bartczak D, Li K, Frankowski KJ, Heck JA, Arnold LA, Schoenen FJ, Frick DN. Identification and analysis of hepatitis C virus NS3 helicase inhibitors using nucleic acid binding assays. Nucleic Acids Res. 2012;40:8607–8621. doi: 10.1093/nar/gks623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Zhou Z, Han L, Karapetyan K, Dracheva S, Shoemaker BA, Bolton E, Gindulyte A, Bryant SH. PubChem’s BioAssay Database. Nucleic Acids Res. 2012;40:D400–D412. doi: 10.1093/nar/gkr1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashem AM, Flaman AS, Farnsworth A, Brown EG, Van Domselaar G, He R, Li X. Aurintricarboxylic acid is a potent inhibitor of influenza A and B virus neuraminidases. PLoS One. 2009;4:e8350. doi: 10.1371/journal.pone.0008350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benchokroun Y, Couprie J, Larsen AK. Aurintricarboxylic acid, a putative inhibitor of apoptosis, is a potent inhibitor of DNA topoisomerase II in vitro and in Chinese hamster fibrosarcoma cells. Biochem Pharmacol. 1995;49:305–313. doi: 10.1016/0006-2952(94)00465-x. [DOI] [PubMed] [Google Scholar]
- 15.Rui H, Xu J, Mehta S, Fang H, Williams J, Dong F, Grimley PM. Activation of the Jak2-Stat5 signaling pathway in Nb2 lymphoma cells by an anti-apoptotic agent, aurintricarboxylic acid. J Biol Chem. 1998;273:28–32. doi: 10.1074/jbc.273.1.28. [DOI] [PubMed] [Google Scholar]
- 16.McCune SA, Foe LG, Kemp RG, Jurin RR. Aurintricarboxylic acid is a potent inhibitor of phosphofructokinase. Biochem J. 1989;259:925–927. doi: 10.1042/bj2590925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsi CJ, Chao Y, Chen CW, Lin WW. Aurintricarboxylic acid protects against cell death caused by lipopolysaccharide in macrophages by decreasing inducible nitric-oxide synthase induction via IkappaB kinase, extracellular signal-regulated kinase, and p38 mitogen-activated protein kinase inhibition. Mol Pharmacol. 2002;62:90–101. doi: 10.1124/mol.62.1.90. [DOI] [PubMed] [Google Scholar]
- 18.Chen CW, Chao Y, Chang YH, Hsu MJ, Lin WW. Inhibition of cytokine-induced JAK-STAT signalling pathways by an endonuclease inhibitor aurintricarboxylic acid. Br J Pharmacol. 2002;137:1011–1020. doi: 10.1038/sj.bjp.0704955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tai CL, Chi WK, Chen DS, Hwang LH. The helicase activity associated with hepatitis C virus nonstructural protein 3 (NS3) J Virol. 1996;70:8477–8484. doi: 10.1128/jvi.70.12.8477-8484.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, Poordad F, Goodman ZD, Sings HL, Boparai N, Burroughs M, Brass CA, Albrecht JK, Esteban R. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1207–1217. doi: 10.1056/NEJMoa1009482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, Focaccia R, Younossi Z, Foster GR, Horban A, Ferenci P, Nevens F, Mullhaupt B, Pockros P, Terg R, Shouval D, van Hoek B, Weiland O, Van Heeswijk R, De Meyer S, Luo D, Boogaerts G, Polo R, Picchio G, Beumont M. Telaprevir for retreatment of HCV infection. N Engl J Med. 2011;364:2417–2428. doi: 10.1056/NEJMoa1013086. [DOI] [PubMed] [Google Scholar]
- 22.Belon CA, Frick DN. In: Hepatitis C: Antiviral Drug Discovery and Development. He Y, Tan SL, editors. Caister Academic Press; 2011. pp. 237–256. [Google Scholar]
- 23.Frick DN, Rypma RS, Lam AM, Gu B. The nonstructural protein 3 protease/helicase requires an intact protease domain to unwind duplex RNA efficiently. J Biol Chem. 2004;279:1269–1280. doi: 10.1074/jbc.M310630200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beran RK, Pyle AM. Hepatitis C viral NS3–4A protease activity is enhanced by the NS3 helicase. J Biol Chem. 2008;283:29929–29937. doi: 10.1074/jbc.M804065200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ndjomou J, Kolli R, Mukherjee S, Shadrick WR, Hanson AM, Sweeney NL, Bartczak D, Li K, Frankowski KJ, Schoenen FJ, Frick DN. Fluorescent primuline derivatives inhibit hepatitis C virus NS3-catalyzed RNA unwinding, peptide hydrolysis and viral replicase formation. Antiviral Res. 2012;96:245–255. doi: 10.1016/j.antiviral.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JL, Morgenstern KA, Griffith JP, Dwyer MD, Thomson JA, Murcko MA, Lin C, Caron PR. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure. 1998;6:89–100. doi: 10.1016/s0969-2126(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 27.Levin MK, Gurjar M, Patel SS. A Brownian motor mechanism of translocation and strand separation by hepatitis C virus helicase. Nat Struct Mol Biol. 2005;12:429–435. doi: 10.1038/nsmb920. [DOI] [PubMed] [Google Scholar]
- 28.Levin MK, Gurjar MM, Patel SS. ATP binding modulates the nucleic acid affinity of hepatitis C virus helicase. J Biol Chem. 2003;278:23311–23316. doi: 10.1074/jbc.M301283200. [DOI] [PubMed] [Google Scholar]
