

Abstract
Congenital hypothyroidism caused by thyroid dysgenesis (CHTD) is a common congenital disorder with a birth prevalence of 1 case in 4000 live births, and up to 8% of individuals with CHTD have co-occurring congenital heart disease. Initially we found nine patients with cardiac and thyroid congenital disorders in our cohort of 158 CHTD patients. To enrich for a rare phenotype likely to be genetically simpler, we selected three patients with a ventricular septal defect for molecular studies. Then, to assess whether rare de novo copy number variants and coding mutations in candidate genes are a source of genetic susceptibility, we used a genome-wide single-nucleotide polymorphism array and Sanger sequencing to analyze blood DNA samples from selected patients with co-occurring CHTD a congenital heart disease. We found rare variants in all three patients, and we selected Netrin-1 as the biologically most plausible contributory factor for functional studies. In zebrafish, ntn1a and ntn1b were not expressed in thyroid tissue, but ntn1a was expressed in pharyngeal arch mesenchyme, and ntn1a-deficient embryos displayed defective aortic arch artery formation and abnormal thyroid morphogenesis. The functional activity of the thyroid in ntn1a-deficient larvae was, however, preserved. Phenotypic analysis of affected zebrafish indicates that abnormal thyroid morphogenesis resulted from a lack of proper guidance exerted by the dysplastic vasculature of ntn1a-deficient embryos. Hence, careful phenotyping of patients combined with molecular and functional studies in zebrafish identify Netrin-1 as a potential shared genetic factor for cardiac and thyroid congenital defects.
Congenital hypothyroidism caused by thyroid dysgenesis (CHTD) is a common congenital disorder with a birth prevalence of 1 case in 4000 live births (1), and up to 8% of individuals with CHTD have co-occurring congenital heart disease (CHD) (2). Incomplete migration of the thyroid resulting in ectopic tissue (sublingual thyroid) is the most common thyroid developmental defect (up to 80%), with athyreosis and orthotopic thyroid hypoplasia being less frequent. Congenital hypothyroidism caused by thyroid dysgenesis exists in the familial (2%) and sporadic (98%) forms (3), with a discordance rate of 92% between monozygotic twins (4) and a female predominance (2); congenital heart disease is also predominantly sporadic (5) with a discordance rate of 90% between monochorionic twins (6).
Germline mutations in thyroid-related transcription factors NKX2.1, FOXE1, and PAX8 have been identified in 3% of patients with sporadic CHTD and no cardiac defects (7, 8). For NKX2.1 and PAX8, all mutations reported to date were monoallelic, and patients presented with orthotopic thyroid gland hypoplasia. Conversely, FOXE1 mutations have been found in the biallelic state in patients presenting with athyreosis, cleft palate, and spiky hair (7). The lack of linkage to these genes in some multiplex families with CHTD points to considerable genetic heterogeneity in this disorder (9). Two other genes, NKX2.5 and HHEX, have been implicated in thyroid and cardiac phenotypes based on human and genetic mouse models (10–12). In transcriptome analysis of ectopic thyroids, none of these transcription factors exhibited decreased expression (13). Altogether these findings underline the importance of identifying new genes associated with CHTD and congenital heart disease (CHD) by considering the following: 1) sporadic de novo germline genetic events [ie, either de novo copy number variants (CNVs) or de novo point mutations], 2) the possibility of multiple hits (de novo or inherited) in modifier genes, 3) the possibility of low-penetrance variants, and 4) somatic epigenetic or genetic events (14).
CNVs have been recognized as a major source of genetic variability (15, 16) and have been shown to confer susceptibility to sporadic diseases (15, 17) such as autism (17) and CHD (18). An association between CHTD and chromosomal variants was demonstrated in two previous studies performed with lower resolution than that used in the present work (19, 20).
Like CHTD, CHD is a sporadic condition (5). An 8% co-occurrence of CHD in CHTD is greater than expected by chance alone, given a global prevalence of CHD of 0.6%–1% (21, 22). Therefore, CHD and CHTD might share common genetic or epigenetic etiologies. A link between cardiovascular and thyroid development has previously been demonstrated in mouse and zebrafish studies revealing coordinated morphogenetic processes (23) as well as the occurrence of thyroid anomalies in mice and zebrafish models with defective cardiovascular development (23, 24). Therefore, we performed a pilot study by selecting patients with co-occurring CHD and CHTD to look for shared genetic factors.
