

Abstract
Although classically considered a WNT signaling intermediary, β-catenin (CTNNB1) can also mediate GnRH induction of gonadotropin β-subunit (Fshb and Lhb) transcription in the murine gonadotrope-like cell line LβT2. Here, we assessed CTNNB1's role in gonadotropin synthesis in vivo. We used a Cre/lox approach to introduce both gain- and loss-of-function mutations in the murine Ctnnb1 gene in gonadotrope cells. Gonadotropin production and fertility were normal in Ctnnb1 knockout mice. Similarly, females harboring a deletion of exon 3 of Ctnnb1, which stabilizes the resulting CTNNB1 protein, showed normal fertility and gonadotropin synthesis. Interestingly, males with the activating CTNNB1-Δexon 3 mutation exhibited 50% reductions in FSH synthesis and secretion, without a corresponding change in LH. This selective regulation of FSH suggested an alteration in the activin/inhibin/follistatin system. Indeed, CTNNB1-Δexon 3 males showed a 60% increase in serum inhibin B levels, and in culture, their pituitaries exhibited a greater sensitivity to exogenous inhibin than controls. At the same time, pituitary, but not testicular, follistatin (Fst) expression was increased significantly in these mice. Castration normalized FSH levels in CTNNB1-Δexon 3 males to those seen in castrated controls. Paradoxically, pituitaries from CTNNB1-Δexon 3 males exhibited greater basal and activin-stimulated FSH synthesis in vitro. Similarly, CTNNB1-Δexon 3 overexpression potentiated activin A-induced murine Fshb promoter activity in LβT2 cells. Together, these results indicate that CTNNB1 is dispensable for gonadotropin synthesis in vivo. However, sustained CTNNB1 signaling potentiates activin-induced Fshb expression in gonadotropes, but this effect is overcome in vivo by enhanced inhibin feedback sensitivity and Fst expression.
The gonadotropins, FSH and LH, are heterodimeric glycoprotein hormones secreted by gonadotrope cells of the anterior pituitary gland. Both are essential regulators of male and female reproductive function, controlling such processes as ovarian follicle development, spermatogenesis, and gonadal steroidogenesis (1, 2). The gonadotropins consist of a common α-subunit (CGA) and distinct β-subunits, FSHβ (encoded by the Fshb gene) and LHβ (encoded by Lhb), which confer biological specificity to each hormone (3). The synthesis and secretion of LH and FSH are regulated by a variety of factors derived from all levels of the reproductive axis, notably hypothalamic GnRH, pituitary activins, and gonadal inhibins and sex steroids (3).
A major mechanism whereby different factors modulate FSH and LH release by the pituitary gland is through the control of Fshb and Lhb transcription. The structures of the Fshb and Lhb promoters are distinct, as are the intracellular signaling molecules and transcription factors that modulate their activity in response to various ligands (reviewed in Refs. 3–7). Interestingly, β-catenin (CTNNB1) was recently identified as a regulator of both Fshb and Lhb transcription in response to GnRH (8, 9). CTNNB1 is a multifunctional protein that notably serves as a mediator of the canonical WNT signaling pathway. In this context, CTNNB1 associates (in the resting state) with a multiprotein complex that includes the scaffold proteins adenomatous polyposis coli and AXIN and several kinases, including glycogen synthase kinase 3β (GSK3β). This complex mediates the phosphorylation of CTNNB1, which effectively targets it for subsequent ubiquitination and degradation by the cellular proteasomal machinery. The binding of a WNT ligand to a receptor of the frizzled family results in a signal that inhibits GSK3β, permitting CTNNB1 to escape the complex in a hypophosphorylated state and to accumulate within the cell. After nuclear translocation, CTNNB1 can bind to various transcription factors, modulating target gene expression in a cell type- and context-specific manner (10, 11).
In the murine gonadotrope-like cell line LβT2 (12), GnRH can induce the nuclear accumulation of CTNNB1 in a manner similar to canonical WNTs (9, 13). Unlike WNT signaling, however, the mechanism requires both protein kinase C and c-Jun N-terminal kinase activity rather than inhibition of GSK3β (9, 13). Within the nucleus, CTNNB1 binds to the transcription factor nuclear receptor subfamily 5, group A, member 1 (NR5A1) (steroidogenic factor-1), thereby favoring the interaction of NR5A1 with early growth response 1 (14). NR5A1 and early growth response 1 interact functionally with paired-like homeodomain transcription factor 1 to form a tripartite complex on the Lhb promoter to mediate responsiveness to GnRH (14). A role for CTNNB1 in the regulation of Fshb in LβT2 cells was also recently demonstrated (9), because CTNNB1 knockdown decreases responsiveness of the Fshb promoter to GnRH. Unlike Lhb, however, CTNNB1's regulation of Fshb does not appear to involve a direct transcriptional mechanism. Rather, CTNNB1 regulates nuclear cofactor Brms1L expression, which in turn mediates GnRH induction of Fshb expression (9).
Although these earlier studies suggested a role for CTNNB1 in the regulation of gonadotropin synthesis, several key questions remain unresolved. Notably, previous investigations have been conducted in vitro, principally in immortalized LβT2 cells. Thus, CTNNB1's role in gonadotropes in vivo is unclear. Here, we used a functional genomics approach (Cre/lox system) to either ablate or constitutively activate CTNNB1 signaling in gonadotropes of mice.
Materials and Methods
Animals
Mice bearing the Ctnnb1tm1Mmt, Ctnnb1tm2Kem, Gnrhrtm1(cre)Uboe, and Gt(ROSA)26Sortm1(EYFP)Cos alleles were obtained, housed, and bred, and their genotypes determined as previously described (15–19). Briefly, the Ctnnb1tm1Mmt allele contains LoxP sites flanking the third exon of the Ctnnb1 gene. Cre-mediated recombination results in an allele that encodes a functional CTNNB1 protein (referred to here as Δex3) that lacks a series of N-terminal serine and threonine residues that are required for its phosphorylation and degradation. The Δex3 protein, therefore, functions as a dominant-stable mutant, accumulating in the cell and activating CTNNB1 signaling in a constitutive manner (20). Ctnnb1tm2Kem is a conventional floxed allele (LoxP sites flanking exons 2–6) that becomes null after Cre-mediated recombination (16). Gnrhrtm1(cre)Uboe (referred to here as GnRH receptor internal ribosome entry site Cre [GRIC]) is a knock-in allele that encodes the wild-type Gnrhr transcript, followed by an internal ribosome entry site and the Cre transgene. GRIC drives Cre expression exclusively in pituitary gonadotrope cells, GnRH receptor expressing neurons, and in the male germ line (21, 22). Therefore, the GRIC allele is introduced from the mother in all crosses, except where noted (see below).
