

Abstract
GnRH release in the median eminence (ME) is the central output for control of reproduction. GnRH processes in the preoptic area (POA) also release GnRH. We examined region-specific regulation of GnRH secretion using fast-scan cyclic voltammetry to detect GnRH release in brain slices from adult male mice. Blocking endoplasmic reticulum calcium reuptake to elevate intracellular calcium evokes GnRH release in both the ME and POA. This release is action potential dependent in the ME but not the POA. Locally applied kisspeptin induced GnRH secretion in both the ME and POA. Local blockade of inositol triphospate-mediated calcium release inhibited kisspeptin-induced GnRH release in the ME, but broad blockade was required in the POA. In contrast, kisspeptin-evoked secretion in the POA was blocked by local gonadotropin-inhibitory hormone, but broad gonadotropin-inhibitory hormone application was required in the ME. Although action potentials are required for GnRH release induced by pharmacologically-increased intracellular calcium in the ME and kisspeptin-evoked release requires inositol triphosphate-mediated calcium release, blocking action potentials did not inhibit kisspeptin-induced GnRH release in the ME. Kisspeptin-induced GnRH release was suppressed after blocking both action potentials and plasma membrane Ca2+ channels. This suggests that kisspeptin action in the ME requires both increased intracellular calcium and influx from the outside of the cell but not action potentials. Local interactions among kisspeptin and GnRH processes in the ME could thus stimulate GnRH release without involving perisomatic regions of GnRH neurons. Coupling between action potential generation and hormone release in GnRH neurons is thus likely physiologically labile and may vary with region.
GnRH neurons form the final common pathway for the central control of reproduction. This is accomplished via episodic secretion of GnRH near portal vessels in the median eminence (ME) to regulate the anterior pituitary (1, 2). In addition to this well-established neuroendocrine output, recent evidence indicates that GnRH is also released in other brain regions, specifically in the preoptic area (POA) (3). This latter observation is consistent with a recent report of perisomatic release in cultured primate GnRH neurons (4) and with reports that GnRH has central neuromodulatory roles in addition to neuroendocrine function (5–9). Central GnRH has been postulated to take part in direct synchronizing of the GnRH neuron network, in local circuit feedback on GnRH neurons, and in altering sex behavior (10–12).
Peptide release has also been observed from the dendrites/soma of magnocellular neuroendocrine neurons located in the supraoptic nucleus. The neuroendocrine function of these neurons is to release vasopressin and oxytocin from terminals in the posterior pituitary (13). Interestingly, dendritic release appears to be regulated independently of secretion to the blood and to use different mechanisms. Specifically, large dense core vesicles containing peptides can be released in an action potential-independent manner that requires increased intracellular Ca2+ levels and can be initiated by signaling arising from ligand interactions with metabotropic receptors (14–17). Parvocellular neuroendocrine neurons, including GnRH neurons, have not been studied in this regard, but many neuromodulators have been reported to regulate GnRH release (18). Of these, kisspeptin is among the most potent activators, and gonadotropin-inhibitory hormone (GnIH) is among the most potent inhibitors. GnRH neurons express the kisspeptin receptor (kiss1r) and are strongly depolarized by kisspeptin (19, 20). Kisspeptin administration increases GnRH release and thereby increases secretion of the pituitary gonadotropin LH (21). GnIH can act via G-protein coupled receptor 147, which is expressed centrally as well as in the pituitary. In brain slices, GnIH inhibits GnRH neuron action potential firing, even in the presence of kisspeptin (22, 23). Whether peptide interactions are also distinct for different brain regions requires a sufficiently sensitive method that can be visually targeted to specific sites. We adapted an electrochemical method, fast-scan cyclic voltammetry (FSCV), for GnRH detection directly in mouse brain slices (3). Using FSCV, both POA and ME release can be detected, allowing studies of the mechanisms underlying release in these regions. Here, we use FSCV to investigate action potential dependence, calcium dependence, and neuropeptidergic (kisspeptin and GnIH) regulation of GnRH secretion in the ME and POA.
Materials and Methods
Animals and brain slice preparation
Adult 40- to 90-day-old gonad-intact male GnRH-enhanced green fluorescent protein (GFP) mice (24) and kisspeptin knockout mice (generous gift of Dr Yee-Ming Chan and Dr Stephanie Seminara, Massachusetts General Hospital) (25) were housed under a 14-hour light, 10-hour dark photoperiod (lights on at 3 am) with Harlan 2916 chow and water available ad libitum. All procedures were approved by the University of Michigan University Committee on the Use and Care of Animals.
Brain slice preparation
All chemicals were purchased from Sigma Chemical Co unless noted. Mice were decapitated, and brain slices were prepared as described (26, 27). All buffers were bubbled with 95%O2/5%CO2 15 minutes before usage. Sagittal 300-μm brain slices were cut using a Leica VT 1200S vibratome (Leica Biosystems) in ice-cold sucrose saline containing: 250mM sucrose, 3.5mM KCl, 26mM NaHCO3, 10mM d-glucose, 1.25mM NaH2PO4, 1.2mM MgSO4, and 3.8mM MgCl2. Slices were incubated 30 minutes at room temperature in a 1:1 mixture of sucrose saline and artificial cerebrospinal fluid (ACSF) containing: 125mM NaCl, 3.5mM KCl, 26mM NaHCO3, 1.25mM NaH2PO4, 2.5mM CaCl2, 1.2mM MgSO4, and 10mM D-glucose (pH 7.4), then transferred to 100% ACSF and incubated 30–300 minutes at room temperature before study. In our experience, brain slices are viable for at least 8 hours.
Fast-scan cyclic voltammetry
Individual slices were transferred to a recording chamber mounted on the stage of an upright microscope (Olympus BX50WI; Opelco). The chamber was perfused continuously with ACSF at a rate of 5–6 mL/min at 31°C–32°C. Slices were stabilized in the chamber for at least 10 minutes before recording. GnRH detection was monitored using carbon fiber microelectrodes (CFMs) manufactured as previously described (28). FSCV recordings were made in voltage-clamp mode of an EPC-10 USB amplifier running Patchmaster (HEKA Elektronik) on a Macintosh Mac Pro computer (Apple Computers). Electrode potential was continuously scanned from 0.5 to 1.45 V at 400 V/s every 100 ms (Figure 1C). GnRH-GFP neurons were identified by brief illumination at 470 nm. CFMs were placed either among GnRH neuron terminals in the ME or near GnRH fiber-fiber appositions in the POA (Figure 1).
Figure 1. Illustration of experimental setup with relative positions of electrode for FSCV and injection sites in the ME (A) and POA (B).
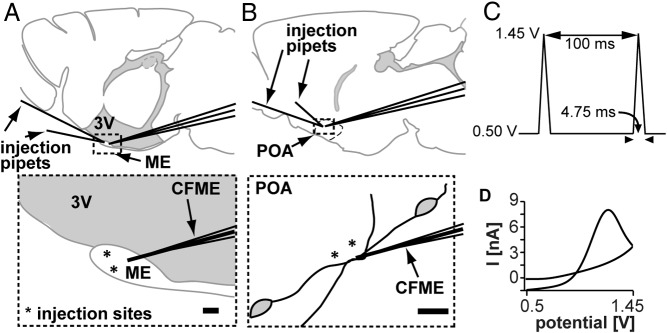
Upper panels show sagittal slice with relative positions of the CFM for FSCV and injection pipets for local drug application. CFM and pipets are separated by 20–30 μm. 3V, third ventricle. Dotted areas are expanded in the lower panels, asterisks in lower panel indicate injection sites. C, FSCV waveform that enables GnRH detection, holding potential = 0.5 V, switching potential = 1.45 V, scan rate = 400V/S. D, An example of CV obtained at the peak current for kisspeptin-evoked GnRH event.
