

Abstract
Insulin may stimulate its own insulin secretion and is a potent growth factor for the pancreatic β-cell. Complications of pregnancy, such as diabetes and intrauterine growth restriction, are associated with changes in fetal insulin concentrations, secretion, and β-cell mass. However, glucose concentrations are also abnormal in these conditions. The direct effect of chronic fetal hyperinsulinemia with euglycemia on fetal insulin secretion and β-cell mass has not been tested. We hypothesized that chronic fetal hyperinsulinemia with euglycemia would increase glucose-stimulated insulin secretion (GSIS) and β-cell mass in the ovine fetus. Singleton ovine fetuses were infused with iv insulin to produce high physiological insulin concentrations, or saline for 7–10 days. The hyperinsulinemic animals also received a direct glucose infusion to maintain euglycemia. GSIS, measured at 133 ± 1 days of gestation, was significantly attenuated in the hyperinsulinemic fetuses (P < .05). There was no change in β-cell mass. The hyperinsulinemic fetuses also had decreased oxygen (P < .05) and higher norepinephrine (1160 ± 438 vs 522 ± 106 pg/mL; P < .005). Acute pharmacologic adrenergic blockade restored GSIS in the hyperinsulinemic-euglycemic fetuses, demonstrating that increased adrenergic signaling mediates decreased GSIS in these fetuses.
Insulin has an essential role in regulating adult β-cell mass (1–4). Furthermore, insulin stimulates its own secretion from isolated adult β-cells (5, 6). In vivo studies in adult humans have confirmed this positive feedback effect on β-cell function, because glucose-stimulated insulin secretion (GSIS) was potentiated 40% after a hyperinsulinemic-euglycemic clamp (7–10). Although these studies implicate insulin as a trophic factor for the mature β-cell, its role in the developing β-cell is unknown. Understanding the trophic role of insulin on the developing β-cell is important, because certain complications of pregnancy, such as diabetes and placental insufficiency, result in increased and decreased, respectively, fetal β-cell mass and insulin secretion (11–14).
Pregnant sheep have been used to model diabetes and placental insufficiency, and findings are consistent with data from clinical studies (15–21). For example, GSIS and β-cells are decreased in fetal sheep after experimental placental insufficiency (19–21). Conversely, pulsatile maternal hyperglycemia increases fetal GSIS and β-cell mass (15, 17). However, each of these models are also characterized by a confounding increase or decrease in fetal glucose concentrations (15, 17, 20, 21). The direct effect of chronic fetal hyperinsulinemia with euglycemia on fetal insulin secretion and β-cell mass remains untested.
We hypothesized that a chronic hyperinsulinemic-euglycemic clamp would increase GSIS and β-cell mass in the normally growing sheep fetus. Our results demonstrate that, contrary to our hypothesis, GSIS was lower and β-cell mass did not change. During this clamp, fetal oxygenation decreased and norepinephrine (NE) concentrations increased. Fetal hypoxia inhibits fetal insulin secretion by stimulating fetal NE, which acts directly on the fetal β-cell to inhibit insulin secretion (22–24). Therefore, we used pharmacological adrenergic inhibition to demonstrate that the lower rate of fetal GSIS during the hyperinsulinemic-euglycemic clamp was due to increased fetal adrenergic signaling.
Materials and Methods
Animal care and surgical procedure
Studies were conducted in pregnant Columbia-Rambouillet sheep, each carrying a singleton late gestation fetus (term, 147 d of gestation [dGA]). Surgery was performed to place fetal catheters into the abdominal aorta and femoral veins and maternal catheters into the femoral artery and vein in 18 animals on 125 ± 0.5 dGA as previously described (25). Animals recovered for at least 5 days before randomization into treatment groups. All animal procedures were in compliance with the guidelines of the United States Department of Agriculture, the National Institutes of Health, and the American Association for the Accreditation of Laboratory Animal Care. The animal care and use protocols were approved by the University of Colorado Institutional Animal Care and Use Committee.
Experimental design
Seventeen animals (one fetus did not survive postoperatively) were randomly assigned to an insulin infusion (INS) group (n = 8) or control (CON) group (n = 9) (Figure 1A). The INS fetuses received a continuous, iv infusion of Humulin R insulin in saline with 0.5% bovine serum albumin (wt/vol) at a rate that was increased over the first 3 days to produce high physiological insulin concentrations; 0.75–1.25 ng/mL compared with 0.25–0.5 ng/mL in this breed of normal fetal sheep (26). A concurrent iv fetal dextrose infusion, 33% (wt/vol), in the insulin concentrations group prevented a fall in glucose concentrations and was adjusted on average 2.5 times daily, with a maximum of 5 times per day. The dextrose infusion rate was increased on average 25 ± 5% (range, 33% decrease to 71% increase) above the current rate based on the fetal and maternal plasma arterial glucose concentration, previous response to dextrose infusion rate changes, and the INS rate. The CON group received a fetal iv infusion of 0.9% NaCl adjusted to match the rate of infusions in the INS group.
Figure 1. Schematic representation of experimental study design.

Diagrams for the chronic 7- to 11-day experiment with the primed continuous variable-rate hyperglycemic clamp (A), and the chronic fetal hyperinsulinemic-euglycemic clamp with the fetal insulin secretion study ± adrenergic blockade (B). ASIS, arginine-stimulated insulin secretion.
Durations of experimental infusions were the same in both groups (8.00 ± 0.50 d INS, 8.44 ± 0.53 CON). The INS in the INS group increased from an initial rate of 34.0 ± 7.1 to 131.3 ± 21.7 mU/h (41.5 mU/h·kg) (Figure 2A) at the end of the infusion period. Insulin concentrations in the INS group were 0.97 ± 0.12 and 0.47 ± 0.08 ng/mL in CON fetuses at the end of the infusion (P < .05) (Figure 2C). To maintain euglycemia in the INS group, the dextrose infusion rate was increased from an initial rate of 3.0 ± 0.4 mg/min to a final rate of 12.9 ± 2.4 mg/min (4.1 ± 1.5 mg/min·kg) at the end of the infusion (Figure 2B).
Figure 2. Fetal insulin and glucose infusion rates and concentrations.

Fetal insulin (A) and glucose (B) infusion rates in the INS group. Fetal arterial plasma insulin (C) and glucose (D) concentrations. Open circles, INS (n = 8); solid squares, CON (n = 9). *, significant difference between INS and CON, P < .05 by mixed models ANOVA. Values are mean ± SE. For these graphs, data from days 8–11 have been reduced to 1 point, as reflected by error bars in the x-axis.