For simplicity, experimental mice bearing the Ctnnb1tm1Mmt and Gnrhrtm1(cre)Uboe alleles are designated “Δex3-GRIC” in the text, whereas those bearing Ctnnb1tm2Kem and Gnrhrtm1(cre)Uboe alleles are designated “knockout (KO)-GRIC”. In most experiments, the Δex3-GRIC genotype was Ctnnb1tm1Mmt/+;Gnrhrtm1(cre)Uboe/+, with relevant controls being Ctnnb1tm1Mmt/+. The KO-GRIC genotype was Ctnnb1tm2Kem/tm2Kem;Gnrhrtm1(cre)Uboe/+, with relevant controls being Ctnnb1tm2Kem/tm2Kem. In experiments designed to analyze male germ cells (ie, Supplemental Figures 1 and 2 and Supplemental Table 1), the Δex3-GRIC genotype was Ctnnb1tm1Mmt/tm1Mmt; Gnrhrtm1(cre)Uboe/+, with relevant controls being Ctnnb1tm1Mmt/tm1Mmt. The germline KO-GRIC genotype was Ctnnb1tm2Kem/−;Gnrhrtm1(cre)Uboe/tm1(cre)Uboe, with relevant controls being Ctnnb1tm2Kem/−. The genotypes for the experiments involving male germ cells were chosen due to their genome being haploid after meiosis. For Δex3-GRIC, 2 copies of the Ctnnb1tm1Mmt allele were required to ensure that all haploid germ cells bore the mutant allele. For KO-GRIC, 2 copies of the Gnrhrtm1(cre)Uboe allele were included so as to increase the probability that all haploid germ cells would express Cre, and hence improve knockout efficiency. The latter also bore a Cre-recombined (−) allele, which was the result of their male parent (whose genotype was Ctnnb1tm2Kem/tm2Kem;Gnrhrtm1(cre)Uboe/+) having recombination of the floxed allele in their germ line.
For mating studies, 8-week-old experimental and control male or female mice were individually paired with an adult C57BL/6 mouse of the opposite sex, and fertility was evaluated over a period of 6 months. The presence of newborn mice was monitored daily starting 20 days after pairing. Pups were counted immediately after birth.
Castration was done under isoflurane (Pharmaceutical Partners of Canada) anesthesia. Briefly, after incision along the linea alba, the testes were exteriorized, the vas deferens and blood vessels ligated using absorbable suture material, and the testes excised. Closure of the abdominal wall was done with absorbable sutures and the skin closed with tissue adhesive. Mice were killed for serum hormone analyses 10 days after surgery.
All procedures were approved by the Institutional Animal Care and Use Committees of the Université de Montréal and McGill University, and conformed to the United States Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Cell culture and plasmids
LβT2 cells (12), a gift from Dr Pamela Mellon (University of California, San Diego), were cultured as previously described (eg, Refs. 23, 24). For reporter assays, cells were seeded at 3 × 105 cells per well in 48-well plates 2–3 days before transfection with Lipofectamine 2000 (Invitrogen). GnRH and recombinant human activin A were obtained from Sigma and R&D Systems, respectively. Recombinant human inhibin A was a generous gift from Dr Teresa Woodruff (Northwestern University, Chicago, Illinois). Reporter assays were performed 3 times in triplicate as previously described (25). The murine −1990/+1 Fshb-luc reporter plasmid was described previously (23). The CTNNB1 and CTNNB1-Δex3 expression constructs were generated by PCR amplification of cDNA derived from pooled pituitary cells of adult maleCtnnb1tm1Mmt/+ mice. The primer pair mCtnnb1.Start.A (CGGGATCCTGGACAATGGCTACTCAAGCTG) and mCtnnb1.Stop.B (GCTCTAGACCTAAAGGACGATTTACAGGTC) was used to generate a wild-type CTNNB1 cDNA, and the primer pair mCtnnb1.δEx3.Start.A (CGGGATCCTGGACAATGGCTACTCAAGATATTGACGGGCAGTATGCAATG) and mCtnnb1.Stop.B was used to generate a Δex3 mutant CTNNB1 cDNA. Both cDNA fragments were ligated into the BamHI and XbaI restriction sites of pcDNA3.0 (Invitrogen) using standard recombinant DNA techniques.
For primary culture experiments, pituitaries were extracted from control or Δex3-GRIC males (>8 wk of age), pooled according to genotype, and enzymatically dispersed. Cells were plated in 24-well plates at a final density of 6–7 × 105 cells per well and cultured as described previously (eg, Refs. 26–29). The day after plating, cells were washed and treated overnight in low serum (2%) medium containing vehicle, activin A, or inhibin A at the indicated concentrations. Medium was collected for analysis of secreted hormones (see below), and cellular RNA was collected for quantitative RT-PCR analysis (see below).
Hormone assays
FSH and LH were measured from spent tissue culture media or from serum by the Ligand Assay and Analysis Core of the University of Virginia Center for Research in Reproduction, using the mouse/rat LH/FSH multiplex assay, or by RIA in instances when FSH alone was measured. Serum inhibin B was measured by ELISA and testosterone by RIA by the same facility. For serum collection, mice were killed by CO2 inhalation, and was blood obtained by cardiac puncture. The blood was left to coagulate for 15 minutes at room temperature and then centrifuged at 3000 rpm for 10 minutes for serum separation. Samples for intratesticular testosterone measurement were prepared by homogenizing 1 testis from each animal in 1 mL of PBS using a Kinematica Polytron homogenizer, sonicating for 60 seconds, and centrifuging to remove cell debris. All samples were stored at −80°C before analysis. Limits of detection were 0.24 ng/mL (LH) and 2.4 ng/mL (FSH) for the LH/FSH multiplex, 3.0 ng/mL for the FSH RIA, 10 pg/mL for the inhibin B ELISA, and 10 ng/dL for the testosterone RIA, respectively. Intra- and interassay coefficients of variation were 5.9%/12.5%, 6.9%/7.5%, 4.7%/7.7%, and 4.4%/6.4% for the LH/FSH multiplex, FSH RIA, inhibin B ELISA, and testosterone RIA, respectively.
Reverse transcription/real-time PCR
Mice were killed by CO2 inhalation, and tissues were removed, snap frozen in liquid N2, and preserved at −80°C until further processing. Total RNA was isolated using the RNeasy Micro kit (QIAGEN) with on-column deoxyribonuclease treatment to remove genomic DNA contamination. The RNA samples (1 μg) were analyzed by RT-quantitative PCR as previously described (eg, Refs. 26, 30) using the primers listed in Supplemental Table 2. Samples were assayed in duplicate or triplicate and analyzed using the 2−ΔΔCt method (31). Gene expression was normalized relative to ribosomal protein L19 (Rpl19).
Determination of recombination efficiency in gonadotropes
For gonadotrope purification, adult (>6 wk old) male and female Ctnnb1tm1Mmt/+;Gnrhrtm1(cre)Uboe/+;Gt(ROSA)26Sortm1(EYFP)Cos/+(Δex3-GRIC;yellow fluorescent protein [YFP]) and Ctnnb1tm2Kem/tm2Kem;Gnrhrtm1(cre)Uboe/+;Gt(ROSA)26Sortm1(EYFP)Cos/+(KO-GRIC;YFP) mice were killed by CO2 inhalation. Cells from dissected pituitaries were isolated and sorted by fluorescence-activated cell sorting into YFP-positive (ie, gonadotropes) and YFP-negative (ie, nongonadotropes) fractions as previously described (27). Analysis of recombination efficiency for the Δex3-GRIC model was done by RT-PCR on RNA isolated from male and female YFP-positive and YFP-negative cell populations as previously described (15, 32). Analysis of recombination efficiency for the KO-GRIC model was done by RT-quantitative polymerase chain reaction analysis of Ctnnb1 mRNA levels in YFP-positive and YFP-negative cell populations (male and female mice were pooled) as described above.