Subjects and Methods
Ethics statement
This study was approved by the Sainte Justine Ethics Committee (Research Ethic Board number 94). All of the parents provided written informed consent.
Subjects
To determine whether rare CNVs are associated with syndromic cases of CHTD, we selected patients with CHTD and CHD. First, the Zoom Endo database (Endocrinology Service, Centre Hospitalier Universitaire Sainte-Justine) containing 158 patients with CHTD (diagnosis established by 99Tc scintigraphy) was merged with a cardiac echocardiography database containing echographies of 20 000 different patients from the same institution. Nine patients were found to overlap, representing a prevalence of 5.5% (9 of 158) for CHD among patients with CHTD, a prevalence consistent with an earlier survey by our group (2). To enrich for a rare phenotype likely to be genetically simpler, we selected three patients with a ventricular septal defect (VSD) for molecular studies. The other six patients, with transient patent ductus arteriosus (PDA) and transient atrial septal defect (ASD) type II (ASD II), were excluded because transient PDA and ASD II are benign and observed in high proportion of otherwise normal newborns; one case (patient 3) also had developmental delay and arthrogryposis (Figure 1). Clinical characteristics for each patient are reported in Table 1. The control cohort consisted of 203 ethnically matched individuals with no evidence of thyroid or heart disease after a medical history review, physical examination, electrocardiogram, and echocardiography. Informed consent was obtained from all participants. Cardiac and endocrinological phenotyping of patients are reported in Table 1, and complete clinical case reports are available in Supplemental Data.
Figure 1. Clinical pictures of patient 3 with CHD, thyroid ectopy, and arthrogryposis.

Presenting patient features are shown; these include down-slanting palpebral fissures, high nasal bridge, hook-shaped nose, small mouth, normal palate, low-set ears with folded helices, and distal arthrogryposis involving fingers and toes (panels A–C). D, For the same patient, the ectopic thyroid gland is revealed by 99mTc sodium pertechnetate scintigraphy.
Table 1.
Phenotypic and Genotypic Characterization of the Three Patients
| Case | Sex | Thyroidal Phenotype | Cardiac Phenotype | Other Phenotype | Direct Sequencing NKX2.5a | CNVb |
Validation | Taq Man Assay Identification | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Copy | Loss/Gain | Chromosome | Start | End | Size, kb | Genes | Classification | ||||||||
| 1 | F | Ectopy | VSD, ASD | ø | R25C | 0c | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| 2 | M | Athyreosis | VSD | ø | wt | 1 | Loss | 16p12.3 | 19 140 333 | 19 216 014 | 76 | SYT17 | Rare Inherited | Confirmed by qPCR | Hs01544221_cn |
| 3 | M | Ectopy | VSD, ASD, PDA | Arthrogryposis | wt | 1 | Loss | 17p13.1 | 8 901 608 | 8 913 177 | 12 | NTN1 | Rare de Novo | Confirmed by qPCR | Hs05487409_cn |
| 1 | Loss | 22q11.21 | 19 046 924 | 19 902 202 | 855 | CRKL | Rare de Novo | Confirmed by qPCR | Hs04079552_cn | ||||||
| 2 | Gain | Y | Karyotype 47XYY | ||||||||||||
Abbreviations: F, female; M, male; N/A, not applicable; PDA, patent ductus arteriosus; qPCR, quantitative PCR; VSD, ventricular septal defect; wt, wild type.
Only positive results for NKX2.5 are presented. No mutations or rare variants were found in NKX2.1, FOXE2, PAX8, or HHEX.
Copy number, chromosome location with the start, end, and length of the rare CNVs with their classification, and major encompassing gene (University of California, Santa Clara, genome browser; HG18 assembly).
No rare deletion or duplication was validated in patient 1.