Before the start of the infusion (d 0) and on the day of the GSIS study, fetal arterial plasma was sampled for insulin, IGF-I, NE, cortisol, glucagon, glucose, lactate, and amino acid concentrations; and fetal arterial whole blood was sampled for pH, partial pressure of carbon dioxide (PaCO2), and partial pressure of oxygen (PaO2), hemoglobin-O2 saturation (SaO2), O2 content, and hematocrit. During the infusion period, fetal arterial plasma was sampled daily for glucose and lactate concentrations. On day 1, every 2–3 days thereafter, and on the day of the GSIS study, fetal arterial blood was sampled for blood gas measurements and plasma insulin concentrations. Maternal arterial blood was sampled before the start of the infusion, every 2–3 days, and on the day of the GSIS study for blood gas measurements, plasma glucose and lactate concentrations.
Insulin secretion studies
A primed, continuous, variable-rate hyperglycemic clamp was used to determine fetal GSIS as previously described (Figure 1B) (21, 27). A continuous transfusion of maternal whole blood into the fetus was started 45 minutes before baseline sampling and maintained for the duration of the study to compensate for blood collection and prevent fetal hypovolemia and/or a fall in fetal hematocrit. The volume of maternal blood transfused was equal to the total amount of fetal blood removed for the GSIS study. All sample times are relative to the start of the fetal glucose infusion (time 0). Baseline plasma glucose and insulin concentrations were determined at −60, −45, −30, and −15 minutes. The hyperglycemic clamp was initiated with a 33% dextrose (wt/vol in saline) bolus (825-mg glucose) into the fetus followed by a variable infusion of 33% dextrose (wt/vol in saline) adjusted to maintain fetal arterial plasma concentration near 45 mg/dL, which elicits 90% maximal insulin concentrations in normal fetal sheep of this breed (26). This dextrose infusion was separate from the chronic infusions and was held constant beginning at minute 45. Fetal arterial plasma samples were collected at 5, 10, 15, 20, 30, 45, 60, 75, 90, and 105 minutes for glucose and insulin concentrations.
To measure glucose-potentiated arginine-stimulated insulin secretion, a bolus of arginine (0.5 mmol/kg estimated fetal weight) in 5 mL of 0.4 mol/L sodium acetate and 0.9% NaCl was injected over 4 minutes into the fetal circulation beginning at 110 minutes. Fetal arterial plasma samples were collected 5, 10, 20, and 30 minutes after the start of the arginine infusion for measurement of insulin and glucagon concentrations. Glucagon was also measured 5 and 20 minutes before the arginine infusion. The chronic insulin, glucose, and saline infusions were continued through the GSIS study and until tissue collection the day after.
In 4 additional animals (Figure 1C), the chronic fetal hyperinsulinemic-euglycemic clamp was performed as described. GSIS was tested with limited blood draws in the absence or presence of combined α- and β-adrenergic receptor blockade with phentolamine (an α-adrenergic antagonist) and propranolol (a β-adrenergic antagonist) infused together. Insulin was sampled during the baseline (−60, −45, −30, and −15) and steady state hyperglycemic period (60, 75, 90, and 105) only. Phentolamine was infused into the fetus at 40 μg min−1 per kg estimated fetal weight−1 after an 870-μg priming dose, and propranolol was infused at 14 μg min−1 per kg estimated fetal weight−1 after a 300-μg priming dose (28–31). GSIS was tested in the absence of combined adrenergic blockade with an infusion of the vehicle (saline) instead of the adrenergic antagonists. Infusion of the adrenergic receptor antagonists or saline was started 45 minutes before the baseline blood sampling and continued throughout the GSIS study. Animals were allowed at least 48 hours between measurements of GSIS. No effects were observed as a result of the order and temporal arrangement of the GSIS studies.
Biochemical analysis
Biochemical analysis was performed as previously described (27). Carbon dioxide, oxygen, pH, and hematocrit were immediately measured on whole blood collected in heparin-coated syringes with the ABL 520 analyzer (Radiometer). Whole blood was collected in EDTA-coated syringes and immediately centrifuged (14 000g) for 3 minutes at 4°C. Glucose and lactate concentrations were measured using the YSI model 2700 select biochemistry analyzer (Yellow Springs Instruments). The remainder of the plasma was stored at −70°C for hormone and amino acid measurements. Insulin concentrations were measured by ELISA (ALPCO Immunoassays; intra- and interassay coefficients of variation, 5.6% and 4.7%, respectively; sensitivity, 0.14 ng/mL). Cortisol concentrations were measured by ELISA (ALPCO Immunoassays; intra- and interassay coefficients of variation, 4.6% and 5.8%, respectively; sensitivity, 1.0 ng/mL). IGF-I concentrations were measured by ELISA (ALPCO Immunoassays; intra- and interassay coefficients of variation, 3.1% and 5.6%, respectively; sensitivity, 0.09 ng/mL). Glucagon concentrations were measured by RIA (intra- and interassay coefficients of variation, 4.8% and 11.7%; sensitivity, 18.5 pg/mL; Millipore). NE concentrations were measured using the following methods. After addition of the internal standard (3,4-dihydroxynorephedrine), plasma NE was purified using the diphenylborate extraction method. The extracted NE was reacted with benzylamine to form a fluorescent derivative. NE and the internal standard derivatives were separated on high performance liquid chromatography using a reverse-phase column (XBridge C-18; Waters) and the Alliance high performance liquid chromatography system equipped with a fluorescence detector (model 2475; Waters). A standard curve was constructed using varying amounts of NE and a constant amount of 3,4-dihydroxynorephedrine. The ratio of the areas of NE to 3,4-dihydroxynorephedrine peaks was used to determine the amount of NE in plasma (intra- and interassay coefficients of variation, 9.2% and 9.0%, respectively; sensitivity, 170 pg/mL). Amino acid concentrations were measured using a Dionex 300 model 4500 amino acid analyzer (Dionex).
Tissue collection
Tissue was collected 18–20 hours after the insulin secretion studies. Maternal sheep and their fetuses were euthanized (iv pentobarbital, 4680 and 940 mg given to the mother and fetus, respectively) and tissues collected. The fetus was removed, blotted dry, and weighed as previously described. The fetal pancreas was dissected free, weighed, and divided (19, 32). The hepatic portion of the pancreas was snap frozen in liquid nitrogen and stored at −80°C for subsequent analysis. The splenic portion of the pancreas was fixed in 4% (wt/vol) paraformaldehyde in PBS overnight, equilibrated with 30% sucrose (wt/vol), and embedded in Optimal Cutting Temperature Freeze Media at −80°C, as previously described (32).