Sperm cell genotyping and seminiferous tubule staging analyses
Cauda epididymides were dissected from 2-month-old Δex3-GRIC and KO-GRIC mice, placed in Petri dishes containing PBS warmed to 37°C, and minced to release their contents. After a 15-minute incubation, the sperm suspension was used for DNA extraction and genotype analyses as previously described (15).
Spermatogenesis was evaluated in Δex3-GRIC and KO-GRIC mice by histopathologic analyses, and any potential spermatogenesis arrest was evaluated by quantifying the expression of markers of specific stages of germ cell development by RT-quantitative polymerase chain reaction (as described above and in the legend of Supplemental Figure 2, primers listed in Supplemental Table 2). A histopathologic analysis of germ cell stages was also done in the Δex3-GRIC model. The stage of the spermatogenesis cycle was determined in 50 randomly selected seminiferous tubules per histologic section (n = 6 for Δex3-GRIC and n = 8 for controls; 1 section per animal) by a veterinary pathologist and based on established morphological criteria (33). For simplicity, certain stages with similar morphological characteristics were grouped together (II–VII and X–XI).
Immunohistochemistry
Immunohistochemical analysis of CTNNB1 expression in testes was done on Bouin's-fixed, paraffin-embedded, 7-μm sections using the VectaStain Elite avidin-biotin complex kit (Vector Labs) as we have done previously (15). The primary antibody was obtained from Cell Signaling Technology (number 9587), and staining was done using the 3,3′-diaminobenzidine peroxidase substrate kit (Vector Labs). No staining was apparent in a negative control omitting the primary antibody (data not shown). Sections were lightly counterstained with hematoxylin before mounting.
Statistical methods
Statistical analyses for multiple comparisons were done using one-, two-, or three-way ANOVA followed by the Tukey's post hoc test (see figure legends for details). Student's t test was used for 2-group comparisons. Data were log transformed before statistical testing. Means were considered significantly different when P < .05. All tests were done using Prism software version 6.0d (GraphPad Software, Inc).
Results
CTNNB1 is dispensable for fertility and gonadotropin synthesis
To determine potential roles of CTNNB1 in gonadotropin synthesis and secretion in vivo, we generated both loss- and gain-of-function mutations in the Ctnnb1 gene in murine gonadotropes using a Cre-lox approach. Mice with the loss-of-function mutation (KO-GRIC) showed normal fertility, gonadal weight, and histology, pituitary Fshb and Lhb expression, and serum FSH levels (Table 1, Supplemental Figures 3 and 4, and data not shown) despite a 94% decrease in Ctnnb1 mRNA levels in purified gonadotrope cells of these mice relative to controls (see determination of recombination efficiency in gonadotropes in Materials and Methods). Furthermore, although we also detected efficient recombination of the floxed alleles in spermatozoa of KO-GRIC males (as described above, the GRIC allele is active in the male germline) (Supplemental Figure 1), no alterations in spermatogenesis were detected by analyses of markers of different stages of germ cell development, histological evaluations of seminiferous tubule stages, evaluations of morphological defects in epididymal spermatozoa, or epididymal sperm cell counts (Supplemental Figure 2, Supplemental Table 1, and data not shown). Thus, CTNNB1 does not appear to play a necessary role in pituitary gonadotropin synthesis or in the later phases of male germ cell development or subsequent function. In contrast, we observed a sex-specific effect in mice with the gain-of-function Ctnnb1 mutation, which is the major focus of the present report.
Table 1.
Fertility Trial
| Control (Ctnnb1tm2Kem/tm2Kem) | KO-GRIC | Control (Ctnnb1tm1Mmt/+) | Δex3-GRIC | |
|---|---|---|---|---|
| Female | ||||
| n | 6 | 5 | 6 | 6 |
| Total litters | 37 | 31 | 35 | 33 |
| Total pups | 252 | 207 | 248 | 231 |
| Litter size | 6.81 ± 0.42 | 6.68 ± 0.47 | 7.09 ± 0.43 | 7.00 ± 0.54 |
| Male | ||||
| n | 4 | 4 | 4 | 4 |
| Total litters | 22 | 20 | 23 | 17 |
| Total pups | 128 | 134 | 159 | 50 |
| Litter size | 5.82 ± 0.43 | 6.70 ± 0.44 | 6.91 ± 0.61 | 2.94 ± 0.39a |
Litter size is expressed as average ± SEM.
Different from Ctnnb1tm1Mmt/+ control, P < .05.
A gain-of-function mutation in Ctnnb1 causes a sex-specific impairment in FSH production
To generate a gonadotrope-specific gain-of-function CTNNB1 mutant strain, we used mice harboring a floxed allele, in which LoxP sites flank the third exon of Ctnnb1. Cre-mediated recombination of this allele results in an in-frame deletion removing critical phosphorylation sites that prime the CTNNB1 protein for ubiquitination and degradation, resulting in a dominant-stable mutant. In this model (except where noted), the mice harbor a single floxed allele along with the GRIC Cre-driver allele (hereafter Δex3-GRIC). We also crossed in a YFP reporter allele to enable purification of gonadotropes and assessment of the efficiency of recombination. RT-PCR analysis of Ctnnb1 mRNA from YFP-positive (gonadotrope) cells from Δex3-GRIC;YFP pituitaries produced 2 bands of approximately equal intensity that represent transcripts derived from the wild-type and Cre-recombined alleles, as would be predicted in the event of efficient recombination (Figure 1A). As expected, the YFP-negative (nongonadotrope) cell fractions were largely devoid of the Cre-recombined Ctnnb1 mRNA species. Therefore, recombination of the floxed exon 3 allele was both efficient and specific in this model.
Figure 1. A male specific FSH synthesis defect occurs in the Δex3-GRIC model.

A, Recombination efficiency in Δex3-GRIC gonadotropes. Pituitary cells from male (left) and female (right) Δex3-GRIC;YFP mice were enzymatically dispersed and fluorescence-activated cell sorting sorted into YFP+ (ie, gonadotropes) and YFP− (ie, nongonadotropes) fractions. RT-PCR analysis of Ctnnb1 mRNA expression was then done using a primer pair that flanks the mRNA sequences encoded by the third exon and that, therefore, generates a smaller PCR product (lower band) after Cre-mediated recombination. As expected, YFP+ Δex3-GRIC;YFP pituitary cells produced 2 mRNA species (ie, derived from the wild-type and the Cre-recombined floxed alleles). PCR products were separated by electrophoresis on 1% agarose gels containing ethidium bromide and photographed under UV illumination. B and C, Pituitary gonadotropin subunit gene expression in adult Δex3-GRIC mice. Six-week-old female (n = 5–7 per genotype) and 8- to 12-week-old male (n = 18–20 per genotype) mice were killed and pituitary glands harvested for measurement of Fshb and Lhb expression by real-time RT-PCR. Serum was also collected for FSH (D) and LH (males only) (E) measurement. Intratesticular testosterone levels were measured from testis homogenates prepared from the same animals (F). Analyses shown in B–D were also done using 6-month-old female Δex3-GRIC mice, yielding similar results (data not shown). RT-PCR analyses were done separately on samples from male and female mice; relative expression, therefore, cannot be compared between genders. Columns, means; error bars, SEM. Significant difference from control is indicated with asterisks (**, P < .01; ***, P < .001, unpaired t test).