Direct sequencing
Blood was obtained from patients and parents using peripheral venipuncture. DNA was extracted using a pureLink genomic DNA minikit (Life Technologies). Exons and intron-exon junctions were sequenced for NKX2.5, HHEX, NKX2.1, and PAX8. The single exon, including the polymorphic region encoding the alanine stretch, was sequenced for FOXE1. Primers and amplification conditions are available upon request.
Post hoc whole-exome sequencing
For the three patients, exome sequencing was performed subsequently at the McGill University and Genome Québec Innovation Center using the Agilent SureSelect oligo capture library and Illumina HiSeq 2 × 100 paired end reads. Details for exome sequencing and variant analysis were performed as described in our previous paper (25).
CNV detection analysis
Samples were genotyped on the Affymetrix genome-wide single-nucleotide polymorphism (SNP) Array 6.0 according to the manufacturer's specifications. To increase specificity, we used a merge procedure of two different algorithms (ie, genotype console software 3.0.2 from Affymetrix and Birdsuite 1.5.5 from the Broad Institute (Cambridge, Massachusetts) to call CNVs, as published previously by our group (18) and as further described in the Supplemental Data.
Quantitative PCR validation
CNVs found by genome-wide SNP array were validated using TaqMan gene copy number assays (Applied Biosystems). Probes were designed using publicly available software (http://www5.appliedbiosystems.com/tools/cnv/). The TaqMan assay identifications are listed in Table 1 and a detailed protocol is provided in the Supplemental Data.
Zebrafish embryo culture
Zebrafish (Danio rerio) embryos were raised at 28.5°C and staged in hours postfertilization (hpf) as described (26). Transgenic zebrafish lines tg(tg:mCherry) (23) and tg(kdrl:EGFP) (27) were used in this study. Embryos were anesthetized in 0.016% tricaine (Sigma), fixed in 4% phosphate-buffered paraformaldehyde (PFA; Sigma) overnight at 4°C, washed in PBS containing 0.1% Tween 20, gradually transferred to 100% methanol, and stored at −20°C until used for in situ hybridization or immunofluorescence analyses. All zebrafish work at the Institute of Interdisciplinary Research in Molecular Human Biology followed protocols approved by the Institutional Animal Care and Use Committee.
Morpholino injections
For inhibition of ntn1a and ntn1b function, zebrafish embryos were injected with morpholino antisense oligonucleotides (MOs) that have previously been validated for their knockdown specificity and efficacy (28–31). To knock down the ntn1a function, 5–6 ng of a splice-blocking MO (sb-MO; 5′-ATGATGGACTTACCGACACATTCGT-3′) were injected as previously described (28–30). To inhibit the ntn1b function, 4–6 ng of a translation-blocking MO (tb-MO; 5′-CGCACGTTACCAAAATCCTTATCAT-3′) were injected as previously described (28, 31). The standard control MO designed by Gene Tools had the following sequence: 5′-CCTCTTACCTCAGTTACAATTTATA-3′. Working solutions of MOs were prepared in 0.12 M KCl containing phenol red and 2–6 nL of MO solution was microinjected into the high yolk of one- to two-cell stage embryos. Inhibition of normal ntn1a mRNA splicing after an sb-MO injection was verified as described (30) (Supplemental Figure 1).
Whole-mount in situ hybridization (WISH)
DNA templates for synthesis of ntn1a, ntn1b, tg, and nkx2.1a riboprobes were generated by PCR (see Supplemental Table 1 for primer sequences). Plasmids for myl7 and kdrl riboprobes have been used as described (32, 33). Single-color WISH was performed essentially as described (34). For dual-color WISH, riboprobes labeled with digoxigenin (DIG) and dinitrophenol were used and sequential alkaline phosphatase staining was performed with BM Purple and Fast Red (Sigma) as described (23). Fluorescent WISH (FISH) using a DIG-labeled riboprobe for tg was performed as described (23). Antibodies used in WISH and FISH experiments are listed in Supplemental Table 2. Stained embryos were postfixed in 4% PFA (Sigma) and embedded in 90% glycerol for whole-mount imaging or in 7% low melting point agarose (Lonza) for vibratome sectioning. Tissue sections at 50–60 μm thickness were cut on a Leica VT1000S vibratome and mounted in Glycergel (Dako). Images of stained sections were acquired using an Axiocam digital camera mounted on an Axioplan 2 microscope (Zeiss).