Analysis of the fetal pancreas
Total RNA was extracted from pulverized pancreas (100 mg), reverse transcribed into cDNA, and real-time quantitative PCR assays for insulin, glucagon, somatostatin, pancreatic polypepetide, pancreatic and duodenal homeobox 1 (PDX-1), GLUT2, Glucokinase, IGF-I, IGF-II, fibroblast growth factor 7 (FGF7), and FGF10 were performed as previously described (33, 34). Samples were analyzed in triplicate for each gene, and the standard curve method of relative quantification was used (35). Genes of interest were normalized to the housekeeping gene S15, which was not different between groups. Results are presented in arbitrary units as fold change relative to the CON group. Pancreatic insulin content was measured as previously described and is expressed as micrograms of insulin per gram of pancreas (wet weight) (33).
Protein was extracted from pulverized pancreas, and 20 μg of protein were separated by PAGE under reduced conditions (5% β-mercaptoethanol). Proteins were then transferred to a polyvinylidene difluoride membrane (Bio-Rad), and Western blotting was performed as previously described (27). All membranes were blocked for 1 hour in PBS with 0.01% Tween 20 (PBST) (Bio-Rad) and 5% nonfat dried milk (wt/vol) for 1 hour at room temperature. The following primary antibodies (Supplemental Table 1) were diluted in PBST with 5% bovine serum albumin (wt/vol): glucokinase (1:1000; Abcam), glucose transporter 2 (GLUT2) (1:1000; Santa Cruz Biotechnology, Inc), IGF-I (1:1000; Abcam), p70 ribosomal protein S6 kinase (1:500; Cell Signaling), and actin (1:20 000; MP Biomedicals). Horseradish peroxidase-conjugated secondary antibodies were diluted in PBST with 5% nonfat dried milk and applied to membranes for 1 hour at room temperature and then washed. Immunocomplexes were detected with enhanced chemiluminescence (Amersham ECL Plus). Densitometry was performed using Scion Image software (Scion Corp). All results were normalized to β-actin to control for loading differences and are expressed relative to the CON group. Antibodies were stripped from the membranes with Restore Western Stripping buffer (Pierce).
Histology of the fetal pancreas
Morphology of the fetal pancreas was analyzed and quantified by an individual blinded to animal treatment group assignments, as previously described (19, 36). Tissue sections were cut at a minimum of 70-μm intervals. Sections were heated to 37°C for 30 minutes and rehydrated with water. Antigen retrieval was performed by microwaving sections in 10 mmol/L citric acid buffer (pH 6.0) to a temperature of 85°C–95°C for 20 minutes, as well as a 10-minute room temperature incubation in Triton X-100 (0.1%, vol/vol) in PBS.
β- and α-Cell area and mass
Sections were blocked with 1.5% normal donkey serum in PBS (vol/vol) for 30 minutes. Guinea pig antiporcine insulin (1:250; Dako), mouse monoclonal antihuman glucagon, (1:500; Sigma), rabbit antihuman somatostatin (1:500; Dako), and rabbit antihuman pancreatic polypeptide (1:500; Dako) were diluted in blocking buffer and sections incubated at 4°C overnight with these primary antibodies (Supplemental Table 1). The day after, immunocomplexes were detected with affinity-purified secondary antiserum (Jackson ImmunoResearch): amino methyl coumarin antiguinea pig IgG (blue), Alexa Flour 594 goat antimouse IgG (red), and cyanine 2 goat antirabbit IgG (green), all diluted 1:500 in blocking buffer for 60 minutes at room temperature. β- and α-Cell area and mass were quantified as previously described (19, 36). Four pancreatic sections were used for each animal. Insulin+, glucagon+, and pancreatic polypeptide+ or somatostatin+ areas were determined for 20 fields of view on 4 pancreatic sections for each animal. Fields of view were selected randomly to represent all portions of the pancreatic section (approximately 1 × 0.75 cm).
Statistical analysis
The statistical analysis was performed using SAS v.9.1 (SAS Institute). Results are expressed as mean ± SE. For repeated measurements, a mixed-model ANOVA was performed to determine effects of treatment group (INS or CON), time, and treatment-time interactions. A term was included to account for repeated measurements made in the same animal, as well as the presence or absence of adrenergic blockade when appropriate. When the overall ANOVA was significant (P < .05), post hoc test comparisons were made using Fisher's protected least squares difference. Measurements made once were compared by Student's t test or Mann-Whitney U test (for nonparametric data, based on and F-Test for heterogeneity of variability). P < .05 was considered significant.
Results
Maternal parameters
Maternal weight before surgery (55.7 ± 5.8 kg INS, 54.2 ± 9.9 kg CON) was not different between groups. Feed (1.57 ± 0.08 kg/d, INS 1.46 ± 0.06 kg/d CON) and water intake (5.34 ± 0.38 L/d INS, 4.29 ± 0.39 L/d CON) during the infusion period also were not different between INS and CON. Maternal glucose, lactate, pH, PaCO2, PaO2, blood O2 content, and SaO2 at the end of the chronic infusion were similar between groups (Table 1). Maternal hematocrit was lower in the INS group but also started lower before the experimental infusions.
Table 1.
Maternal and Fetal Arterial Glucose, Acid Base Balance, Blood Gases, and Hormones
| CON | INS | |
|---|---|---|
| Maternal | ||
| Glucose, mg/dL | 70.0 ± 3.3 | 72.6 ± 2.7 |
| pH | 7.44 ± 0.01 | 7.46 ± 0.01 |
| PaCO2, mm Hg | 34.6 ± 0.8 | 34.3 ± 0.6 |
| Lactate, mmol/L | 0.66 ± 0.05 | 1.05 ± 0.16 |
| PaO2, mm Hg | 90.2 ± 1.0 | 91 ± 2.2 |
| Hemoglobin-SaO2 % | 96.3 ± 0.8 | 97.5 ± 0.5 |
| O2 content, mmol/L | 5.9 ± 0.1 | 5.5 ± 0.2 |
| Hct, % | 31.7 ± 0.8 | 28.9 ± 0.8a |
| Fetal | ||
| pH | 7.34 ± 0.00 | 7.34 ± 0.01 |
| PaCO2, mm Hg | 51.8 ± 1.2 | 55.3 ± 0.9 |
| Lactate, mmol/L | 2.0 ± 0.12 | 1.7 ± 0.11 |
| Hct, % | 35.4 ± 1.0 | 35.0 ± 2.6 |
| IGF-I, ng/mL | 116.6 ± 10.5 | 151.5 ± 12.6a |
| Cortisol, ng/mL | 9.08 ± 1.13 | 9.38 ± 2.76 |
| NE, pg/mL | 522 ± 106 | 1160 ± 438b |
| Glucagon, pg/mL | 44.34 ± 26.91 | 42.84 ± 6.18 |
Abbreviations; Hct, hematocrit; IGF1, insulin like growth factor 1.