Like KO-GRIC females, Δex3-GRIC females exhibited normal fertility (Table 1). In contrast, Δex3-GRIC males sired litters approximately half the size of those produced by controls. This result was not unexpected, however, because the GRIC allele also drives Cre expression in male germ cells (21) and transmission of the Cre-recombined Δex3 allele to progeny through the germ line is embryonically lethal (34). Indeed, genotype analyses of epididymal spermatozoa from Δex3-GRIC males showed efficient recombination of the allele (Supplemental Figure 1). We killed 3 wild-type females 9.5 days after mating to Δex3-GRIC males and observed multiple resorbing embryos in their uteri (n = 9 out of 22 total). We, therefore, conclude that the reduced fertility observed in Δex3-GRIC males was principally due to their transmission of the embryonically lethal recombined exon 3 allele to (ostensibly half of) their offspring.
Ovarian and uterine weights were comparable between Δex3-GRIC and control females (Supplemental Figure 4, right panels). In contrast, testis and seminal vesicle weights of Δex3-GRIC males were slightly, but significantly, lower than controls (Supplemental Figure 4). Because these findings were suggestive of androgen insufficiency, we also measured intratesticular testosterone levels. The results confirmed that Δex3-GRIC males had markedly reduced intratesticular testosterone relative to age-matched controls (Figure 1F). Despite this, no overt effects on testis histopathology, spermatogenesis, or epididymal sperm counts were observed (Supplemental Figure 2, Supplemental Table 1, and data not shown).
Fshb and mRNA levels were then measured in pituitary glands from Δex3-GRIC mice, along with circulating gonadotropin levels. The abundance of neither transcript was altered in female Δex3-GRIC mice, and serum FSH levels were also comparable with controls (Figure 1, B–D). A slight decrease in Lhb was observed in pituitary glands from Δex3-GRIC males, but this did not translate into an alteration in serum LH (Figure 1, C and E). Conversely, a marked decrease in both pituitary Fshb and serum FSH was observed in Δex3-GRIC males (Figure 1, B and D). These data indicate that removal of exon 3 from a single Ctnnb1 allele in murine gonadotropes leads to a male-specific impairment in Fshb expression and FSH secretion in vivo.
CTNNB1-Δex3 potentiates Fshb expression in vitro
To determine whether the observed impairment in FSH production by Δex3-GRIC males reflected a pituitary intrinsic effect, we cultured pituitaries from these and control mice and examined both Fshb mRNA expression and FSH release. Basal Fshb production in these cultures depends on endogenous signaling by an activin-like molecule and can also be potentiated by exogenous activins (eg, Refs. 26, 35). Given the selective effect of the mutation on FSH in vivo, we hypothesized that pituitaries in these mice might have reduced activin sensitivity. In complete contrast to this prediction, however, pituitary cultures from Δex3-GRIC males showed higher basal and activin A-stimulated (1nM for 24 h) Fshb mRNA expression relative to controls (Figure 2A). Lhb mRNA levels, on the other hand, did not differ between genotypes or treatments. We also observed a trend towards enhanced FSH (but not LH) secretion in the spent media from Δex3-GRIC cells after activin A treatment relative to controls (P = .068) (Figure 2B). We obtained similar results in immortalized murine gonadotrope-like LβT2 cells transfected with a murine Fshb promoter reporter (Figure 3). Transfection of expression vectors encoding either full-length CTNNB1 (Figure 3A) or CTNNB1 lacking exon 3 (Δex3) (Figure 3B) potentiated activin A-stimulated reporter activity. Wild-type or mutant CTNNB1 also potentiated GnRH-stimulated reporter activity, and addition of activin A and GnRH together resulted in a synergistic effect. Unexpectedly, the Δex3 mutant was somewhat less potent than the wild-type CTNNB1 in trans-activating the Fshb promoter. Although the reasons for this could not be determined, the Δex3 mutant might not have been as highly expressed as wild type, might have less intrinsic transactivation activity, or both. Nonetheless, these results indicate that overexpressed or stabilized CTNNB1 potentiates activin action in gonadotropes or gonadotrope-like cells in vitro.
Figure 2. CTNNB1 potentiates activin-stimulated Fshb expression in gonadotrope cells.

Cells were cultured from pituitary glands of male Δex3-GRIC or control mice and treated or not with 1nM activin A. After 24 hours, cells were harvested for analysis of Fshb or Lhb mRNA levels by real-time RT-PCR (A), and spent media were collected for analysis of FSH and LH (B). Experiments were performed 3 times, in triplicate. Columns, means of the 3 experimental replicates; error bars, SEM. Columns labeled with different letters were statistically different (P < .05, two-way ANOVA with Tukey's post hoc test).
Figure 3. CTNNB1 potentiates GnRH- and activin-stimulated Fshb promoter activity in LβT2 cells.
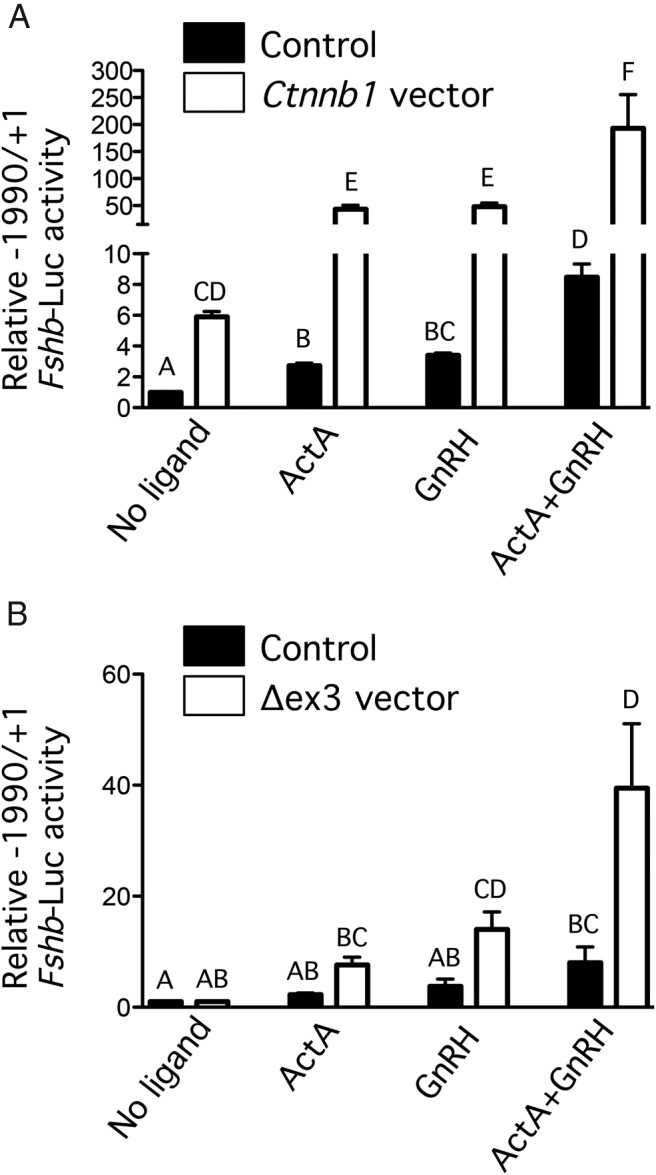
LβT2 cells were transfected with the indicate murine Fshb promoter luciferase construct and an expression vector encoding wild-type (A) or Δex3 mutant (B) CTNNB1, or the empty pcDNA 3.0 vector as a control, before treatment with GnRH (100nM), activin (1nM), neither, or both. Luciferase activity values are expressed relative to the empty vector/no ligand control. Experiments were performed 3 times, in triplicate. Columns, means of the 3 experimental replicates; error bars, SEM. Columns labeled with different letters were statistically different (P < .05, three-way ANOVA with Tukey's post hoc test).