Whole-mount immunofluorescence
Whole-mount immunofluorescence (WIF) staining was performed essentially as described (23). Specifications and sources of primary and secondary antibodies used to detect green fluorescent protein (GFP), mCherry, cardiac troponin T, and T4 in zebrafish embryos are provided in Supplemental Table 2. After WIF staining, specimens were incubated in 4′,6′-diamino-2-phenylindole (DAPI) to label cell nuclei and postfixed in 4% PFA. Combined FISH and WIF staining was performed as described (23) Confocal images were acquired using an LSM 510 confocal microscope (Zeiss). Three-dimensional reconstruction of confocal stacks was performed using Zen 2010 D software (Zeiss).
Statistical analyses
Data sets from thyroid cell number measurements in zebrafish were first analyzed for normal distribution (Kolmogorov-Smirnov test) and homogeneity of variances (Bartlett's test). If measurements of a response attribute, or a log-transformation of it, were found to be normally distributed with equal variances, then an unpaired Student's t test was used for pair-wise comparison of stage-matched experimental groups. Statistical analyses were performed using the software package GraphPad Prism 4.0 (GraphPad). Differences were considered significant at P < .05.
Results
Clinical ascertainment
By merging data from our endocrinology and cardiology clinical databases, we identified nine patients with congenital hypothyroidism caused by thyroid dysgenesis (CHTD) and congenital heart defects (CHD). From these nine, a subset of three patients was selected for molecular studies, based on the clinical severity (ie, patients with ventricular septum defects). Parental DNAs were available for all three patients.
Direct sequencing of HHEX, NKX2.1, NKX2.5, FOXE1, and PAX8 and post hoc whole-exome sequencing
To exclude mutations in genes known to be associated with isolated CHTD (NKX2.1, FOXE1, and PAX8) or with combined thyroid and cardiac defects (HHEX, NKX2.5), we sequenced these genes in all three patients. No candidate pathogenic mutations were detected in HHEX, NKX2.1, FOXE1, and PAX8. All patients were homozygous for the FOXE1 14 alanine stretch polymorphism (35). Subsequently, we performed whole-exome sequencing as described elsewhere (25) and no additional pathogenic variants were found in these patients.
Identification of a rare inherited heterozygous variant of NKX2.5 in patient 1
In patient 1, we identified a c.73C>T transition in NKX2.5 (rs28936670, minor allele frequency (0.003), resulting in a pArg25Cys change. This variant was inherited from the unaffected father (Table 1). No rare deletion or duplication was validated in that patient.
CNV detection
CNV detection in patients and their parents was performed using the Affymetrix genome-wide SNP Array 6.0. All detected variants were not found in 203 ethnically matched controls. No rare CNV were found in patient 1. CNV found in patients 2 and 3 are described below.
Identification of a rare inherited deletion of synaptotagmin-17 (SYT17) in patient 2
In patient 2, we identified a deletion of 76 kb encompassing SYT17 and the undefined locus UNQ5810 on chromosome 16p13.2 (Table 1). This variant was inherited from the unaffected father. SYT17 is ubiquitously expressed, with high levels in the thyroid and heart. SYT17 belongs to the group of synaptotagmin-soluble N-ethylmaleimide sensitive fusion factor attachment protein receptor interactors, although its possible involvement in CTHD and CHD have not been examined to date.
Identification of two rare de novo CNVs and a 47, XYY karyotype in patient 3
In patient 3 (Figure 1), we identified two rare de novo CNVs as well as a 47, XYY karyotype (Table 1). The first de novo deletion encompassed a total of 855 kb of sequence on chromosome 22q11, corresponding to an atypical 22q11 deletion syndrome (36). This interval contains 33 genes that would thus be haploinsufficient, including CRKL. Somewhat surprisingly, a routine FISH performed with the usual TUPLE1 (TUP-like enhancer of split gene 1) probe revealed no 22q11 deletion. However, FISH using BAC RP11-801020 confirmed the deletion in the distal DiGeorge syndrome (DGS) region in the index case, whereas parental FISH results were normal. The second CNV was a de novo 12-kb deletion between the second and third exons of netrin-1 (NTN1) on chromosome 17p13.1. NTN1 is a laminin-related secreted protein that acts as an axon guidance molecule during neural development (37).