Experimental design
The general design of experiments was as follows. The voltage protocol for GnRH detection was run for 10–15 minutes after CFMs were placed in the tissue before collecting data to allow recordings to stabilize. Release was recorded continuously for the duration of each experiment, during which a variety of treatments were applied by brief (10–20 s) local injection (3–5 μL from a 25-μL Hamilton syringe) via a pipette placed 20–30 μm from the CFM using a micromanipulator (Figure 1, A and B) or by bath treatment. Intervals and timing of these treatments are specified for each study below. At the end of each recording, the viability of slice and ability to detect GnRH were confirmed applying 20mM KCl 20 minutes after the final experimental treatment. Only slices that released GnRH in response to KCl are included in the analysis (∼90%).
First, we investigated the Ca2+ and action potential dependence of GnRH secretion in ME vs POA. Local injection of thapsigargin (5μM; Tocris Biosciences) or cyclopiazonic acid (10μM CPA) was used to elevate cytoplasmic calcium levels (29, 30). Both drugs block Ca2+ reuptake into the endoplasmic reticulum by inhibiting sarcoendoplasmic reticulum calcium transport ATPase (SERCA) pumps, and brief applications as used here tend to increase intracellular calcium. To test whether multiple releases could be induced, thapsigargin or CPA was injected at 20-minute intervals. To test the requirement for action potential generation, thapsigargin was injected under control conditions, then the slice was bathed with 0.5μM tetrodotoxin (TTX) (Calbiochem) for 5–10 minutes to block sodium channel-dependent action potentials and release in response to thapsigargin injection tested again. To test the requirement for kisspeptin release, the ability of CPA to induce GnRH release in the ME was tested in kisspeptin knock-out (KO) mice. POA studies were not done in kisspeptin KO mice because GFP is required to target the CFMs in the POA and GFP is not expressed in GnRH neurons in this mouse line.
Next, to test effects of neuromodulators on GnRH release from the POA and ME, 10nM kisspeptin (Phoenix Pharmaceuticals) alone or in combination with 1μM GnIH (Phoenix) was locally injected into the slice. Kisspeptin alone was similarly injected during bath application of 1μM GnIH. The interval between treatments was 10–15 minutes.
Next, we investigated the role of intracellular Ca2+ mobilization in kisspeptin-evoked GnRH release in the POA and ME. We blocked inositol trisphosphate (IP3)-mediated release of Ca2+ from the endoplasmic reticulum with xestospongin C (XC) (20μM Tocris). Kisspeptin was injected locally 3 times at 10- to 15-minute intervals: under control conditions (kisspeptin only), with local injection of XC, and after washout of XC. In some studies in the POA, kisspeptin was locally injected under control conditions and during bath application of XC (5μM). To test possible off-target effects of XC on kisspeptin-evoked secretion, we used Cd2+ to test whether blockade of Ca2+ channels would prevent kisspeptin from stimulating GnRH release. Kisspeptin was locally administered in both the POA and the ME under control conditions and in the presence of bath-applied 200μM Cd2+. This was followed by 20 minutes of washout period, and then 20mM KCl was used to test slice viability.
To determine whether kisspeptin-evoked GnRH release in the ME was action potential dependent, 3 local kisspeptin injections were performed at 10- to 15-minute intervals: under control conditions, then in the presence of bath-applied TTX (0.5μM, 3–4 min before second kisspeptin injection), and then in the presence of both TTX and the calcium channel blocker Cd2+ (200μM 3–4 min before third kisspeptin injection).
Analysis
FSCV data were converted to general text format using Igor Pro (Wavemetrics) and then analyzed using Demon software (generously provided by Jordan Yorgason, Rodrigo Espana, and Sara Jones, Wake Forest University Health Sciences) as described previously (3, 28). Cyclic voltammograms (CVs) were background subtracted by averaging 10 background scans. An example of the CV curve obtained at the peak current for kisspeptin-evoked GnRH release is shown in Figure 1D. To verify the identity of a spontaneous release peak as GnRH, 5 control CVs collected after GnRH was injected into a slice were averaged. Each putative GnRH CV was correlated with this average and was considered to be GnRH if R2 ≥ 0.8. This threshold was set to allow for electrode variability. A total of 92% of CVs passed this test. Changes in GnRH concentration were estimated based on CFM calibration in 5μM GnRH.
Data were transferred to Prism5 for statistical analysis (GraphPad Software). Parameters measured included release event duration, GnRH concentration change, lag to GnRH release after each pharmacological treatment, and percentage of treatments resulting in evoked GnRH secretion. Data were analyzed by parametric or nonparametric ANOVA tests as dictated by data distribution followed by two-tailed post hoc analyses. Data are presented as mean ± SEM, and P < .05 was considered significant.
Results
Intracellular Ca2+-evoked GnRH release is action potential dependent only in the ME
Mechanisms leading to neurosecretion can differ between dendritic/somatic and terminal regions (31). We used brief exposure to 5μM thapsigargin injected locally into the ME or POA as illustrated in Figure 1, A and B, to elicit release. Thapsigargin blocks Ca2+ reuptake into the endoplasmic reticulum by inhibiting SERCA pumps, and brief applications as used here tend to increase intracellular calcium (14, 32, 33). Thapsigargin was first injected under control conditions and then after blocking sodium-dependent action potentials with TTX (0.5μM, timeline; Figure 2A). Thapsigargin reliably induced GnRH release in both the POA (n = 7 of 7 trials) (Figure 2, B and F) and ME (n = 5 of 5 trials) (Figure 2, C and F) under control conditions. Thapsigargin-induced release persisted in the POA with similar magnitude and duration after treating the slice with TTX to block action potentials. (Figure 2, B and F). In marked contrast, blocking action potentials inhibited thapsigargin-induced GnRH release in the ME (Figure 2, C and F).
Figure 2. Increasing intracellular Ca2+ by blocking SERCA pumps elicits GnRH release in the POA and ME but requires action potentials only in the ME.

A, Experimental timeline for B and C. B and C, Representative examples of thapsigargin (Tp)-evoked GnRH release shown as a current heat map during the control period (left) and TTX treatment (right) in the POA (B) and ME (C). D, top, Experimental timeline. Bottom, Repeated injection of Tp under control conditions induces repeated GnRH release in the ME. E, Mean ± SEM peak GnRH release amplitude (left) shown as change in concentration and release duration (right). Numbers within bars indicate sample size; ' indicates second injection; “ indicates third injection of CPA, accordingly. F, Mean ± SEM percent responding to Tp (left), concentration change (center), and release duration (right). Gray bars show POA, black bars show ME; lower case letters indicate P < .05 χ2 test. G, GnRH release in response to CPA is not blocked in kisspeptin (K) KO mice. Mean ± SEM percent responding to CPA or kisspeptin (K) (left), concentration change (center), and release duration (right); asterisks indicate P < .05 with Student's t test.
To test whether the lack of response to a second application of thapsigargin in the ME was due to inability of thapsigargin to elicit repeated releases, thapsigargin (Figure 2, D and E) or CPA, a drug with the same mechanism of action (Figure 2E), was repeatedly injected under control conditions (ie, no TTX). Both thapsigargin (n = 3) and CPA (n = 4) elicited similar repeated GnRH release in 100% of trials in the ME with no decrement in either amplitude or duration. Neither GnRH concentration change nor duration of release in response to thapsigargin differed between regions (Figure 2F). The requirement for action potentials for increased intracellular calcium induced by blocking endoplasmic reticulum reuptake to induce GnRH release suggested the possibility that Ca2+ was elevated in a cell upstream of the GnRH neuron itself and that upstream cell needed to generate action potentials to release a subsequent mediator to induce GnRH release. To test the hypothesis that this intermediate mediator was kisspeptin, the ability of CPA to induce GnRH release in the ME was tested in kisspeptin KO mice (in the absence of TTX). CPA repeatedly induced GnRH release in kisspeptin knockout mice in the ME (Figure 2G), although GnRH concentration change in response to CPA was lower than that in response to kisspeptin (Figure 2G). Together, these data suggest that GnRH secretion in response to increased intracellular Ca2+ can be action potential independent in the POA.