All values are mean ± SE and were obtained at the end of the chronic experimental infusion.
P < .05 CON (n = 9) vs INS (hyperinsulinemic, n = 8) by mixed models ANOVA.
P < .005 CON (n = 9) vs INS (hyperinsulinemic, n = 8) by mixed models ANOVA.
Fetal metabolites, blood gases, and hormones during 7–11 days of INS
Fetal arterial plasma glucose concentrations in the INS group were not statistically different from CON (Figure 2D). Fetal arterial pH, PaCO2, lactate, and hematocrit did not change over time and were similar between groups (Table 1). PaO2, blood O2 content, and SaO2 were lower in the INS group (P < .05) (Figure 3).
Figure 3. Fetal oxygenation.
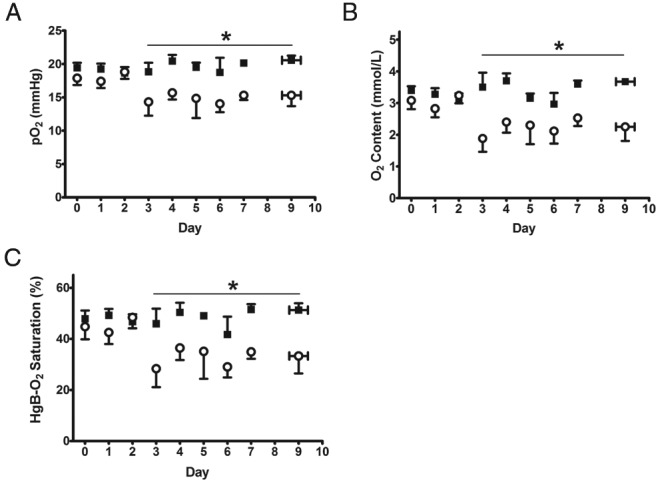
Fetal arterial pO2 (A), O2 content (B), and SaO2 (C) are shown as mean ± SE. Open circles, INS (n = 8); solid squares, CON (n = 9). *, significant difference between INS and CON, P < .05 by mixed models ANOVA. Values are mean ± SEM. For these graphs data from days 8–11 have been reduced to 1 point, as reflected by error bars in the x-axis.
Fetal arterial plasma cortisol and glucagon concentrations were similar between groups (Table 1). IGF-I and NE concentrations at the end of the experimental infusion were significantly higher in the INS group (P < .05) (Table 1). Several fetal arterial plasma amino acid concentrations were lower in the INS fetuses than in the CON group (Supplemental Table 1).
In vivo insulin secretion after 7–11 days of INS
Acute GSIS was tested at the end of the chronic hyperinsulinemic-euglycemic clamp with a primed, continuous, variable-rate fetal hyperglycemic clamp. During the fetal hyperglycemic clamp, insulin concentrations in the CON group increased over 200% above baseline (P < .0001), but GSIS was absent in the INS group (Figure 4). Glucose concentrations and the acute dextrose infusion rate (9.70 ± 3.43 mg/min·kg INS, 11.66 ± 4.41 mg/min·kg CON) during the clamp were similar between the 2 groups. Arginine-stimulated insulin secretion and arginine-stimulated glucagon secretion were similar between the 2 groups (maximum arterial plasma insulin concentrations 1.33 ± 0.20 ng/mL INS, 1.52 ± 0.06 ng/mL CON; maximum arterial plasma glucagon concentrations 45.4 ± 4.1 pg/mL INS, 63.6 ± 4.1 pg/mL CON).
Figure 4. Fetal insulin secretion at the end of the treatment period.
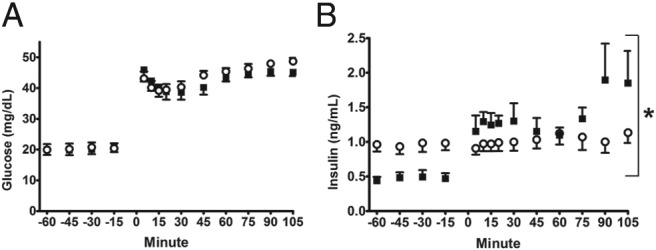
Mean ± SE fetal arterial plasma glucose (A) and insulin (B) concentrations are shown relative to the start of the primed continuous variable-rate hyperglycemic clamp (time, 0 min). Open circles, INS (n = 8); solid squares, CON (n = 9). *, significant difference between INS and CON, P < .05 by mixed models ANOVA.
In order to determine whether increased fetal adrenergic signaling was responsible for decreased GSIS in the INS group, we performed the acute primed, continuous, variable-rate fetal hyperglycemic clamp in a subset of INS fetuses with and without acute pharmacological adrenergic inhibition (Figure 1C). GSIS was restored in the hyperinsulinemic fetuses that received adrenergic blockade during the hyperglycemic clamp (117% increase from baseline, P < .05) (Figure 5). Glucose concentrations, pH, and blood gases were not different in these fetuses compared with the original INS group and did not change with adrenergic blockade (data not shown).
Figure 5. GSIS is restored with adrenergic blockade in INS fetuses.
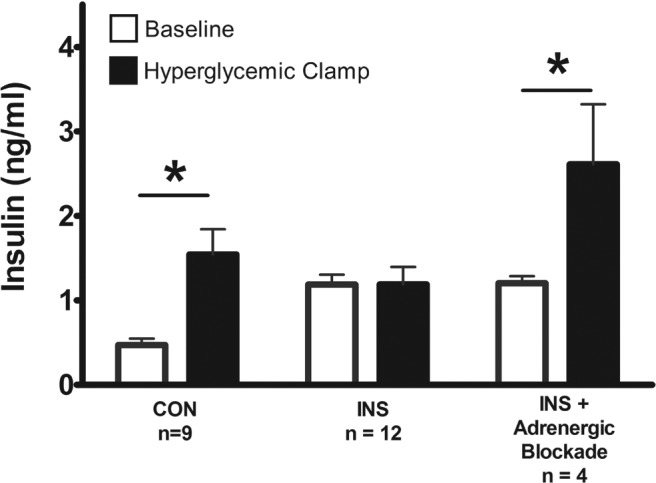
Mean ± SE baseline and hyperglycemic clamp fetal insulin concentrations during the primed continuous variable-rate hyperglycemic clamp are shown for CON fetuses and for INS fetuses with and without adrenergic blockade. *, significant difference between baseline and hyperglycemic clamp insulin concentrations, P < .05 by mixed models ANOVA.