Constitutively stable CTNNB1 increases gonadotrope sensitivity to inhibins
Given the decreased pituitary Fshb expression and FSH secretion in Δex3-GRIC males in vivo (Figure 1), the greater basal and activin A-induced Fshb expression in pituitary cells cultured from the same strains (Figure 2) appeared paradoxical. We, therefore, hypothesized that endocrine factors present in vivo, but absent in vitro, might explain these discrepant findings. In particular, we questioned whether increased inhibin B production/secretion (a testicular effect) and/or responsiveness to inhibins (a pituitary effect) might explain the impaired FSH production in Δex3-GRIC males. Because Cre is expressed in male germ cells in mice harboring the GRIC allele (21), we needed to rule in or out a testicular effect. Although inhibin B is the product of Sertoli cells, it was possible that stabilization of CTNNB1 in germ cells might have indirectly affected inhibin production in adjacent somatic cells. It was also possible that ectopic Cre activity in Sertoli cells could have affected inhibin production. To explore the latter possibility, we examined whether CTNNB1 expression was altered in the somatic cells of testes from Δex3-GRIC mice using immunohistochemistry. As indicated in Figure 4A, CTNNB1 expression in Δex3-GRIC testes was indistinguishable from controls in both germ and somatic cell populations. We next examined testicular expression of the subunits comprising inhibin B (ie, Inha and Inhbb) but did not observe any differences between genotypes (Figure 4B). Moreover, we did not detect increases in the inhibin βA subunit (Inhba) or total follistatin (Fst) mRNA in testes of Δex3-GRIC mice (Figure 4B), suggesting that de novo inhibin A or increased testicular Fst production likely did not account for the impairment in FSH in these mice. Interestingly, although there were no changes in inhibin subunit mRNA expression, we did observe a significant, although relatively modest (60%), elevation of serum inhibin B levels in Δex3-GRIC relative to control mice (Figure 4C).
Figure 4. Characterization of Δex3-GRIC testes.

A, Immunohistochemical analysis of CTNNB1 protein expression in adult Δex3-GRIC and control testes. B, Real-time RT-PCR analyses of Fst, Inha, Inhba, and Inhbb mRNA levels in the testes of adult Δex3-GRIC and control mice (n = 5/genotype). C, Serum inhibin B levels in adult male Δex3-GRIC and control mice (n = 15/genotype). Columns, means; error bars, SEM. Significant difference from control is indicated with an asterisk (*, P < .05, unpaired t test).
To determine whether the increase in inhibin B fully accounted for the substantial decrease in pituitary Fshb/FSH observed in Δex3-GRIC males, we compared FSH levels in castrated mice. Castration led to increases in LH, but not FSH, levels in control mice relative to intact animals (Figure 5). In Δex3-GRIC mice, however, both LH and FSH increased significantly after castration. Indeed, serum FSH in castrated Δex3-GRIC mice increased to levels that were statistically indistinguishable from castrated controls. These data, therefore, indicated that a secreted testicular factor, perhaps inhibin B, suppresses FSH to a greater extent in male Δex3-GRIC than control mice.
Figure 5. Castration restores FSH secretion in the Δex3-GRIC males.
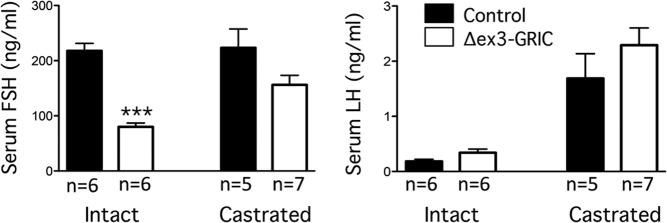
The indicated numbers of 12-week-old Δex3-GRIC and control males were bilaterally castrated or not (“intact”) and killed 10 days later for serum FSH and LH analyses. Columns, means; error bars, SEM. Significant difference from control is indicated with asterisks (***, P < .001, unpaired t test).
These data did not, however, indicate whether the elevation in inhibin B secretion or enhanced pituitary sensitivity to this inhibin B (or some combination of the 2) explained the more than 50% suppression of FSH production in Δex3-GRIC mice. To address the latter possibility, we treated cultured pituitary cells from adult male Δex3-GRIC and control mice with graded concentrations of inhibin A (recombinant inhibin B was unavailable to us) for 24 hours and quantified Fshb mRNA levels by real-time RT-PCR. As shown in Figure 6, cells from Δex3-GRIC were significantly more sensitive to inhibin A antagonism than were cells from controls. The lowest concentration tested (0.1nM) did not significantly affect Fshb mRNA levels in cells from control mice but reduced Fshb to approximately 33% of untreated levels in cells from Δex3-GRIC mice. Higher doses of inhibin A suppressed Fshb in cells of both genotypes, but a significantly greater effect was obtained in the cells from Δex3-GRIC mice at all concentrations tested. Therefore, increased inhibin feedback (mediated by both increased serum levels and gonadotrope responsiveness) appeared to contribute to the suppression of Fshb/FSH in male Δex3-GRIC mice.
Figure 6. Ex vivo inhibin sensitivity assay.
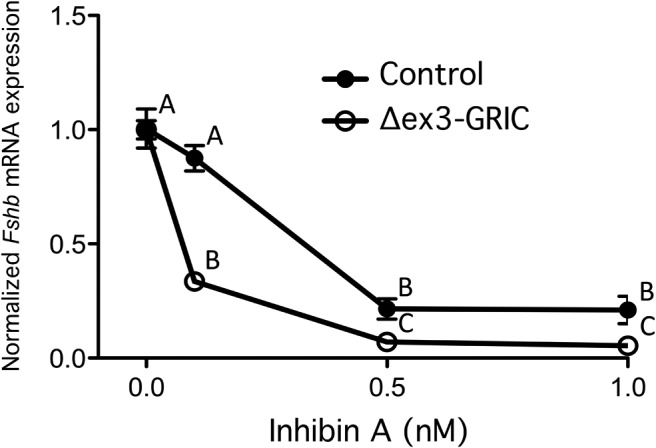
Real-time RT-PCR analyses were done on primary cultured pituitary cells from male Δex3-GRIC and control mice to assess the ability of graded doses of inhibin A to suppress Fshb mRNA expression. Because untreated Δex3-GRIC cells express higher intrinsic levels of Fshb than controls (as seen in Figure 2A), data were normalized for each genotype relative to the untreated samples, so as to better demonstrate differences in inhibin sensitivity. Error bars, SEM. Data points labeled with different letters were statistically different (P < .05, two-way ANOVA with Tukey's post hoc test).