Local GnIH blocks kisspeptin-induced GnRH release in the POA, but bath GnIH application is required in the ME
We next examined the effects of 2 established modulators of GnRH neuron action potential firing, kisspeptin and GnIH, on GnRH release (19, 22). GnRH release in response to 10nM kisspeptin was monitored in both the ME and POA when given alone, then after local injection of 1μM GnIH, and finally after bath application of GnIH (Figure 3A). To minimize the effects of kisspeptin on the neuronal network upstream of GnRH neurons, kisspeptin was always applied locally through brief injection directly to the slice. In both the POA (n = 7 of 7) and ME (n = 6 of 7), kisspeptin potently and repeatedly induced GnRH secretion (Figure 3). There were no differences in either event duration or concentration change between regions or with repeated application (Figure 3, D–F). In the POA, local coapplication of GnIH blocked GnRH release in 5 of 7 trials. In contrast, local GnIH failed to block kisspeptin-induced GnRH release in all 6 trials in the ME. This suggests either that GnIH receptors are not near the drug application site in the ME or that GnIH is ineffective in blocking GnRH release in this region. To test whether widespread application of GnIH could block GnRH release induced by local kisspeptin injection, GnIH was bath applied. Bath-applied GnIH completely blocked kisspeptin-evoked GnRH secretion in both brain regions, indicating that this treatment is effective and supporting the hypothesis that the receptors are not located within the effective local drug application zone within the ME (n = 6 each) (Figure 3, D–F). It is also possible that differences between locally- and bath-applied treatments may reflect structural differences between the 2 brain regions that alter diffusion rates/spatial range of locally-applied pharmacological agents, but it is difficult to approach this problem experimentally.
Figure 3. Neuropeptide modulation of GnRH secretion in the ME and POA.
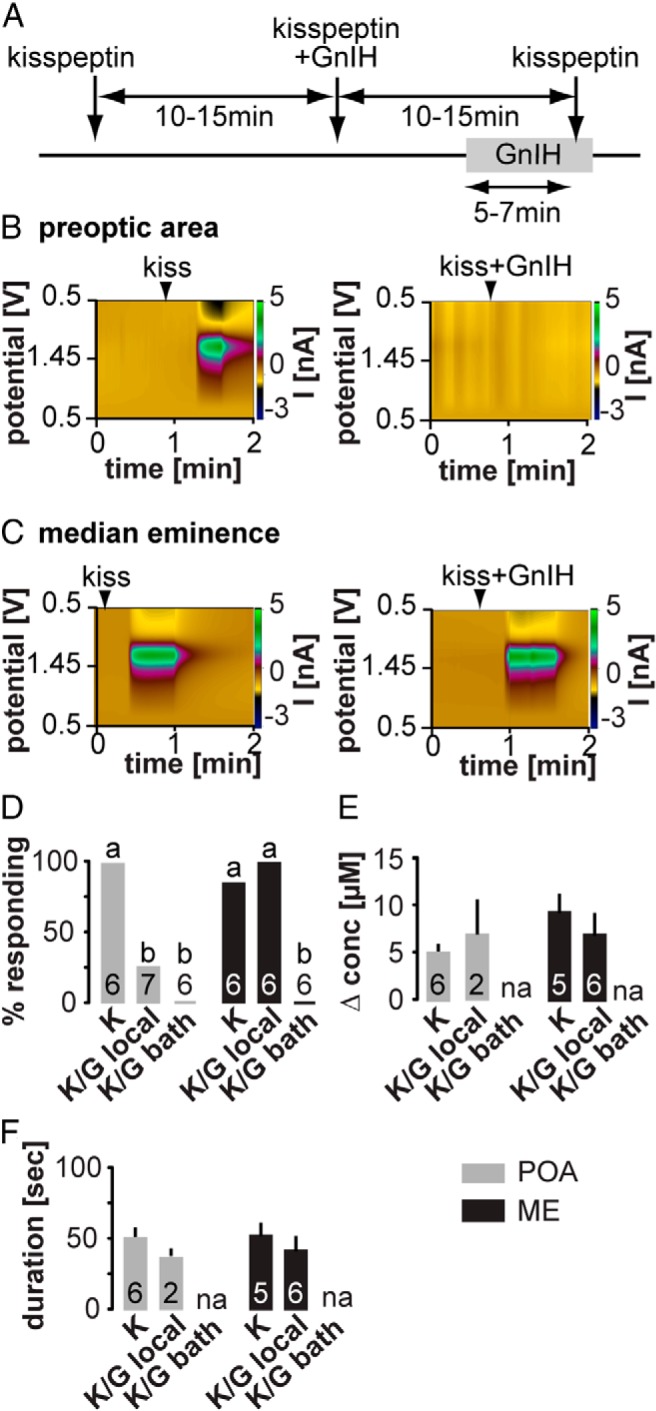
A, Experimental timeline: arrows indicate local application, gray bars indicate bath application. B and C, Representative examples of local kisspeptin-evoked GnRH secretion in the POA (B) and ME (C). Left panel shows response to local injection of kisspeptin alone in each region, right panel shows response to local injection of both kisspeptin and GnIH (ie, first 2 injection arrows in time line). D–F, Mean ± SEM percent of cells responding to kisspeptin (D), peak GnRH release amplitude (E) shown as change in concentration, and release duration (F). Gray bars show POA, black bars show ME, lower case letters indicate P < .05 χ2 test.
Kisspeptin-evoked GnRH secretion requires both mobilization of intracellular Ca2+ stores and extracellular Ca2+ influx
In the next set of experiments, we investigated the intersection between neuromodulators and intracellular calcium mobilization. Kisspeptin acts via a Gq-coupled receptor, depleting phosphatidylinositol 4,5-bisphosphate (PIP2) and activating diacylglycerol and IP3 production (34); any of these mechanisms might alter GnRH release. To test the hypothesis that kisspeptin-induced GnRH secretion requires release of Ca2+ from intracellular stores, we used XC to block IP3-dependent Ca2+ release (Figure 4A). Local administration of 20μM XC largely prevented kisspeptin from eliciting GnRH secretion in the ME (release observed in 1 of 6 cases) but not POA (release observed in 5 of 5 cases) (Figure 4). To test the possibility that XC targets in the POA are distal to the kisspeptin application site, XC (5μM) was bath applied. Bath-applied XC fully blocked kisspeptin-induced GnRH secretion in all 7 slices tested (Figure 4, F–H). These results suggest that action of intracellular Ca2+ mobilization in kisspeptin-evoked GnRH secretion is required in both the ME and POA. Moreover, the spatial distribution of XC targets (most likely endoplasmic reticulum), kisspeptin receptors and/or their downstream mediators, and GnRH release sites differ between the POA and ME. One of the caveats of XC is that the same concentrations that block IP3 release from the ER may have off-target effects, including inhibition of voltage-dependent Ca2+ and K+ channels (35). Inhibition of the latter would likely depolarize membrane potential and increase probability of release, thus an off-target inhibition of voltage-gated K+ channels by XC would tend to have the opposite effect to that observed here. Of interest, kisspeptin inhibits voltage-gated potassium channels in GnRH neurons (36), which may be related to its stimulation of GnRH release in the present study. To test whether blockade of cell-membrane Ca2+ channels without a direct effect on intracellular Ca2+ stores would prevent kisspeptin from inducing GnRH secretion, we locally applied kisspeptin in both the POA and the ME of slices bathed in ACSF containing 200μM Cd2+, a blocker of Ca2+ channels. Under these conditions, kisspeptin still stimulated GnRH release in the ME, but release was of lower amplitude (Figure 4, I and J). This suggests that XC works in the ME most likely through blocking IP3-dependent Ca2+ release. In the POA, however, Cd2+ prevented kisspeptin-induced GnRH release (Figure 4I). This suggests further regional differences between the POA and the ME but leaves open the possibility that in the POA XC blocks kisspeptin-induced release via both intracellular and cell-membrane mechanisms. The compound 2-aminoethoxydiphenylborane, which among its actions can block IP3-mediated Ca2+ release from stores, could not be tested in our experimental setting due to its profound fouling of the CFM that prevented GnRH detection (data not shown).