Characteristics of the fetal pancreas after 7–11 days of INS
Fetal pancreatic β-cell area and mass were not different between the INS and CON fetuses (Table 2). Pancreatic insulin and glucokinase mRNA were significantly lower in INS fetuses, as were pancreatic insulin and glucokinase protein (P < .05) (Table 2 and Figure 6). Protein expression of pancreatic GLUT2 and IGF-I were significantly higher in INS fetuses (P < .05) (Figure 6). However, pancreatic PDX-1, GLUT2, glucagon, somatostatin, pancreatic polypeptide, IGF-I, IGF-II, FGF7, and FGF10 were not different (Table 2). α-Cell mass and area, as well as the somatostatin+/pancreatic polypeptide+ area, were also similar between groups (Table 2).
Table 2.
Characteristics of the Fetal Pancreas
| CON | INS | |
|---|---|---|
| β-Cell mass, mg | 62 ± 11 | 53 ± 5 |
| Insulin-positive area, % | 2.13 ± 0.22 | 2.00 ± 0.20 |
| α-Cell mass, mg | 17 ± 3 | 15 ± 1 |
| Glucagon-positive area, % | 0.59 ± 0.04 | 0.57 ± 0.04 |
| Pancreatic insulin content (μg/gm) | 264.6 ± 33.8 | 167.3 ± 27.6a |
| Insulin mRNA (ratio) | 1.00 ± 0.22 | 0.47 ± 0.11a |
| Glucagon mRNA (ratio) | 1.00 ± 0.11 | 0.74 ± 0.16 |
| Somatostatin mRNA (ratio) | 1.00 ± 0.15 | 0.87 ± 0.19 |
| Pancreatic polypeptide mRNA (ratio) | 1.00 ± 0.14 | 1.00 ± 0.32 |
| Glucokinase mRNA (ratio) | 1.00 ± 0.06 | 0.50 ± 0.08a |
| GLUT2 mRNA (ratio) | 1.00 ± 0.09 | 1.07 ± 0.10 |
| PDX-1 mRNA (ratio) | 1.00 ± 0.19 | 0.95 ± 0.12 |
| IGF-I mRNA (ratio) | 1.00 ± 0.09 | 1.10 ± 0.12 |
| IGF-II mRNA (ratio) | 1.00 ± 0.09 | 1.44 ± 0.22 |
| FGF-7 mRNA (ratio) | 1.00 ± 0.14 | 1.29 ± 0.39 |
| FGF-10 mRNA (ratio) | 1.00 ± 0.04 | 1.01 ± 0.09 |
All values are mean ± SE.
P < .05 CON (n = 9) vs INS (hyperinsulinemic, n = 8) by Student's t test or Mann-Whitney test.
Figure 6. Glucokinase is lower and GLUT2 and IGF-I are higher in INS fetal pancreases.
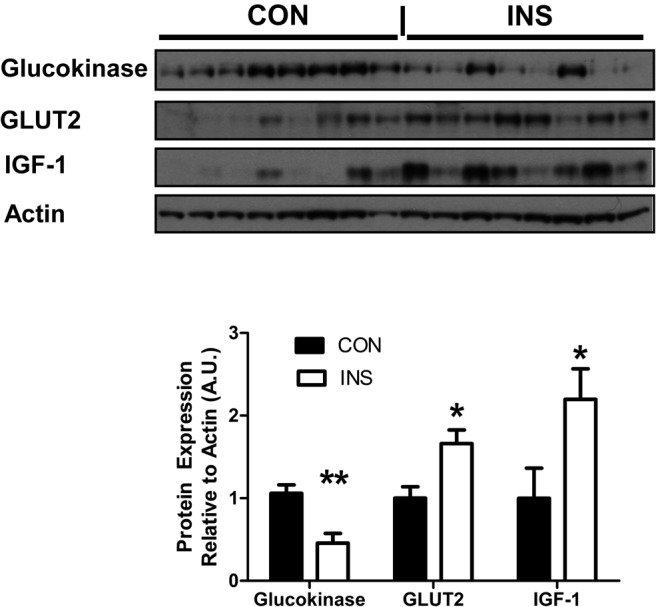
Glucokinase, GLUT2, and IGF-I were measured by Western blotting in fetal pancreases in CON (n = 8) and INS (n = 8) groups. Western blottings are shown with densitometry quantification. There was significantly less (**, P < .01) glucokinase and more (*, P < .05) GLUT2 and IGF-I in INS pancreases. IGF-1, insulin like growth factor 1.
Fetal and organ weights after 7–11 days of INS
Fetal gestational age, weights, organ weights, lengths, and placental weights at the end of the study were the similar between CON and INS fetuses (Supplemental Table 1). Fetal organ weights relative to total fetal weight also were not different between groups (data not shown).
Discussion
In the present study, we show that a chronic (7–11 d; approximately 125–134 dGA) and continuous fetal hyperinsulinemic-euglycemic clamp suppresses fetal GSIS despite normal β-cell mass. Furthermore, we found decreased oxygen concentrations, along with increased NE concentrations in the hyperinsulinemic fetuses, raising the possibility that increased adrenergic signaling may be responsible for decreased GSIS in the INS fetuses. Because of this possibility, we then showed that acute pharmacological adrenergic blockade restored insulin secretion in the INS fetuses, demonstrating that increased adrenergic signaling is, indeed, the mechanism responsible for decreased GSIS.
The relationship between increased adrenergic signaling and decreased fetal insulin secretion has been demonstrated previously. Acute experimental fetal hypoxemia in pregnant sheep decreased fetal GSIS and increased fetal catecholamine concentrations. After α-adrenergic blockade, however, GSIS was increased, similar to our results (29, 37). Similar results were found in fetal sheep under normal and hypoxemic conditions after surgical removal of the fetal adrenal medulla. In normal fetuses, hypoxemia inhibited GSIS, but in the demedullated fetuses, catecholamine concentrations did not increase during hypoxemia, and GSIS was unaffected by oxygen concentrations (24). Other fetal sheep models with chronic hypoxemia and associated increased NE concentrations also have decreased GSIS (38, 39). Furthermore, GSIS was tested with and without adrenergic blockade in a model of severe placental insufficiency characterized by fetuses with higher NE, lower oxygen, and lower GSIS. After acute adrenergic blockade, GSIS was fully restored, demonstrating the importance of adrenergic signaling for suppressing insulin secretion during chronic fetal hypoxemia (30). Our current results confirm these previous studies and show that adrenergic inhibition of GSIS occurs in the setting of euglycemia with hyperinsulinemia.