According to the current model, inhibins suppress FSH by competitively binding to the activin type II receptor (ACVR2A) on pituitary gonadotropes. The coreceptor betaglycan (a product of the Tgfbr3 gene) significantly increases the affinity of inhibin for ACVR2A. We, therefore, examined expression of these receptors in whole pituitaries. Both Acvr2a and Tgfbr3 mRNA levels were slightly, but significantly, elevated in Δex3-GRIC mice relative to controls (Figure 7, A and B), which might contribute to enhanced inhibin sensitivity in vivo. However, in vitro, these differences are no longer apparent (data not shown), suggesting that the enhanced sensitivities of cultured gonadotropes to activin and inhibin do not depend on changes in the expression of these receptors.
Figure 7. Gene expression of Acvr2 (A), Tgfbr3 (B), Fst (C), and Fst288 (D) in Δex3-Gric pituitary cells.

Real-time RT-PCR analyses of the indicated transcripts in the pituitary glands of adult Δex3-GRIC (n = 18) and control males (n = 18). Columns, means; error bars, SEM. Significant difference from control is indicated with asterisks (**, P < .01; ***, P < .001, unpaired t test).
Follistatin is up-regulated in pituitaries of CTNNB1 gain of function mice
Finally, although Fst expression was unaltered in testes of Δex3-GRIC mice, Fst was recently shown to be stimulated by CTNNB1-Δex3 in developing mouse embryos. We, therefore, investigated levels of total Fst and Fst288 mRNA in pituitaries of control and Δex3-GRIC males. Remarkably, both transcripts were increased by 3-fold in the latter (Figure 7, C and D), suggesting that Fst antagonism of activin signaling in the pituitary might contribute to the FSH suppression in vivo. Interestingly, Fst mRNA levels did not differ in cultured pituitary cells between the 2 genotypes (data not shown; see Discussion below).
Discussion
CTNNB1 has long been thought of as a key effector of the canonical WNT signaling pathway, acting to transduce WNT signals by translocating to the nucleus and interacting with different classes of transcription factors (10, 11). More recent studies, however, have widened our view of CTNNB1 signaling, as it clearly functions in a WNT-independent manner to transduce and modulate a variety of biological signals (36–38). In LβT2 cells, GnRH stabilizes CTNNB1, which subsequently enhances the transcriptional activity of both Lhb and Fshb (9, 14). Our results, however, indicate that these cell line data might not fully or accurately predict CTNNB1's role in vivo, as mice harboring a gonadotrope-specific loss-of-function mutation in Ctnnb1 show normal fertility and gonadotropin subunit expression. At a minimum, other mechanisms may compensate for the loss of CTNNB1 in gonadotropes of these mice. However, the results may simply reflect that CTNNB1 does not play a necessary role in GnRH-stimulated gonadotropin β-subunit expression in vivo. In contrast to the results seen with knockout animals, we unexpectedly observed a robust and sex-specific suppression of FSH in mice with a gain-of-function mutation in Ctnnb1.
Male, but not female, mice harboring a deletion of exon 3 in one Ctnnb1 allele exhibit a 50% reduction in FSH. The molecular basis for this effect is not entirely clear at present but appears to reflect, at least in part, modulation of the activin/inhibin/follistatin system. Both in vivo and ex vivo observations suggest that gonadotropes of Δex3-GRIC males exhibit enhanced sensitivity to inhibin. Because inhibins are selective regulators of FSH synthesis and secretion, this may help explain why FSH, but not LH, is reduced in these mice. At the same time, these data may also contribute to our understanding of the sex-specific nature of the effect. That is, although male mice make 1 form of inhibin (inhibin B), females make 2 (inhibin A and B). FSH levels are higher in male than in female mice (and rats) (eg, Figure 1D). This sex difference could reflect the greater suppressive effect of 2 inhibins in females vs only 1 in males. Consistent with this idea, postgonadectomy increases in FSH are typically greater in female than in male mice (eg, Refs. 39–42). Indeed, in the current study, FSH levels did not increase at all in control males after bilateral orchidectomy. Therefore, even if the pituitaries of Δex3-GRIC females show enhanced inhibin sensitivity as seen in males, this might be masked in vivo by the already potent inhibition of FSH by the combined actions of inhibins A and B.
Another potential contributing factor to the sex-specific effect may be the elevated serum inhibin B observed in Δex3-GRIC males (inhibin levels were not measured in females because they showed no fertility or FSH impairments). The basis for this increase is unclear as: 1) inhibin subunit expression is unaltered in the testes of Δex3-GRIC mice; and 2) inhibin B is a product of Sertoli cells, which do not express the Cre enzyme in our model. Nonetheless, it is possible that unintended effects of recombination in the testes of Δex3-GRIC males contributed to the sex difference in FSH regulation, because Cre is not expressed in ovaries of GRIC mice. Likewise, the basis for the decrease in intratesticular testosterone levels observed in the Δex3-GRIC model cannot be easily explained, because LH is regarded as the principal gonadotropin regulator of testosterone synthesis, and levels of this hormone appear normal in these mice. Although FSH clearly plays a role in male reproductive physiology, its specific role as a regulator of testicular androgen synthesis in mice remains unclear (43). Although male Fshr knockout mice have markedly reduced testosterone levels, testosterone synthesis in Fshb knockout mice is largely unaffected (43). Whether the reduction in intratesticular testosterone levels in the Δex3-GRIC males is related to the FSH phenotype or may represent an unintended, testis-autonomous effect remains to be determined.
Although enhanced inhibin sensitivity provides one potential explanation for the suppressed FSH in Δex3-GRIC males, we have not yet established the underlying mechanism. Our ability to do so is hampered to some extent by our incomplete understanding of mechanisms of inhibin action, particularly in vivo. According to the current model, which is based largely on in vitro observations, inhibins act by competitively antagonizing activin signaling. More specifically, in the presence of a coreceptor, betaglycan (or transforming growth factor, beta receptor III), inhibins bind the ACVR2 with high affinity, impairing activin binding (44). This in turn leads to a net loss of FSH, because autocrine/paracrine activins are generally regarded as necessary drivers of FSH synthesis (6). We, therefore, hypothesized that altered (enhanced) Tgfbr3 expression might underlie the suppressed FSH in Δex3-GRIC males, and we observed a small but significant increase in Tgfbr3 mRNA in Δex3-GRIC pituitaries. Tgfbr3 is expressed in multiple cell types. Therefore, it is possible that we might have observed an even greater increase if our analysis had been limited to gonadotropes. That said, Tgfbr3 levels did not differ in cultured pituitary cells between genotypes (data not shown) calling into question the necessity for increased betaglycan expression for enhanced inhibin sensitivity.