Figure 4. Pharmacological blockade of inositol triphospate-dependent intracellular Ca2+ release reduces kisspeptin-induced GnRH secretion.
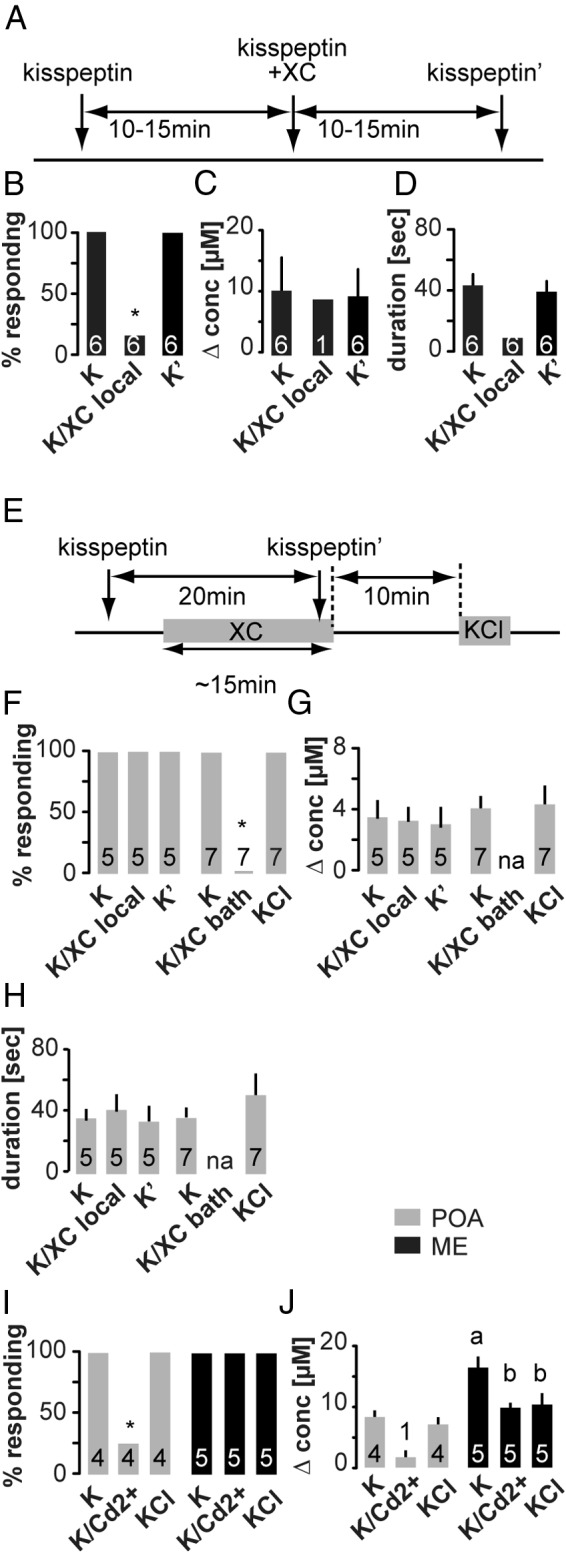
A, Experimental timeline for B–D and left portion of F–H; arrows indicate local, gray bars indicate bath application. B–D, Mean ± SEM percent of cells responding to kisspeptin in the ME (B), peak GnRH release amplitude (C) shown as change in concentration, and release duration (D). Numbers within bars indicate sample size, asterisks indicate P < .05 χ2 test. E. Experimental timeline for right portion of F–H, arrows indicate local, and gray bars indicate bath application. F–H, Mean ± SEM percent of cells responding to kisspeptin in the POA (F), peak GnRH release amplitude (G) shown as change in concentration, and release duration (H). I and J, Mean ± SEM percent of cells responding to kisspeptin or KCl and peak GnRH release amplitude (J) shown as change in concentration. Gray bars show POA, black bars show ME. Numbers within bars indicate sample size, asterisks indicate P < .05 χ2 test. Lower case letters indicate P < .05 repeated measures ANOVA with Dunnett's multiple comparison test.
Finally, we investigated whether kisspeptin-induced release requires generation of action potentials. This was tested only in the ME, because release induced by increasing intracellular Ca2+ was only action potential dependent in this region. Kisspeptin (10nM) was locally injected into the ME before and during treatment with TTX (Figure 5A). In 10 of 10 slices, kisspeptin still evoked GnRH secretion when action potentials were blocked (Figure 5, B–D). To test whether blockade of cell-membrane voltage-dependent Ca2+ channels in addition to action potentials inhibits kisspeptin-induced GnRH release, we repeated the experiment in the presence of 200μM Cd2+. This treatment blocked kisspeptin induction of GnRH secretion in the ME (n = 5 of 5) (Figure 5, B–D), suggesting that Ca2+ from both intracellular stores and outside of the cell underlies kisspeptin-induced GnRH secretion.
Figure 5. Blockade of both action potentials and soma membrane voltage-gated calcium channels prevents kisspeptin-induced GnRH release in the ME.
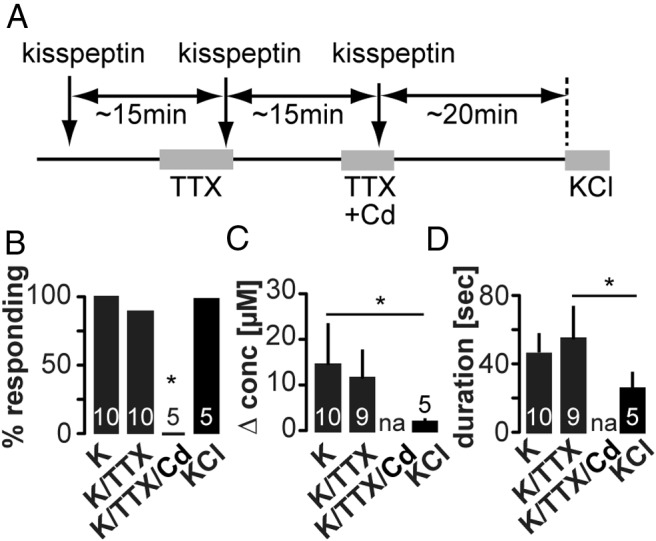
A, Experimental timeline; arrows indicate local, and gray bars indicate bath application. B–D, Mean ± SEM percent of cells responding to kisspeptin in the ME (B), peak GnRH release amplitude (C) shown as change in concentration, and release duration (D). Numbers within bars indicate sample size, asterisks indicate P < .05 χ2 test (B), one-way ANOVA with Tukey's multiple comparison test (C and D).