Experimental fetal hyperinsulinemia, independent of fetal glucose concentrations, has previously been associated with the development of fetal hypoxia after acute (h) and chronic (d) insulin infusion, with 2 proposed mechanisms, one being increased fetal oxygen consumption and the other decreased umbilical blood flow with decreased placental oxygen transfer (40–46). Fetal oxygen consumption is fairly constant (40, 41, 43, 45, 46), but there are conflicting results with respect to the effects of hyperinsulinemia on umbilical blood flow, with most acute studies finding no difference (40, 41, 43, 46). Studies are currently underway using the same fetal hyperinsulinemic-euglycemic clamp to test the effects on umbilical blood flow, fetal oxygen consumption, and placental oxygen consumption and transfer to the fetus. Regardless of the mechanism, the results reported here support the possibility that fetal hypoxemia in fetuses of diabetic mothers may partly be due to direct effects of increased fetal insulin concentrations (47).
Although the current experiments with acute adrenergic blockade show the importance of adrenergic signaling for suppressing GSIS after a chronic fetal hyperinsulinemic-euglycemic clamp, we also measured other factors that have the potential to inhibit GSIS. Neither cortisol nor glucagon concentrations were different between groups and therefore cannot explain decreased GSIS. Many of the fetal arterial plasma amino acid concentrations were lower in the INS group, a result seen in several other experimental fetal insulin clamps of varying duration (41, 45). This is likely due to elevated insulin concentrations and not to the associated increase in catecholamines or decrease in oxygen, both of which tend to increase fetal amino acid concentrations (48–50). Amino acids, especially arginine, lysine, and the branched chain amino acids (leucine, isoleucine, and valine) stimulate fetal insulin secretion (21, 25, 40), and therefore, decreased concentrations of amino acids in the INS group may have contributed to decreased GSIS. The branched chain amino acids, especially leucine, also stimulate β-cell replication (51), although the lower concentrations of the branched chain amino acids did not negatively impact β-cell mass in the INS fetuses as might have been expected. The fetal hyperinsulinemic-euglycemic clamp also produced a significant increase in circulating and pancreatic fetal IGF-I concentrations. In adults, IGF-I is a potent inhibitor of GSIS; thus, its increase in the INS fetuses also might have contributed to decreased insulin secretion, augmenting the inhibitory effect on GSIS of the increased adrenergic signaling (52, 53).
We also measured fetal pancreatic β-cell area and mass and found no differences between the 2 groups to explain decreased insulin secretion in the INS group. Consistent with these results pancreatic mRNA expression of IGFs and FGFs implicated in the regulation of β-cell mass were not changed, although fetal pancreatic IGF-I was higher in the INS group. PDX-1 mRNA was not different between groups, but pancreatic glucokinase and insulin were lower in the INS group. However, the functional significance of decreased pancreatic insulin and glucokinase for GSIS appears to be minimal, because acute adrenergic blockade is sufficient to restore glucose responsiveness in the INS group. Pancreatic GLUT2 was higher in the INS group, which also would not be expected to result in suppression of GSIS. We did not find any maternal changes that could explain decreased fetal insulin secretion or fetal hypoxia in the INS group. Although there was a lower maternal hematocrit in INS group, maternal oxygen content and PaO2 were not significantly different and therefore unlikely to have been responsible for the fetal hypoxemia.
Several limitations of our study should be acknowledged. First, although not statistically significant, INS fetuses had lower plasma arterial glucose concentrations on days 3 and 4 of the experiment. However, by day 5 and for the duration of the study, glucose concentrations were equivalent, and therefore, negative effects of the transient hypoglycemia were likely mitigated (54). A second limitation is that the variability in the length of experimental infusions might have introduced variability in our results. However, the gestational age at time of study was tightly controlled, and we found no differences in GSIS within the INS group based on length of infusion. A third limitation of our study is that our experimental design was not able to distinguish the relative negative impact of hyperinsulinemia from hypoxemia on increased NE concentrations and decreased insulin secretion in the INS group.
In conclusion, we found that a chronic (7–11 d; approximately 125–134 dGA) fetal hyperinsulinemic-euglycemic clamp is associated with the development of fetal hypoxemia, increased catecholamine release, and decreased GSIS. This suppression of GSIS was reversed by adrenergic blockade, implicating increased NE and adrenergic signaling as the mechanism responsible for decreased GSIS in the hyperinsulinemic fetus. These adverse effects on insulin secretion were independent of any change in fetal glucose concentrations or β-cell mass. These data provide further evidence for the role of adrenergic signaling in suppressing fetal insulin secretion in pregnancies complicated by fetal hypoxia.
Acknowledgments
This work was supported by a Pilot and Feasibility Award from the Diabetes and Endocrinology Research Center, University of Colorado (NIH Grant P30DK57516) as well as NIH Grants R01DK088139 and K08HD060688 (to P.J.R., Principal Investigator [PI]). L.D.B. was supported by National Institutes of Health Building Interdisciplinary Careers in Women's Health Scholar Award K12 HD057022 (L.D.B., PI) and Children's Hospital Colorado Research Institute. S.R.T. was supported by the National Institutes of Health Grant K01-DK090199. W.W.H. was supported by NIH Grants T32007186-32 (PI, PD), K12HD068372 (Program Director), and NIH-NCATS UL1TR001082, TL1TR001081, and KL2TR001080 (Co-Director), and a Grand Challenges Exploration Grant from the Bill and Melinda Gates Foundation (OPP1061082). Histological services were provided by the University of Colorado Cancer Center (P30CA046934).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CON
- control
- dGA
- days of gestation
- FGF7
- fibroblast growth factor 7
- GLUT2
- glucose transporter 2
- GSIS
- glucose-stimulated insulin secretion
- INS
- insulin infusion
- NE
- norepinephrine
- PaCO2
- partial pressure of carbon dioxide
- PaO2
- partial pressure of oxygen
- PBST
- PBS with 0.01% Tween 20
- PDX-1
- pancreatic and duodenal homeobox 1
- SaO2
- O2 saturation.