- 29.Gu M, Rice CM. Three conformational snapshots of the hepatitis C virus NS3 helicase reveal a ratchet translocation mechanism. Proc Natl Acad Sci U S A. 2010;107:521–528. doi: 10.1073/pnas.0913380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen CS, Chiou CT, Chen GS, Chen SC, Hu CY, Chi WK, Chu YD, Hwang LH, Chen PJ, Chen DS, Liaw SH, Chern JW. Structure-based discovery of triphenylmethane derivatives as inhibitors of hepatitis C virus helicase. J Med Chem. 2009;52:2716–2723. doi: 10.1021/jm8011905. [DOI] [PubMed] [Google Scholar]
- 31.Belon CA, High YD, Lin TI, Pauwels F, Frick DN. Mechanism and specificity of a symmetrical benzimidazolephenylcarboxamide helicase inhibitor. Biochemistry. 2010;49:1822–1832. doi: 10.1021/bi901974a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frick DN, Banik S, Rypma RS. Role of divalent metal cations in ATP hydrolysis catalyzed by the hepatitis C virus NS3 helicase: magnesium provides a bridge for ATP to fuel unwinding. J Mol Biol. 2007;365:1017–1032. doi: 10.1016/j.jmb.2006.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frick DN, Rypma RS, Lam AM, Frenz CM. Electrostatic analysis of the hepatitis C virus NS3 helicase reveals both active and allosteric site locations. Nucleic Acids Res. 2004;32:5519–5528. doi: 10.1093/nar/gkh891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 35.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 36.Moustakas DT, Lang PT, Pegg S, Pettersen E, Kuntz ID, Brooijmans N, Rizzo RC. Development and validation of a modular, extensible docking program: DOCK 5. J Comput Aided Mol Des. 2006;20:601–619. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 37.Frick DN, Banik S, Rypma RS. Modulation of the hepatitis C virus NS3 helicase activity by divalent metal cations. Hepatology. 2006;44:342A–342A. [Google Scholar]
- 38.Suzich JA, Tamura JK, Palmer-Hill F, Warrener P, Grakoui A, Rice CM, Feinstone SM, Collett MS. Hepatitis C virus NS3 protein polynucleotide-stimulated nucleoside triphosphatase and comparison with the related pestivirus and flavivirus enzymes. J Virol. 1993;67:6152–6158. doi: 10.1128/jvi.67.10.6152-6158.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lam AM, Keeney D, Eckert PQ, Frick DN. Hepatitis C virus NS3 ATPases/helicases from different genotypes exhibit variations in enzymatic properties. J Virol. 2003;77:3950–3961. doi: 10.1128/JVI.77.7.3950-3961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turnbull WB, Daranas AH. On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J Am Chem Soc. 2003;125:14859–14866. doi: 10.1021/ja036166s. [DOI] [PubMed] [Google Scholar]
- 41.Preugschat F, Averett DR, Clarke BE, Porter DJ. A steady-state and pre-steady-state kinetic analysis of the NTPase activity associated with the hepatitis C virus NS3 helicase domain. J Biol Chem. 1996;271:24449–24457. doi: 10.1074/jbc.271.40.24449. [DOI] [PubMed] [Google Scholar]
- 42.Yao N, Hesson T, Cable M, Hong Z, Kwong AD, Le HV, Weber PC. Structure of the hepatitis C virus RNA helicase domain. Nat Struct Biol. 1997;4:463–467. doi: 10.1038/nsb0697-463. [DOI] [PubMed] [Google Scholar]
- 43.Cho HS, Ha NC, Kang LW, Chung KM, Back SH, Jang SK, Oh BH. Crystal structure of RNA helicase from genotype 1b hepatitis C virus. A feasible mechanism of unwinding duplex RNA. J Biol Chem. 1998;273:15045–15052. doi: 10.1074/jbc.273.24.15045. [DOI] [PubMed] [Google Scholar]
- 44.Sikora B, Chen Y, Lichti CF, Harrison MK, Jennings TA, Tang Y, Tackett AJ, Jordan JB, Sakon J, Cameron CE, Raney KD. Hepatitis C virus NS3 helicase forms oligomeric structures that exhibit optimal DNA unwinding activity in vitro. J Biol Chem. 2008;283:11516–11525. doi: 10.1074/jbc.M708125200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jennings TA, Mackintosh SG, Harrison MK, Sikora D, Sikora B, Dave B, Tackett AJ, Cameron CE, Raney KD. NS3 helicase from the hepatitis C Virus can function as a monomer or oligomer depending on enzyme and substrate concentrations. J Biol Chem. 2009;284:4806–4814. doi: 10.1074/jbc.M805540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li K, Frankowski KJ, Belon CA, Neuenswander B, Ndjomou J, Hanson AM, Shanahan MA, Schoenen FJ, Blagg BS, Aube J, Frick DN. Optimization of Potent Hepatitis C Virus NS3 Helicase Inhibitors Isolated from the Yellow Dyes Thioflavine S and Primuline. J Med Chem. 2012;55:3319–3330. doi: 10.1021/jm300021v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cushman M, Wang PL, Chang SH, Wild C, De Clercq E, Schols D, Goldman ME, Bowen JA. Preparation and anti-HIV activities of aurintricarboxylic acid fractions and analogues: direct correlation of antiviral potency with molecular weight. J Med Chem. 1991;34:329–337. doi: 10.1021/jm00105a052. [DOI] [PubMed] [Google Scholar]