Discussion
Pulsatile GnRH secretion from the ME into the pituitary portal vasculature is a crucial driver of reproductive function, and local release of GnRH in the POA may serve a variety of neuromodulatory roles. Little is known, however, about the mechanisms underlying release in the ME vs POA. Here, we used a FSCV, a localized detector of GnRH release (3, 37), in combination with localized delivery of neuromodulators and/or agents to alter directly intracellular signaling. We demonstrate both similarities and differences in the region-specific regulation of GnRH release that may be attributable to relative location of cell surface receptors and GnRH release sites, as well as to signaling to and within the GnRH neurons.
Electron microscopic studies indicate that GnRH is localized to large dense-core vesicles in the soma, dendrites, axons, and terminals of GnRH neurons (38–41). We hypothesized that, consistent with other neurosecretory systems, increased intracellular Ca2+ is necessary for the release of those vesicles and tested this by brief local injection of thapsigargin or CPA to block Ca2+ reuptake into the endoplasmic reticulum. Thapsigargin has been reported to rapidly increase intracellular calcium levels in immortalized GnRH neurons (42, 43). Blocking SERCA pumps with either thapsigargin or CPA to elevate intracellular Ca2+ rapidly and repeatedly evoked GnRH release in both the POA and ME. Because treatments were delivered near the site of GnRH measurement, their effective zone is spatially limited. It is not possible, however, to determine whether treatments act directly upon GnRH neurons, on their afferents, on local glia or a combination of these. Signals between neurons would likely require action potentials in the presynaptic cell to release an intermediate signal, thus blocking action potential firing may reveal clues as to the site of action. Further, thapsigargin was reported to increase action potential firing in immortalized GnRH neurons (43). We thus determined whether SERCA blocker-induced GnRH release required action potentials. In the POA, GnRH release was action potential independent. This suggests that GnRH release is most likely evoked by SERCA inhibition directly in GnRH neurons in the somato-dendritic region of these cells. Action potential-independent modification of neuromodulator release from presynaptic neurons cannot, however, be completely excluded (44).
In contrast to the POA, action potentials were required for thapsigargin-induced GnRH release in the ME. This indicates that propagation of a signal is needed either between an afferent neuron and the GnRH neuron, or to connect more distant elements within the GnRH neuron itself (in the case of SERCA blockers, endoplasmic reticulum, and GnRH release sites). With regard to signaling from an afferent neuron, CPA-evoked GnRH release persisted in the ME of kisspeptin KO mice. This indicates that kisspeptin is not required for this response, although the increased amplitude of GnRH release in response to kisspeptin vs CPA in kisspeptin KO animals may imply a role for kisspeptin in enhancing this response. Overall, the present observation that blocking action potentials blocks the ability of elevating intracellular calcium with SERCA blockers to induce GnRH release in the ME is not attributable to a failure of kisspeptin release from afferent neurons. This does not eliminate the possibility that SERCA blockers are increasing action potential firing and subsequent neurosecretion from an afferent neuron. In this regard, activation of neurokinin 3 receptors also induces GnRH release in the ME of kisspeptin KO mice (37). Neurokinin 3 receptors are the high-affinity binding site for neurokinin B (45), a peptide that is coexpressed in some kisspeptin neurons (46) and shown to be important for fertility (47). Further, tanycytes surrounding GnRH terminals contain endoplasmic reticulum (40, 41). Thus indirect actions via either neurons or glia are possible explanations for the action potential dependence of SERCA blocker-induced GnRH release in the ME. These data support and extend previous reports of differences between central/dendritic and peripheral/terminal release in the magnocellular neuroendocrine system releasing oxytocin and vasopressin. Dendritic release of these peptides can be elicited independent of action potential firing, whereas secretion from axon terminals is typically activity dependent (44). Dendritic release involves mobilization of Ca2+ from thapsigargin-sensitive stores for both oxytocin and vasopressin (16) and further calcium influx due to activation of voltage-sensitive Ca2+ channels for vasopressin (16, 17). For the GnRH system, coordination possibly provided by action potential-driven release in the ME may be critical for generating the pulsatile pattern required by the pituitary.
The physiological activator of increased intracellular calcium required for dendritic release in magnocellular neurons is proposed to be ligand-receptor interactions (31). In GnRH neurons, kisspeptin is a potent stimulator of action potential firing (19) and hormone release into the pituitary portal vasculature via the ME (48). The kisspeptin receptor (kiss1r) is coupled to Gq and signals via PIP2 depletion and IP3 (49, 50). We demonstrate here that GnRH secretion can be evoked by kisspeptin in an IP3-dependent manner not only in the ME (51) but also in the POA. In the POA, however, it was necessary to block IP3-mediated calcium release over a wider area to inhibit kisspeptin-induced GnRH release. This suggests the kisspeptin receptor and intracellular Ca2+ stores are further apart in the GnRH neuron processes in the perisomatic region than they are in the GnRH neuron terminals in the ME, although it is also possible that regional differences in drug diffusion contribute to these observations. Further, the localized nature of both kisspeptin application and GnRH measurement used in the present study indicates that, in both the POA and ME, kisspeptin receptors and sites of GnRH release are likely overlapping (Figure 6). Interestingly, local injection of kisspeptin repeatedly induced GnRH release of short duration with no decrement under the conditions used in the present studies. This is in contrast to reports of desensitization of GnRH neuron action potential firing with repeated bath application of kisspeptin, even when bath application was a single drop to the slice recording chamber with a high flow rate (52, 53). Further, the action potential firing increases in those studies lasted several minutes (typically >20 min), compared with relatively brief pulses of release in the present study lasting under 2 minutes. This may be due to a lower concentration and smaller volume and therefore smaller mass of kisspeptin being delivered in a more localized manner in the present study. Although clearly effective in eliciting GnRH release, the amount of kisspeptin used in the present study may not induce desensitization and also may be more easily washed away. With regard to the latter, studies of kisspeptin signaling in cultured cells indicate that intracellular elevations in Ca2+ are prolonged unless kisspeptin is removed from the medium (54). It is also possible that the bath application methods used in the previous studies more broadly engaged additional parts of the upstream network, thereby resulting in prolonged stimulation of GnRH neuron activity. Because GnRH neurons express kiss1R, kisspeptin action is likely, at least in part, directly on GnRH neurons. Kisspeptin can, however, also excite GnRH neurons indirectly by increasing excitatory GABAergic as well as glutamatergic transmission to these cells (52, 55–57).
Figure 6. Possible models for the differential regulation of GnRH release in the POA and ME.
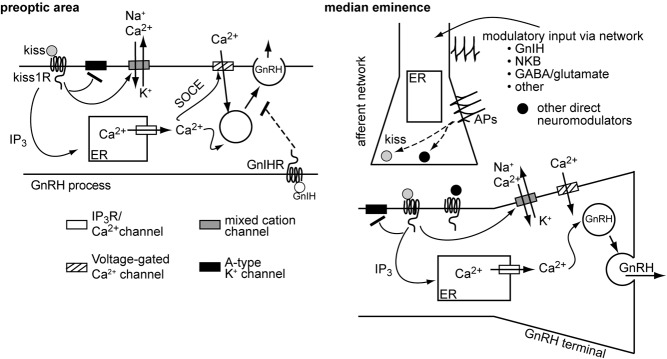
In the POA (left), GnRH release can occur without action potentials and can be locally inhibited by GnIH. We postulate all the elements necessary for control may be contained locally within the GnRH process itself. It is important to point out that influences from afferent neurons are not excluded. For example, kisspeptin clearly influences release, likely via direct action on the neuronal process. In the ME (right), although kisspeptin can induce GnRH release in an action potential-independent manner, suggesting that the GnRH neuron itself does not need to generate spikes for secretion, regulation of GnRH release also appeared to depend more on the upstream network, because 1) elevating intracellular Ca2+ by blocking endoplasmic reticulum (ER) reuptake was unable to induce GnRH release in the absence of action potentials; 2) GnIH did not inhibit GnRH release locally, suggesting that it is sculpting secretory pattern via the network; and 3) it is likely that endogenous release of kisspeptin and other neuromodulators is action potential dependent. We hypothesize that by injecting kisspeptin locally the requirement for GnRH neuron action potential firing was bypassed and kisspeptin directly stimulated GnRH release from the GnRH terminal. SOCE, store-operated calcium entry.