Netrin1 mRNA expression in zebrafish embryos
Because Netrin1 is implicated in the regulation of various developmental processes including angiogenesis, nonneuronal cell migration, and epithelial morphogenesis (38, 39), we decided to use zebrafish embryos as a model to characterize the expression and function of Netrin1 with respect to thyroid and cardiovascular development. In zebrafish, two paralogous homologs of human NTN1 are expressed, ntn1a (40) and ntn1b (41). Zebrafish ntn1a and ntn1b act as axon guidance molecules, and ntn1a has also been implicated in vascular development (30, 42). We first used WISH to examine spatiotemporal patterns of ntn1a and ntn1b expression in the thyroid/pharyngeal region for which detailed expression data were not yet available. For this purpose, embryos and larvae were fixed at various developmental stages throughout their development between 24 and 100 hpf. Thyroid specification in zebrafish occurs around 24 hpf (43) and the thyroid primordium can be stained by WISH using a nkx2.1a riboprobe (Figure 2A). For ntn1a, we did not detect any notable expression in the thyroid/pharyngeal region of zebrafish embryos at 24, 26, 30, and 34 hpf (Figure 2B and data not shown). Robust ntn1a expression became detectable in the pharyngeal region from 36 hpf onwards (Figure 2, C–E). Dual-color WISH revealed pharyngeal ntn1a expression domains rostral and lateral to the thyroid primordium, but ntn1a was not expressed in the thyroid primordium itself (Figure 2, F–H). Instead, ntn1a was expressed in the pharyngeal mesenchyme surrounding the aortic arch arteries (Figure 2, I and J). For ntn1b, we detected, if any, only a very weak staining of the pharyngeal region at 24, 26, 30, 34, 36, 38, 46, 48, 55, and 60 hpf (Figure 2, K–N, and data not shown). Robust ntn1b expression became detectable at 72 hpf in lateral pharyngeal regions (Figure 2O). Dual-color WISH showed no detectable ntn1b expression in thyroid cells at any stage examined (Figure 2, P–R, and data not shown), and vibratome sections of stained embryos revealed no ntn1b mRNA expression in the pharyngeal arch region (Figure 2S). However, between 46 and 55 hpf, the thyroid primordium was transiently apposed to ntn1b-expressing cardiac tissue (Figure 2, N and T, and data not shown).
Figure 2. Expression of ntn1a mRNA and ntn1b mRNA during zebrafish development.

A–E, During early thyroid development (from 24 to 36 hpf), ntn1a mRNA is not expressed in the region of the thyroid primordium (see thyroidal nkx2.1a expression domain marked by arrowhead in panel A). Pharyngeal expression of ntn1a is detectable in whole-mount embryos from 36 to 72 hpf (see arrowheads in panels C–E). F–J, Vibratome sections of dual-color-stained embryos revealed that ntn1a expression in the pharyngeal region does not include the thyroid primordium (marked by nkx2.1a staining, arrowhead in panels F–H). Parasagittal sections (see panels I and J) show strong ntn1a expression in the pharyngeal mesenchyme (arrows in panel I) surrounding the aortic arch arteries (marked by kdrl staining and indicated as numbers 1, 3, 4, 5, and 6 in panel J). Sagittal (panels F and G), transverse (panel H), and parasagittal sections (panels I and J) are shown. K–O, ntn1b is very weakly expressed in the pharyngeal region between 24 and 60 hpf (panels K–M). From 46 to 60 hpf, a transient weak ntn1b expression was detected in the zebrafish heart (asterisk in panel N). At later stages, ntn1b is expressed in lateral pharyngeal regions (arrowheads in panel O). P–T, Vibratome sections of dual-color-stained embryos revealed that ntn1b expression is absent in the thyroid primordium (marked by nkx2.1a staining, arrowheads in panels P–R) at all stages examined. Very weak, diffuse ntn1b staining was present in parasagittal sections (see panel S) at the level of the pharyngeal arches. Sagittal sections (see panel T) show that thyroid cells are near ntn1b-expressing cardiac tissue between 46 and 55 hpf. Sagittal (panels P, Q, and T), transverse (panel R), and parasagittal sections (panel S) are shown. Scale bar, 100 μm (A–E and (K–O); 50 μm (F–J and P–T).