References
- 1. Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic β cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96(3):329–339. [DOI] [PubMed] [Google Scholar]
- 2. Beith JL, Alejandro EU, Johnson JD. Insulin stimulates primary β-cell proliferation via Raf-1 kinase. Endocrinology. 2008;149(5):2251–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Johnson JD, Bernal-Mizrachi E, Alejandro EU, et al. Insulin protects islets from apoptosis via Pdx1 and specific changes in the human islet proteome. Proc Natl Acad Sci USA. 2006;103(51):19575–19580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muller D, Jones PM, Persaud SJ. Autocrine anti-apoptotic and proliferative effects of insulin in pancreatic β-cells. FEBS Lett. 2006;580(30):6977–6980. [DOI] [PubMed] [Google Scholar]
- 5. Aspinwall CA, Lakey JR, Kennedy RT. Insulin-stimulated insulin secretion in single pancreatic β cells. J Biol Chem. 1999;274(10):6360–6365. [DOI] [PubMed] [Google Scholar]
- 6. Luciani DS, Johnson JD. Acute effects of insulin on β-cells from transplantable human islets. Mol Cell Endocrinol. 2005;241(1–2):88–98. [DOI] [PubMed] [Google Scholar]
- 7. Anderwald C, Tura A, Grassi A, et al. Insulin infusion during normoglycemia modulates insulin secretion according to whole-body insulin sensitivity. Diabetes Care. 2011;34(2):437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bouche C, Lopez X, Fleischman A, et al. Insulin enhances glucose-stimulated insulin secretion in healthy humans. Proc Natl Acad Sci USA. 2010;107(10):4770–4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Halperin F, Lopez X, Manning R, Kahn CR, Kulkarni RN, Goldfine AB. Insulin augmentation of glucose-stimulated insulin secretion is impaired in insulin-resistant humans. Diabetes. 2012;61(2):301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lopez X, Cypess A, Manning R, O'Shea S, Kulkarni RN, Goldfine AB. Exogenous insulin enhances glucose-stimulated insulin response in healthy humans independent of changes in free fatty acids. J Clin Endocrin Metab. 2011;96(12):3811–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gatford KL, Simmons RA. Prenatal programming of insulin secretion in intrauterine growth restriction. Clin Obstet Gynecol. 2013;56(3):520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Holemans K, Aerts L, Van Assche FA. Lifetime consequences of abnormal fetal pancreatic development. J Physiol. 2003;547(pt 1):11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nicolini U, Hubinont C, Santolaya J, Fisk NM, Rodeck CH. Effects of fetal intravenous glucose challenge in normal and growth retarded fetuses. Horm Metab Res. 1990;22(8):426–430. [DOI] [PubMed] [Google Scholar]
- 14. Reusens B, Remacle C. Programming of the endocrine pancreas by the early nutritional environment. Int J Biochem Cell Biol. 2006;38(5–6):913–922. [DOI] [PubMed] [Google Scholar]
- 15. Carver TD, Anderson SM, Aldoretta PW, Hay WW., Jr Effect of low-level basal plus marked “pulsatile” hyperglycemia on insulin secretion in fetal sheep. Am J Physiol. 1996;271(5 pt 1):E865–E871. [DOI] [PubMed] [Google Scholar]
- 16. Fetita LS, Sobngwi E, Serradas P, Calvo F, Gautier JF. Consequences of fetal exposure to maternal diabetes in offspring. J Clin Endocrin Metab. 2006;91(10):3718–3724. [DOI] [PubMed] [Google Scholar]
- 17. Frost MS, Zehri AH, Limesand SW, Hay WW, Jr, Rozance PJ. Differential effects of chronic pulsatile versus chronic constant maternal hyperglycemia on fetal pancreatic β-cells. J Pregnancy. 2012;2012:812094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Green AS, Rozance PJ, Limesand SW. Consequences of a compromised intrauterine environment on islet function. J Endocrinol. 2010;205(3):211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Limesand SW, Jensen J, Hutton JC, Hay WW., Jr Diminished β-cell replication contributes to reduced β-cell mass in fetal sheep with intrauterine growth restriction. Am J Physiol Regul Integr Comp Physiol. 2005;288(5):R1297–R1305. [DOI] [PubMed] [Google Scholar]
- 20. Limesand SW, Rozance PJ, Macko AR, Anderson MJ, Kelly AC, Hay WW., Jr Reductions in insulin concentrations and β-cell mass precede growth restriction in sheep fetuses with placental insufficiency. Am J Physiol Endocrinol Metab. 2013;304(5):E516–E523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Limesand SW, Rozance PJ, Zerbe GO, Hutton JC, Hay WW., Jr Attenuated insulin release and storage in fetal sheep pancreatic islets with intrauterine growth restriction. Endocrinology. 2006;147(3):1488–1497. [DOI] [PubMed] [Google Scholar]
- 22. Chen X, Green AS, Macko AR, Yates DT, Kelly AC, Limesand SW. Enhanced insulin secretion responsiveness and islet adrenergic desensitization after chronic norepinephrine suppression is discontinued in fetal sheep. Am J Physiol Endocrinol Metab. 2014;306(1):E58–E64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Straub SG, Sharp GW. Evolving insights regarding mechanisms for the inhibition of insulin release by norepinephrine and heterotrimeric G proteins. Am J Physiol Cell Physiol. 2012;302(12):C1687–C1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yates DT, Macko AR, Chen X, et al. Hypoxaemia-induced catecholamine secretion from adrenal chromaffin cells inhibits glucose-stimulated hyperinsulinaemia in fetal sheep. J Physiol. 2012;590(pt 21):5439–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rozance PJ, Limesand SW, Hay WW., Jr Decreased nutrient-stimulated insulin secretion in chronically hypoglycemic late-gestation fetal sheep is due to an intrinsic islet defect. Am J Physiol Endocrinol Metab. 2006;291(2):E404–E411. [DOI] [PubMed] [Google Scholar]