It is also possible that the robust and prolonged increase in GnRH neuron firing rate in response to kisspeptin may not generate prolonged neurosecretion. Of further interest in this regard, blocking action potential firing did not prevent kisspeptin from eliciting GnRH secretion in the ME without concomitant blockade of Ca2+ influx via Cd2+-sensitive channels. The apparent discrepancy between action potential dependence of GnRH release evoked by thapsigargin and action potential-independent release evoked by kisspeptin in the ME can be explained in a couple of ways. First, IP3-mediated release of calcium subsequent to kiss1r activation would not be blocked by preventing action potential firing if kisspeptin were acting on kiss1r expressed by GnRH neurons, providing further evidence for direct action of kisspeptin on GnRH neurons to evoke GnRH release. Second, kisspeptin activates mechanisms in addition to intracellular calcium mobilization in GnRH neurons, including reducing A-type potassium currents (36) and increasing transient receptor potential channel 4 (TRPC4) currents (58, 50) and nonspecific cation currents (59). Third, depletion of PIP2 from the plasma membrane can modulate a variety of ion channel activities (60–65) that could ultimately change secretory output.
Another neuropeptide postulated to be involved in control of GnRH neuron activity is GnIH. It inhibits both electrical activity of GnRH neurons (22, 66) and secretion of gonadotropic hormones from the pituitary (67). GnIH was also recently implicated in prepubertal suppression of FSH levels and LH response to GnRH (68, 69). These findings suggest that one mechanism underlying GnIH inhibition of LH secretion may be via suppression of GnRH release. We demonstrate that GnIH counteracts the stimulatory effect of kisspeptin on GnRH release in both the POA and ME, in contrast to the tight overlap of kisspeptin receptors and GnRH release. However, there were region-specific differences in spatial distribution of GnIH targets. In the ME, local coapplication of GnIH with kisspeptin did not prevent kisspeptin-induced GnRH secretion, suggesting no or insufficient activation of GnIH receptors near kisspeptin receptors in this region (Figure 6). Consistent with this observation, immunohistochemistry has demonstrated little to no GnIH immunopositive fibers in the ME (70–72). This suggests that GnIH receptors may not be present in the ME. In the POA, however, local GnIH effectively prevented kisspeptin-evoked GnRH release in most cases, suggesting more proximal location of kisspeptin receptors and GnIH receptors in this region (Figure 6). Consistent with this observation, some GnRH neurons have been reported to express the putative GnIH receptor G-protein coupled receptor 147 (73). This observation contrasts somewhat with previous electrophysiological studies demonstrating that GnIH inhibits firing only in a subset of GnRH neurons (66, 22). GnIH could block GnRH release via mechanisms not involving firing inhibition, another possible indication of uncoupling between GnRH neuron action potential generation and GnRH release. Moreover, bath application of GnIH suppressed kisspeptin-evoked release in both POA and ME. This suggests that GnIH may prevent kisspeptin-induced release of GnRH via upstream networks.
Location-dependent regulation of GnRH release in the POA vs ME suggests regional differences in GnRH neuron processes, particularly in the arrangement of neuromodulator receptors, intracellular signaling systems, and GnRH release sites in these 2 regions (Figure 6). The limited electrophysiological studies performed in distal GnRH processes approaching the ME suggest that these processes receive fast synaptic transmission (74). A similar arrangement is likely for neuromodulatory inputs and is indeed suggested by the differential regulation of GnRH release observed in the present study. Such distinct regulatory mechanisms allow GnRH release in each region to be independent of the other. This facilitates the fine-tuning of the GnRH output to serve a variety of different physiological functions both within the brain and exiting the brain as neuroendocrine output to the pituitary.
Acknowledgments
We thank Elizabeth Wagenmaker and Dr Laura Burger for expert technical assistance, Dr R. Anthony DeFazio and Dr Carol F. Elias for editorial comments, and Dr Seminara and Dr Chan for sharing the kisspeptin knockout mice.
This work was supported by the National Institute of Health/Eunice Kennedy Shriver National Institute of Child Health and Human Development Grant R01 HD34860.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ACSF
- artificial cerebrospinal fluid
- CFM
- carbon fiber microelectrode
- CPA
- cyclopiazonic acid
- CV
- cyclic voltammogram
- FSCV
- fast-scan cyclic voltammetry
- GFP
- green fluorescent protein
- GnIH
- gonadotropin-inhibitory hormone
- IP3
- inositol trisphosphate
- kiss1r
- kisspeptin receptor
- KO
- knock-out
- ME
- median eminence
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- POA
- preoptic area
- SERCA
- sarcoendoplasmic reticulum calcium transport ATPase
- TTX
- tetrodotoxin
- XC
- xestospongin C.
References
- 1. Clarke IJ, Cummins JT. The temporal relationship between gonadotropin releasing hormone (GnRH) and luteinizing hormone (LH) secretion in ovariectomized ewes. Endocrinology. 1982;111(5):1737–1739. [DOI] [PubMed] [Google Scholar]
- 2. Moenter SM, Brand RM, Midgley AR, Karsch FJ. Dynamics of gonadotropin-releasing hormone release during a pulse. Endocrinology. 1992;130(1):503–510. [DOI] [PubMed] [Google Scholar]
- 3. Glanowska KM, Venton BJ, Moenter SM. Fast scan cyclic voltammetry as a novel method for detection of real-time gonadotropin-releasing hormone release in mouse brain slices. J Neurosci. 2012;32(42):14664–14669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fuenzalida LC, Keen KL, Terasawa E. Colocalization of FM1–43, Bassoon, and GnRH-1: GnRH-1 release from cell bodies and their neuroprocesses. Endocrinology. 2011;152(11):4310–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DePaolo LV, King RA, Carrillo AJ. In vivo and in vitro examination of an autoregulatory mechanism for luteinizing hormone-releasing hormone. Endocrinology. 1987;120(1):272–279. [DOI] [PubMed] [Google Scholar]
- 6. Xu C, Xu XZ, Nunemaker CS, Moenter SM. Dose-dependent switch in response of gonadotropin-releasing hormone (GnRH) neurons to GnRH mediated through the type I GnRH receptor. Endocrinology. 2004;145(2):728–735. [DOI] [PubMed] [Google Scholar]
- 7. Xu C, Roepke TA, Zhang C, Rønnekleiv OK, Kelly MJ. Gonadotropin-releasing hormone (GnRH) activates the m-current in GnRH neurons: an autoregulatory negative feedback mechanism? Endocrinology. 2008;149(5):2459–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen P, Moenter SM. GABAergic transmission to gonadotropin-releasing hormone (GnRH) neurons is regulated by GnRH in a concentration-dependent manner engaging multiple signaling pathways. J Neurosci Off J Soc Neurosci. 2009;29(31):9809–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han SK, Lee K, Bhattarai JP, Herbison AE. Gonadotrophin-releasing hormone (GnRH) exerts stimulatory effects on GnRH neurons in intact adult male and female mice. J Neuroendocrinol. 2010;22(3):188–195. [DOI] [PubMed] [Google Scholar]