ntn1a knockdown causes defective cardiovascular and thyroid development
Although ntn1a-morphants have been reported for nervous system and vascular development (28, 30, 31), no data have been available concerning thyroid development in ntn1a-deficient embryos. To test whether ntn1a is required for normal thyroid development, we knocked down ntn1a function in zebrafish embryos using a previously validated ntn1a sb-MO (28–30). Injection of 5–6 ng ntn1a sb-MO efficiently prevented normal ntn1a mRNA splicing and closely recapitulated previously described effects patterns on the trunk vasculature (Supplemental Figure 1). All results reported below have been obtained using this MO concentration.
To examine early thyroid and cardiac development in ntn1a-deficient embryos, we performed dual-color WISH for cardiac and thyroid markers in 28- and 55-hpf embryos. WISH staining of embryos with the myocardium-specific myl7 probe revealed cardiac laterality defects in ntn1a-morphants. The first bilateral symmetry breaking event during zebrafish heart development is a leftward displacement of the cardiac cone, a process called cardiac jogging, which results in a leftward positioning of the venous pole relative to the midline. The direction of cardiac jogging is regulated by left-right signaling (44), and a normal leftward positioning of the venous pole (left jogging) was observed in noninjected (NI) embryos (NI-controls) and embryos injected with control-MO (MO-controls) at 28 hpf (Figure 3, A and B). In contrast, the direction of cardiac jogging was randomized in ntn1a-morphants (Figure 3U) with 42%, 28%, and 30% of ntn1a-morphants displaying left heart jogging (Figure 3C), a no-jog phenotype (Figure 3D), and right heart jogging (Figure 3E), respectively. A second important event involved in establishing laterality of the zebrafish heart is the process of cardiac looping occurring between 36 and 48 hpf (44). When examined at 55 hpf, hearts of NI-controls (Figure 3F) and MO-controls (Figure 3G) showed correct D-looping (ventricle positioned right to the atrium), whereas ntn1a-morphants displayed abnormal heart looping (Figure 3V). Only 47% of ntn1a-morphants showed D-looped hearts (Figure 3H), 26% had unlooped midline hearts (Figure 3I) and 27% showed a reversed looping with the ventricle positioned left to the atrium (Figure 3J).
Figure 3. ntn1a-deficient embryos display defects in thyroid and cardiac development.

A–D, Dual-color WISH of nkx2.1a and myl7 expression in NI embryos, embryos injected with control morpholino (co-MO), and embryos injected with ntn1a MO (ntn1a-MO). myl7 staining of the heart tube revealed normal leftward positioning of the venous pole (left jog) in NI and co-MO embryos, whereas heart jogging was randomized in ntn1a-morphants (panels C–E). Thyroidal nkx2.1a expression (arrowhead) was not different between experimental groups at 28 hpf. Dorsal views are shown, anterior is to the top. F–O, Dual-color WISH of tg and myl7 expression in 55-hpf embryos (frontal view). myl7 staining showed correct heart looping (D loop) with the ventricle (V) positioned right to the atrium (A) in NI and co-MO embryos (see panels F and G). ntn1a-morphants displayed randomization of heart looping (see panels H–J) including midline hearts and hearts with reversed looping (L loop). In addition, ntn1a-morphants have irregularly shaped thyroid primordia (arrowheads in panels F–J). In contrast to the compact and slightly ovoid thyroid primordium of control embryos (see panels K and L), ntn1a-morphants displayed unilaterally (see panels M and O) or bilaterally (see panels N) expanded thyroid tissue. Magnified views of the thyroid region in panels K and L are not necessarily from the same embryos as shown in panels F–J. P–T, WISH analysis of tg expression at 80 hpf showed a normal progressive expansion of thyroid tissue along the anterior-posterior axis in control embryos (see panels P and Q). In ntn1a-morphants, thyroid tissue was disorganized and often mislocated away from the midline (see panels R, S, and T). Ventral views are shown, anterior is to the top. U and V, Quantification of the number of embryos displaying defects of heart tube jogging at 28 hpf (panel V) and heart looping at 55 hpf (panel U) as determined by myl7 staining. Results are presented as the percentage of embryos displaying a particular phenotype, and N denotes the total number of specimen analyzed for each treatment group. Scale bar, 100 μm (A–J), 50 μm (K–T).