- 26. Green AS, Macko AR, Rozance PJ, et al. Characterization of glucose-insulin responsiveness and impact of fetal number and sex difference on insulin response in the sheep fetus. Am J Physiol Endocrinol Metab. 2011;300(5):E817–E823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maliszewski AM, Gadhia MM, O'Meara MC, Thorn SR, Rozance PJ, Brown LD. Prolonged infusion of amino acids increases leucine oxidation in fetal sheep. Am J Physiol Endocrinol Metab. 2012;302(12):E1483–E1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Danielson L, McMillen IC, Dyer JL, Morrison JL. Restriction of placental growth results in greater hypotensive response to α-adrenergic blockade in fetal sheep during late gestation. J Physiol. 2005;563(pt 2):611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jackson BT, Cohn HE, Morrison SH, Baker RM, Piasecki GJ. Hypoxia-induced sympathetic inhibition of the fetal plasma insulin response to hyperglycemia. Diabetes. 1993;42(11):1621–1625. [DOI] [PubMed] [Google Scholar]
- 30. Leos RA, Anderson MJ, Chen X, Pugmire J, Anderson KA, Limesand SW. Chronic exposure to elevated norepinephrine suppresses insulin secretion in fetal sheep with placental insufficiency and intrauterine growth restriction. Am J Physiol Endocrinol Metab. 2010;298(4):E770–E778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sperling MA, Christensen RA, Ganguli S, Anand R. Adrenergic modulation of pancreatic hormone secretion in utero: studies in fetal sheep. Pediatr Res. 1980;14(3):203–208. [DOI] [PubMed] [Google Scholar]
- 32. Cole L, Anderson M, Antin PB, Limesand SW. One process for pancreatic β-cell coalescence into islets involves an epithelial-mesenchymal transition. J Endocrinol. 2009;203(1):19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gadhia MM, Maliszewski AM, O'Meara MC, et al. Increased amino acid supply potentiates glucose-stimulated insulin secretion but does not increase β-cell mass in fetal sheep. Am J Physiol Endocrinol Metab. 2013;304(4):E352–E362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen X, Rozance PJ, Hay WW, Jr, Limesand SW. Insulin-like growth factor and fibroblast growth factor expression profiles in growth-restricted fetal sheep pancreas. Exp Biol Med (Maywood). 2012;237(5):524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wong ML, Medrano JF. Real-time PCR for mRNA quantitation. BioTechniques. 2005;39(1):75–85. [DOI] [PubMed] [Google Scholar]
- 36. Rozance PJ, Limesand SW, Zerbe GO, Hay WW., Jr Chronic fetal hypoglycemia inhibits the later steps of stimulus-secretion coupling in pancreatic β-cells. Am J Physiol Endocrinol Metab. 2007;292(5):E1256–E1264. [DOI] [PubMed] [Google Scholar]
- 37. Jackson BT, Piasecki GJ, Cohn HE, Cohen WR. Control of fetal insulin secretion. Am J Physiol Regul Integr Comp Physiol. 2000;279(6):R2179–R2188. [DOI] [PubMed] [Google Scholar]
- 38. Owens JA, Gatford KL, De Blasio MJ, Edwards LJ, McMillen IC, Fowden AL. Restriction of placental growth in sheep impairs insulin secretion but not sensitivity before birth. J Physiol. 2007;584(pt 3):935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Simonetta G, Rourke AK, Owens JA, Robinson JS, McMillen IC. Impact of placental restriction on the development of the sympathoadrenal system. Pediatr Res. 1997;42(6):805–811. [DOI] [PubMed] [Google Scholar]
- 40. Brown LD, Rozance PJ, Barry JS, Friedman JE, Hay WW., Jr Insulin is required for amino acid stimulation of dual pathways for translational control in skeletal muscle in the late-gestation ovine fetus. Am J Physiol Endocrinol Metab. 2009;296(1):E56–E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown LD, Rozance PJ, Thorn SR, Friedman JE, Hay WW., Jr Acute supplementation of amino acids increases net protein accretion in IUGR fetal sheep. Am J Physiol Endocrinol Metab. 2012;303(3):E352–E364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carson BS, Philipps AF, Simmons MA, Battaglia FC, Meschia G. Effects of a sustained insulin infusion upon glucose uptake and oxygenation of the ovine fetus. Pediatr Res. 1980;14(2):147–152. [DOI] [PubMed] [Google Scholar]
- 43. Hay WW, Jr, DiGiacomo JE, Meznarich HK, Hirst K, Zerbe G. Effects of glucose and insulin on fetal glucose oxidation and oxygen consumption. Am J Physiol. 1989;256(6 pt 1):E704–E713. [DOI] [PubMed] [Google Scholar]
- 44. Lavezzi JR, Thorn SR, O'Meara MC, et al. Increased fetal insulin concentrations for one week fail to improve insulin secretion or -cell mass in fetal sheep with chronically reduced glucose supply. Am J Physiol Regul Integr Comp Physiol. 2012;304(1):R50–R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Milley JR, Rosenberg AA, Philipps AF, Molteni RA, Jones MD, Jr, Simmons MA. The effect of insulin on ovine fetal oxygen extraction. Am J Obstet Gynecol. 1984;149(6):673–678. [DOI] [PubMed] [Google Scholar]
- 46. Simmons MA, Jones MD, Jr, Battaglia FC, Meschia G. Insulin effect on fetal glucose utilization. Pediatr Res. 1978;12(2):90–92. [DOI] [PubMed] [Google Scholar]
- 47. Taricco E, Radaelli T, Rossi G, et al. Effects of gestational diabetes on fetal oxygen and glucose levels in vivo. BJOG. 2009;116:1729–1735. [DOI] [PubMed] [Google Scholar]
- 48. Jones CT, Ritchie JW. The metabolic and endocrine effects of circulating catecholamines in fetal sheep. J Physiol. 1978;285:395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Milley JR. Ovine fetal metabolism during norepinephrine infusion. Am J Physiol. 1997;273(2 pt 1):E336–E347. [DOI] [PubMed] [Google Scholar]
- 50. Milley JR. Ovine fetal leucine kinetics and protein metabolism during decreased oxygen availability. Am J Physiol. 1998;274(4 pt 1):E618–E626. [DOI] [PubMed] [Google Scholar]
- 51. McDaniel ML, Marshall CA, Pappan KL, Kwon G. Metabolic and autocrine regulation of the mammalian target of rapamycin by pancreatic β-cells. Diabetes. 2002;51(10):2877–2885. [DOI] [PubMed] [Google Scholar]
- 52. Leahy JL, Vandekerkhove KM. Insulin-like growth factor-I at physiological concentrations is a potent inhibitor of insulin secretion. Endocrinology. 1990;126(3):1593–1598. [DOI] [PubMed] [Google Scholar]
- 53. Porksen N, Hussain MA, Bianda TL, et al. IGF-I inhibits burst mass of pulsatile insulin secretion at supraphysiological and low IGF-I infusion rates. Am J Physiol. 1997;272(3 pt 1):E352–E358. [DOI] [PubMed] [Google Scholar]
- 54. Limesand SW, Hay WW., Jr Adaptation of ovine fetal pancreatic insulin secretion to chronic hypoglycaemia and euglycaemic correction. J Physiol. 2003;547:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]