- 10. Terasawa E. Control of luteinizing hormone-releasing hormone pulse generation in nonhuman primates. Cell Mol Neurobiol. 1995;15(1):141–164. [DOI] [PubMed] [Google Scholar]
- 11. Caraty A, Delaleu B, Chesneau D, Fabre-Nys C. Sequential role of e2 and GnRH for the expression of estrous behavior in ewes. Endocrinology. 2002;143(1):139–45. [DOI] [PubMed] [Google Scholar]
- 12. Martinez-Fuentes AJ, Hu L, Krsmanovic LZ, Catt KJ. Gonadotropin-releasing hormone (GnRH) receptor expression and membrane signaling in early embryonic GnRH neurons: role in pulsatile neurosecretion. Mol Endocrinol. 2004;18(7):1808–1817. [DOI] [PubMed] [Google Scholar]
- 13. Castel M, Morris J, Belenky M. Non-synaptic and dendritic exocytosis from dense-cored vesicles in the suprachiasmatic nucleus. Neuroreport. 1996;7(2):543–547. [DOI] [PubMed] [Google Scholar]
- 14. Lambert RC, Dayanithi G, Moos FC, Richard P. A rise in the intracellular Ca2+ concentration of isolated rat supraoptic cells in response to oxytocin. J Physiol. 1994;478(pt 2):275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dayanithi G, Widmer H, Richard P. Vasopressin-induced intracellular Ca2+ increase in isolated rat supraoptic cells. J Physiol. 1996;490(pt 3):713–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ludwig M, Sabatier N, Bull PM, Landgraf R, Dayanithi G, Leng G. Intracellular calcium stores regulate activity-dependent neuropeptide release from dendrites. Nature. 2002;418(6893):85–89. [DOI] [PubMed] [Google Scholar]
- 17. Ludwig M, Bull PM, Tobin VA, et al. Regulation of activity-dependent dendritic vasopressin release from rat supraoptic neurones. J Physiol. 2005;564(2):515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gore AC. GnRH: The Master Molecule of Reproduction. Norwell, MA: Kluwer Academic Publishers; 2002. [Google Scholar]
- 19. Pielecka-Fortuna J, Chu Z, Moenter SM. Kisspeptin acts directly and indirectly to increase gonadotropin-releasing hormone neuron activity and its effects are modulated by estradiol. Endocrinology. 2008;149(4):1979–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herbison AE, de Tassigny Xd, Doran J, Colledge WH. Distribution and postnatal development of Gpr54 gene expression in mouse brain and gonadotropin-releasing hormone neurons. Endocrinology. 2010;151(1):312–321. [DOI] [PubMed] [Google Scholar]
- 21. Smith JT, Li Q, Yap KS, et al. Kisspeptin is essential for the full preovulatory LH surge and stimulates GnRH release from the isolated ovine median eminence. Endocrinology. 2011;152(3):1001–1012. [DOI] [PubMed] [Google Scholar]
- 22. Wu M, Dumalska I, Morozova E, van den Pol AN, Alreja M. Gonadotropin inhibitory hormone inhibits basal forebrain vGluT2-gonadotropin-releasing hormone neurons via a direct postsynaptic mechanism. J Physiol. 2009;587(pt 7):1401–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Umatani C, Abe H, Oka Y. Neuropeptide RFRP inhibits the pacemaker activity of terminal nerve GnRH neurons. J Neurophysiol. 2013;109(9):2354–2363. [DOI] [PubMed] [Google Scholar]
- 24. Suter KJ, Song WJ, Sampson TL, et al. Genetic targeting of green fluorescent protein to gonadotropin-releasing hormone neurons: characterization of whole-cell electrophysiological properties and morphology. Endocrinology. 2000;141(1):412–419. [DOI] [PubMed] [Google Scholar]
- 25. Lapatto R, Pallais JC, Zhang D, et al. Kiss1−/− mice exhibit more variable hypogonadism than Gpr54−/− mice. Endocrinology. 2007;148(10):4927–4936. [DOI] [PubMed] [Google Scholar]
- 26. Nunemaker CS, DeFazio RA, Moenter SM. Estradiol-sensitive afferents modulate long-term episodic firing patterns of GnRH neurons. Endocrinology. 2002;143(6):2284–2292. [DOI] [PubMed] [Google Scholar]
- 27. Chu Z, Moenter SM. Endogenous activation of metabotropic glutamate receptors modulates GABAergic transmission to gonadotropin-releasing hormone neurons and alters their firing rate: a possible local feedback circuit. J Neurosci Off J Soc Neurosci. 2005;25(24):5740–5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mundroff ML, Wightman RM. Amperometry and cyclic voltammetry with carbon fiber microelectrodes at single cells. Curr Protoc Neurosci Editor Board Jacqueline N Crawley Al. 2002;Chapter 6:Unit 6.14. [DOI] [PubMed] [Google Scholar]
- 29. Shoback D, Chen TH, Pratt S, Lattyak B. Thapsigargin stimulates intracellular calcium mobilization and inhibits parathyroid hormone release. J Bone Miner Res Off J Am Soc Bone Miner Res. 1995;10(5):743–750. [DOI] [PubMed] [Google Scholar]
- 30. Badaoui A, Huchet-Cadiou C, Léoty C. Effects of cyclopiazonic acid on membrane currents, contraction and intracellular calcium transients in frog heart. J Mol Cell Cardiol. 1995;27(11):2495–2505. [DOI] [PubMed] [Google Scholar]
- 31. Ludwig M, Leng G. Dendritic peptide release and peptide-dependent behaviours. Nat Rev Neurosci. 2006;7(2):126–136. [DOI] [PubMed] [Google Scholar]
- 32. Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca(2+)-ATPases. Trends Pharmacol Sci. 1998;19(4):131–135. [DOI] [PubMed] [Google Scholar]
- 33. Beck A, Nieden RZ, Schneider HP, Deitmer JW. Calcium release from intracellular stores in rodent astrocytes and neurons in situ. Cell Calcium. 2004;35(1):47–58. [DOI] [PubMed] [Google Scholar]
- 34. Bianco SD, Kaiser UB. Molecular biology of the kisspeptin receptor: signaling, function, and mutations. Adv Exp Med Biol. 2013;784:133–158. [DOI] [PubMed] [Google Scholar]
- 35. Ozaki H, Hori M, Kim YS, et al. Inhibitory mechanism of xestospongin-C on contraction and ion channels in the intestinal smooth muscle. Br J Pharmacol. 2002;137(8):1207–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pielecka-Fortuna J, DeFazio RA, Moenter SM. Voltage-gated potassium currents are targets of diurnal changes in estradiol feedback regulation and kisspeptin action on gonadotropin-releasing hormone neurons in mice. Biol Reprod. 2011;85(5):987–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gaskins GT, Glanowska KM, Moenter SM. Activation of neurokinin 3 receptors stimulates GnRH release in a location-dependent but kisspeptin-independent manner in adult mice. Endocrinology. 2013;154(11):3984–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lehman MN, Karsch FJ, Robinson JE, Silverman AJ. Ultrastructure and synaptic organization of luteinizing hormone-releasing hormone (LHRH) neurons in the anestrous ewe. J Comp Neurol. 1988;273(4):447–458. [DOI] [PubMed] [Google Scholar]
- 39. Goldsmith PC, Thind KK, Perera AD, Plant TM. Glutamate-immunoreactive neurons and their gonadotropin-releasing hormone-neuronal interactions in the monkey hypothalamus. Endocrinology. 1994;134(2):858–868. [DOI] [PubMed] [Google Scholar]
- 40. Prevot V, Dutoit S, Croix D, Tramu G, Beauvillain JC. Semi-quantitative ultrastructural analysis of the localization and neuropeptide content of gonadotropin releasing hormone nerve terminals in the median eminence throughout the estrous cycle of the rat. Neuroscience. 1998;84(1):177–191. [DOI] [PubMed] [Google Scholar]
- 41. Prevot V, Croix D, Bouret S, et al. Definitive evidence for the existence of morphological plasticity in the external zone of the median eminence during the rat estrous cycle: implication of neuro-glio-endothelial interactions in gonadotropin-releasing hormone release. Neuroscience. 1999;94(3):809–819. [DOI] [PubMed] [Google Scholar]