In light of the suppressed FSH observed in Δex3-GRIC males in vivo, we were surprised to observe enhanced basal and activin-stimulated Fshb expression in their pituitaries in vitro. As endocrine inhibins are removed in this in vitro context, one might have predicted a normalization of FSH synthesis, if enhanced inhibin feedback from the testes alone explained the in vivo observations. Indeed, this is what we observed after castration in vivo. The fact that Fshb levels are enhanced in these cultured cells, however, suggests a compensatory process. That is, the pituitary might increase activin production and/or sensitivity to offset its potentiated responsiveness to inhibins. We did not, however, observe changes in pituitary activin β-subunit expression in vivo or in Acvr2 expression in cultured cells (data not shown). Nor did we detect an increase in the number of FSH-producing gonadotropes (data not shown), as one might have predicted a priori in cells expressing the dominant-stable CTNNB1 (20, 45). In addition, if compensatory mechanisms are at play in this model, it is unclear whether enhanced activin signaling is a cause or consequence of increased inhibin sensitivity. That is, in LβT2 cells, which are never exposed to endocrine inhibins, overexpression of either wild-type or the dominant-stable CTNNB1 increases both basal and activin-stimulated Fshb transcription. Therefore, increased inhibin action may in fact represent the compensatory process in these mice.
Finally, we also observed a potent up-regulation of Fst expression in pituitaries of Δex3-GRIC males. Fst is produced by pituitary gonadotropes and folliculostellate cells (46). Given that the genetic manipulation used here was gonadotrope specific, it is likely that the increase in follistatin production was within this cell type. If the increase mRNA correlates with increased protein expression, we would expect robust antagonism of local activin action on FSH synthesis. If this is indeed a mechanism underlying FSH suppression in our model, why is the effect gender-specific? Although we do not have a definitive answer, there may be a role for testosterone. That is, T stimulates Fst expression in CTNNB1-dependent fashion in some contexts and the androgen receptor and CTNNB1 physically interact (47, 48). Therefore, although Δex3-CTNNB1 alone may be insufficient to stimulate Fst expression, it may potentiate the effects of T. Consistent with this idea, the increase in Fst mRNA levels is lost in cultured cells where T is low or absent (data not shown), and FSH levels increase after castration in Δex3-GRIC mice (unfortunately, we do not have pituitaries from the castrated mice [in Figure 5] to enable an assessment of Fst expression therein). Future investigations will be needed to determine whether T, androgen receptor, and Δex3-CTNNB1 functionally interact to regulate Fst in gonadotropes.
In summary, we report that CTNNB1 appears dispensable for gonadotropin synthesis and fertility in mice in vivo. We also observe that a dominant-stable form of CTNNB1 impairs FSH synthesis in male mice in vivo while at the same time enhancing its synthesis, particularly in response to activins, in vitro. Although the dominant-stable CTNNB1 appears to increase inhibin sensitivity, increase pituitary Fst expression in vivo, and enhance mechanisms of activin signaling, the underlying mechanisms remain to be fully elucidated. Whether such an increase in CTNNB1 signaling can occur in gonadotropes in specific physiological (or pathophysiological) contexts remains unknown but is plausible and worthy of investigation.
Acknowledgments
We thank Ms Meggie Girard and Dr Paolete Soto for technical assistance with mouse colony management and genotype analyses, Dr Jérôme Fortin for reviewing a draft of the original manuscript, Dr Pamela Mellon for the LβT2 cells, Dr Teresa Woodruff for the inhibin A, and Ken McDonald for assistance with FACS. D.B. is the Canada Research Chair in Ovarian Molecular Biology and Functional Genomics.
This work was supported by operating grants MOP-123447 (to D.J.B./D.B.) and MOP-102508 (to D.B.) from the Canadian Institutes of Health Research. V.K. was supported by a Merit Scholarship from the Ministère de l'Éducation, des Loisirs et des Sports. U.B. is supported by SFB894. The University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core is supported by the National Institutes of Health Eunice Kennedy Shriver National Institute of Child Health and Human Development (Specialized Cooperative Centers Program in Reproduction and Infertility Research) Grant U54-HD28934.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ACVR2A
- activin type II receptor
- CTNNB1
- β-catenin
- GRIC
- GnRH receptor internal ribosome entry site Cre
- GSK3β
- glycogen synthase kinase 3β
- KO
- knockout
- NR5A1
- nuclear receptor subfamily 5, group A, member 1
- YFP
- yellow fluorescent protein.
References
- 1. Ascoli M, Fanelli F, Segaloff DL. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev. 2002;23:141–174. [DOI] [PubMed] [Google Scholar]
- 2. Simoni M, Gromoll J, Nieschlag E. The follicle-stimulating hormone receptor: biochemistry, molecular biology, physiology, and pathophysiology. Endocr Rev. 1997;18:739–773. [DOI] [PubMed] [Google Scholar]
- 3. Burger LL, Haisenleder DJ, Dalkin AC, Marshall JC. Regulation of gonadotropin subunit gene transcription. J Mol Endocrinol. 2004;33:559–584. [DOI] [PubMed] [Google Scholar]
- 4. Melamed P, Kadir MN, Wijeweera A, Seah S. Transcription of gonadotropin β subunit genes involves cross-talk between the transcription factors and co-regulators that mediate actions of the regulatory hormones. Mol Cell Endocrinol. 2006;252:167–183. [DOI] [PubMed] [Google Scholar]
- 5. Thackray VG. Fox tales: regulation of gonadotropin gene expression by forkhead transcription factors. Mol Cell Endocrinol. 2014;385:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bernard DJ, Fortin J, Wang Y, Lamba P. Mechanisms of FSH synthesis: what we know, what we don't, and why you should care. Fertil Steril. 2010;93:2465–2485. [DOI] [PubMed] [Google Scholar]
- 7. Jorgensen JS, Quirk CC, Nilson JH. Multiple and overlapping combinatorial codes orchestrate hormonal responsiveness and dictate cell-specific expression of the genes encoding luteinizing hormone. Endocr Rev. 2004;25:521–542. [DOI] [PubMed] [Google Scholar]
- 8. Gardner S, Stavrou E, Rischitor PE, Faccenda E, Pawson AJ. Targeting mediators of Wnt signalling pathways by GnRH in gonadotropes. J Mol Endocrinol. 2010;44:195–201. [DOI] [PubMed] [Google Scholar]
- 9. Wang Q, Chikina M, Zaslavsky E, Pincas H, Sealfon SC. β-Catenin regulates GnRH-induced FSHβ gene expression. Mol Endocrinol. 2013;27:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boyer A, Goff AK, Boerboom D. WNT signaling in ovarian follicle biology and tumorigenesis. Trends Endocrinol Metab. 2010;21:25–32. [DOI] [PubMed] [Google Scholar]
- 11. Lapointe E, Boerboom D. WNT signaling and the regulation of ovarian steroidogenesis. Front Biosci (Schol Ed). 2011;3:276–285. [DOI] [PubMed] [Google Scholar]
- 12. Alarid ET, Windle JJ, Whyte DB, Mellon PL. Immortalization of pituitary cells at discrete stages of development by directed oncogenesis in transgenic mice. Development. 1996;122:3319–3329. [DOI] [PubMed] [Google Scholar]