- 42. Charles AC, Hales TG. Mechanisms of spontaneous calcium oscillations and action potentials in immortalized hypothalamic (GT1–7) neurons. J Neurophysiol. 1995;73(1):56–64. [DOI] [PubMed] [Google Scholar]
- 43. Van Goor F, Krsmanovic LZ, Catt KJ, Stojilkovic SS. Coordinate regulation of gonadotropin-releasing hormone neuronal firing patterns by cytosolic calcium and store depletion. Proc Natl Acad Sci USA. 1999;96(7):4101–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ludwig M, Callahan MF, Morris M. Effects of tetrodotoxin on osmotically stimulated central and peripheral vasopressin and oxytocin release. Neuroendocrinology. 1995;62(6):619–627. [DOI] [PubMed] [Google Scholar]
- 45. Krause JE, Staveteig PT, Mentzer JN, et al. Functional expression of a novel human neurokinin-3 receptor homolog that binds [3H]senktide and [125I-MePhe7]neurokinin B, and is responsive to tachykinin peptide agonists. Proc Natl Acad Sci USA. 1997;94(1):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goodman RL, Lehman MN, Smith JT, et al. Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin A and neurokinin B. Endocrinology. 2007;148(12):5752–5760. [DOI] [PubMed] [Google Scholar]
- 47. Topaloglu AK, Semple RK. Neurokinin B signalling in the human reproductive axis. Mol Cell Endocrinol. 2011;346(1–2):57–64. [DOI] [PubMed] [Google Scholar]
- 48. Constantin S, Caraty A, Wray S, Duittoz AH. Development of gonadotropin-releasing hormone-1 secretion in mouse nasal explants. Endocrinology. 2009;150(7):3221–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kotani M, Detheux M, Vandenbogaerde A, et al. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem. 2001;276(37):34631–34636. [DOI] [PubMed] [Google Scholar]
- 50. Zhang C, Bosch MA, Rønnekleiv OK, Kelly MJ. Kisspeptin activation of TRPC4 channels in female GnRH neurons requires PIP2 depletion and cSrc kinase activation. Endocrinology. 2013;154(8):2772–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. d'Anglemont de Tassigny X, Fagg LA, Carlton MB, Colledge WH. Kisspeptin can stimulate gonadotropin-releasing hormone (GnRH) release by a direct action at GnRH nerve terminals. Endocrinology. 2008;149(8):3926–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pielecka-Fortuna J, Moenter SM. Kisspeptin increases γ-aminobutyric acidergic and glutamatergic transmission directly to gonadotropin-releasing hormone neurons in an estradiol-dependent manner. Endocrinology. 2010;151(1):291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dumalska I, Wu M, Morozova E, Liu R, van den Pol A, Alreja M. Excitatory effects of the puberty-initiating peptide kisspeptin and group I metabotropic glutamate receptor agonists differentiate two distinct subpopulations of gonadotropin-releasing hormone neurons. J Neurosci. 2008;28(32):8003–8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Min L, Soltis K, Reis AC, et al. Dynamic kisspeptin receptor trafficking modulates kisspeptin-mediated calcium signaling. Mol Endocrinol. 2014;28(1):16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. DeFazio RA, Heger S, Ojeda SR, Moenter SM. Activation of A-type γ-aminobutyric acid receptors excites gonadotropin-releasing hormone neurons. Mol Endocrinol. 2002;16(12):2872–2891. [DOI] [PubMed] [Google Scholar]
- 56. Herbison AE, Moenter SM. Depolarising and hyperpolarising actions of GABA(A) receptor activation on gonadotrophin-releasing hormone neurones: towards an emerging consensus. J Neuroendocrinol. 2011;23(7):557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Constantin S, Piet R, Iremonger K, et al. GnRH neuron firing and response to GABA in vitro depend on acute brain slice thickness and orientation. Endocrinology. 2012;153(8):3758–3769. [DOI] [PubMed] [Google Scholar]
- 58. Zhang C, Roepke TA, Kelly MJ, Rønnekleiv OK. Kisspeptin depolarizes gonadotropin-releasing hormone neurons through activation of TRPC-like cationic channels. J Neurosci. 2008;28(17):4423–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu X, Lee K, Herbison AE. Kisspeptin excites gonadotropin-releasing hormone neurons through a phospholipase C/calcium-dependent pathway regulating multiple ion channels. Endocrinology. 2008;149(9):4605–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hilgemann DW, Ball R. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science. 1996;273(5277):956–959. [DOI] [PubMed] [Google Scholar]
- 61. Baukrowitz T, Schulte U, Oliver D, et al. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 1998;282(5391):1141–1144. [DOI] [PubMed] [Google Scholar]
- 62. Shyng SL, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998;282(5391):1138–1141. [DOI] [PubMed] [Google Scholar]
- 63. Chuang HH, Prescott ED, Kong H, et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411(6840):957–962. [DOI] [PubMed] [Google Scholar]
- 64. Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002;35(3):507–520. [DOI] [PubMed] [Google Scholar]
- 65. Wu L, Bauer CS, Zhen XG, Xie C, Yang J. Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature. 2002;419(6910):947–952. [DOI] [PubMed] [Google Scholar]
- 66. Ducret E, Anderson GM, Herbison AE. RFamide-related peptide-3, a mammalian gonadotropin-inhibitory hormone ortholog, regulates gonadotropin-releasing hormone neuron firing in the mouse. Endocrinology. 2009;150(6):2799–2804. [DOI] [PubMed] [Google Scholar]
- 67. Ubuka T, Son YL, Tobari Y, Tsutsui K. Gonadotropin-inhibitory hormone action in the brain and pituitary. Front Endocrinol. 2012;3:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. León S, García-Galiano D, Ruiz-Pino F, et al. Physiological roles of gonadotropin-inhibitory hormone signaling in the control of mammalian reproductive axis: studies in the NPFF1 receptor null mouse. Endocrinology. 2014;155(8):2953–2965. [DOI] [PubMed] [Google Scholar]
- 69. Glanowska KM, Burger LL, Moenter SM. Regulation of high frequency GnRH release in early prepubertal mice. Program of the 96th Annual Meeting of The Endocrine Society Chicago, IL; 2014:OR30–3. [Google Scholar]
- 70. Ukena K, Tsutsui K. Distribution of novel RFamide-related peptide-like immunoreactivity in the mouse central nervous system. Neurosci Lett. 2001;300(3):153–156. [DOI] [PubMed] [Google Scholar]
- 71. Kriegsfeld LJ, Mei DF, Bentley GE, et al. Identification and characterization of a gonadotropin-inhibitory system in the brains of mammals. Proc Natl Acad Sci USA. 2006;103(7):2410–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rizwan MZ, Porteous R, Herbison AE, Anderson GM. Cells expressing RFamide-related peptide-1/3, the mammalian gonadotropin-inhibitory hormone orthologs, are not hypophysiotropic neuroendocrine neurons in the rat. Endocrinology. 2009;150(3):1413–1420. [DOI] [PubMed] [Google Scholar]
- 73. Rizwan MZ, Poling MC, Corr M, et al. RFamide-related peptide-3 receptor gene expression in GnRH and kisspeptin neurons and GnRH-dependent mechanism of action. Endocrinology. 2012;153(8):3770–3779. [DOI] [PubMed] [Google Scholar]
- 74. Herde MK, Iremonger KJ, Constantin S, Herbison AE. GnRH neurons elaborate a long-range projection with shared axonal and dendritic functions. J Neurosci. 2013;33(31):12689–12697. [DOI] [PMC free article] [PubMed] [Google Scholar]