- 13. Gardner S, Maudsley S, Millar RP, Pawson AJ. Nuclear stabilization of β-catenin and inactivation of glycogen synthase kinase-3β by gonadotropin-releasing hormone: targeting Wnt signaling in the pituitary gonadotrope. Mol Endocrinol. 2007;21:3028–3038. [DOI] [PubMed] [Google Scholar]
- 14. Salisbury TB, Binder AK, Grammer JC, Nilson JH. Maximal activity of the luteinizing hormone β-subunit gene requires β-catenin. Mol Endocrinol. 2007;21:963–971. [DOI] [PubMed] [Google Scholar]
- 15. Boyer A, Hermo L, Paquet M, Robaire B, Boerboom D. Seminiferous tubule degeneration and infertility in mice with sustained activation of WNT/CTNNB1 signaling in sertoli cells. Biol Reprod. 2008;79:475–485. [DOI] [PubMed] [Google Scholar]
- 16. Brault V, Moore R, Kutsch S, et al. Inactivation of the β-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. [DOI] [PubMed] [Google Scholar]
- 17. Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. [DOI] [PubMed] [Google Scholar]
- 18. Srinivas S, Watanabe T, Lin CS, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wen S, Schwarz JR, Niculescu D, et al. Functional characterization of genetically labeled gonadotropes. Endocrinology. 2008;149:2701–2711. [DOI] [PubMed] [Google Scholar]
- 20. Harada N, Tamai Y, Ishikawa T, et al. Intestinal polyposis in mice with a dominant stable mutation of the β-catenin gene. EMBO J. 1999;18:5931–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wen S, Ai W, Alim Z, Boehm U. Embryonic gonadotropin-releasing hormone signaling is necessary for maturation of the male reproductive axis. Proc Natl Acad Sci USA. 2010;107:16372–16377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wen S, Götze IN, Mai O, Schauer C, Leinders-Zufall T, Boehm U. Genetic identification of GnRH receptor neurons: a new model for studying neural circuits underlying reproductive physiology in the mouse brain. Endocrinology. 2011;152:1515–1526. [DOI] [PubMed] [Google Scholar]
- 23. Bernard DJ. Both SMAD2 and SMAD3 mediate activin-stimulated expression of the follicle-stimulating hormone β subunit in mouse gonadotrope cells. Mol Endocrinol. 2004;18:606–623. [DOI] [PubMed] [Google Scholar]
- 24. Lamba P, Santos MM, Philips DP, Bernard DJ. Acute regulation of murine follicle-stimulating hormone β subunit transcription by activin A. J Mol Endocrinol. 2006;36:201–220. [DOI] [PubMed] [Google Scholar]
- 25. Wang Y, Fortin J, Lamba P, et al. Activator protein-1 and smad proteins synergistically regulate human follicle-stimulating hormone β-promoter activity. Endocrinology. 2008;149:5577–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fortin J, Boehm U, Weinstein MB, Graff JM, Bernard DJ. Follicle-stimulating hormone synthesis and fertility are intact in mice lacking SMAD3 DNA binding activity and SMAD2 in gonadotrope cells. FASEB J. 2014;28:1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fortin J, Kumar V, Zhou X, et al. NR5A2 regulates Lhb and Fshb transcription in gonadotrope-like cells in vitro, but is dispensable for gonadotropin synthesis and fertility in vivo. PLoS One. 2013;8:e59058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tran S, Zhou X, Lafleur C, et al. Impaired fertility and FSH synthesis in gonadotrope-specific Foxl2 knockout mice. Mol Endocrinol. 2013;27:407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ho CC, Zhou X, Mishina Y, Bernard DJ. Mechanisms of bone morphogenetic protein 2 (BMP2) stimulated inhibitor of DNA binding 3 (Id3) transcription. Mol Cell Endocrinol. 2011;332:242–252. [DOI] [PubMed] [Google Scholar]
- 30. Lapointe E, Boyer A, Rico C, et al. FZD1 regulates cumulus expansion genes and is required for normal female fertility in mice. Biol Reprod. 2012;87:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 32. Boyer A, Paquet M, Laguë MN, Hermo L, Boerboom D. Dysregulation of WNT/CTNNB1 and PI3K/AKT signaling in testicular stromal cells causes granulosa cell tumor of the testis. Carcinogenesis. 2009;30:869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Creasy DM. Evaluation of testicular toxicity in safety evaluation studies: the appropriate use of spermatogenic staging. Toxicol Pathol. 1997;25:119–131. [DOI] [PubMed] [Google Scholar]
- 34. Kemler R, Hierholzer A, Kanzler B, et al. Stabilization of β-catenin in the mouse zygote leads to premature epithelial-mesenchymal transition in the epiblast. Development. 2004;131:5817–5824. [DOI] [PubMed] [Google Scholar]
- 35. Rejon CA, Ho CC, Wang Y, Zhou X, Bernard DJ, Hébert TE. Cycloheximide inhibits follicle-stimulating hormone β subunit transcription by blocking de novo synthesis of the labile activin type II receptor in gonadotrope cells. Cell Signal. 2013;25:1403–1412. [DOI] [PubMed] [Google Scholar]
- 36. Haq S, Michael A, Andreucci M, et al. Stabilization of β-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc Natl Acad Sci USA. 2003;100:4610–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lade AG, Monga SP. β-Catenin signaling in hepatic development and progenitors: which way does the WNT blow? Dev Dyn. 2011;240:486–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lu Z, Hunter T. Wnt-independent β-catenin transactivation in tumor development. Cell Cycle. 2004;3:571–573. [PubMed] [Google Scholar]
- 39. Kumar TR, Low MJ. Gonadal steroid hormone regulation of human and mouse follicle stimulating hormone β-subunit gene expression in vivo. Mol Endocrinol. 1993;7:898–906. [DOI] [PubMed] [Google Scholar]
- 40. Kovacic N, Parlow AF. Alterations in serum FSH-LH ratios in relation to the estrous cycle, pseudopregnancy, and gonadectomy in the mouse. Endocrinology. 1972;91:910–915. [DOI] [PubMed] [Google Scholar]
- 41. Naik SI, Young LS, Charlton HM, Clayton RN. Pituitary gonadotropin-releasing hormone receptor regulation in mice. II: females. Endocrinology. 1984;115:114–120. [DOI] [PubMed] [Google Scholar]
- 42. Naik SI, Young LS, Charlton HM, Clayton RN. Pituitary gonadotropin-releasing hormone receptor regulation in mice. I: males. Endocrinology. 1984;115:106–113. [DOI] [PubMed] [Google Scholar]
- 43. Sairam MR, Krishnamurthy H. The role of follicle-stimulating hormone in spermatogenesis: lessons from knockout animal models. Arch Med Res. 2001;32:601–608. [DOI] [PubMed] [Google Scholar]
- 44. Lewis KA, Gray PC, Blount AL, et al. Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature. 2000;404:411–414. [DOI] [PubMed] [Google Scholar]
- 45. Gaston-Massuet C, Andoniadou CL, Signore M, et al. Increased wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci USA. 2011;108:11482–11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kaiser UB, Lee BL, Carroll RS, Unabia G, Chin WW, Childs GV. Follistatin gene expression in the pituitary: localization in gonadotropes and folliculostellate cells in diestrous rats. Endocrinology. 1992;130:3048–3056. [DOI] [PubMed] [Google Scholar]
- 47. Yang F, Li X, Sharma M, et al. Linking β-catenin to androgen-signaling pathway. J Biol Chem. 2002;277:11336–11344. [DOI] [PubMed] [Google Scholar]
- 48. Singh R, Bhasin S, Braga M, et al. Regulation of myogenic differentiation by androgens: cross talk between androgen receptor/β-catenin and follistatin/transforming growth factor-β signaling pathways. Endocrinology. 2009;150:1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]