Although cardiac jogging and subsequent cardiac looping are discrete processes, the direction of cardiac jogging is generally considered a good predictor for cardiac looping phenotypes (45). Our experiments were not designed to address specifically the relationship between jogging directionality and subsequent cardiac looping for individual embryos, but the observed frequencies of jogging and looping anomalies in ntn1a-morphants are consistent with a model in which left jogging (42%) is expected to be accompanied by D looping (47%), right jogging (30%) by reversed looping (27%), and a no-jog phenotype (28%) would result in unlooped midline hearts (26%).
WISH staining of the thyroid marker nkx2.1a in 28 hpf embryos did not reveal gross differences in size, shape, and location of the thyroid primordium between NI-controls (n = 47), MO-controls (n = 104), and ntn1a-morphants (n = 116) (Figure 3, A–E). At 55 hpf, however, 48% of ntn1a-morphants (n = 46 of 95) presented an aberrant thyroid morphology. In contrast to the compact midline thyroids of NI-controls (n = 43 of 45) and MO-controls (n = 83 of 86), thyroid tissue of ntn1a-morphants were not limited to the midline and showed irregular lateral expansions (Figure 3, M–O). Laterally expanding thyroid tissue was observed at similar frequencies on the left (n = 17 of 95) and the right side (n = 19 of 95); bilateral thyroid expansions were less frequent (n = 10 of 95). At approximately 55–60 hpf, the thyroid starts to expand along the pharyngeal midline and tg staining of 80 hpf control embryos showed the progressive anterior-posterior (AP) expansion of thyroid tissue (Figure 3, P and Q). In most ntn1a-morphants (n = 52 of 80), however, this AP expansion was defective, and ntn1a-morphants displayed irregularly positioned clusters of thyroid cells close to the heart outflow tract (OFT) (Figure 3, R and S) as well as laterally misplaced thyroid tissue (Figure 3T). No thyroid or cardiac laterality defects were detected in embryos injected with a tb-MO targeting ntn1b (data not shown).
The irregular thyroid morphologies of ntn1a-morphants resembled thyroid phenotypes previously observed in zebrafish embryos with defects in pharyngeal vasculature morphogenesis (23). Using transgenic tg(kdrl:EGFP) embryos expressing enhanced green fluorescent protein (EGFP) in endothelial cells, we detected gross malformations of the pharyngeal vasculature after injection of ntn1a sb-MO but not control-MO or ntn1b tb-MO (Figure 4, A–C). Similar to mammalian embryos, the zebrafish aortic arch artery (AA) network consists of paired bilateral arteries that connect the heart OFT to the dorsal aortae. Confocal microscopy of 55 hpf tg(kdrl:EGFP) embryos and three-dimensional (3D) reconstruction of the pharyngeal vasculature revealed a spectrum of defects in AA morphogenesis in 63% of ntn1a-morphants (Figure 4, D–H). Although the AA1 and the branchial AAs 3–6 were clearly formed in 55-hpf control embryos, perturbed AA morphogenesis in ntn1a-morphants ranged from AA hypoplasia to severe underdevelopment of the entire AA system (Figure 4, K–M). AA malformations in ntn1a-morphants were further accompanied by hypobranchial artery (HA) dysplasia and failure to form a paired ventral aorta connecting each AA to the heart OFT.
Figure 4. Defective morphogenesis of pharyngeal vessels is associated with aberrant localization of thyroid tissue in ntn1a-deficient embryos.
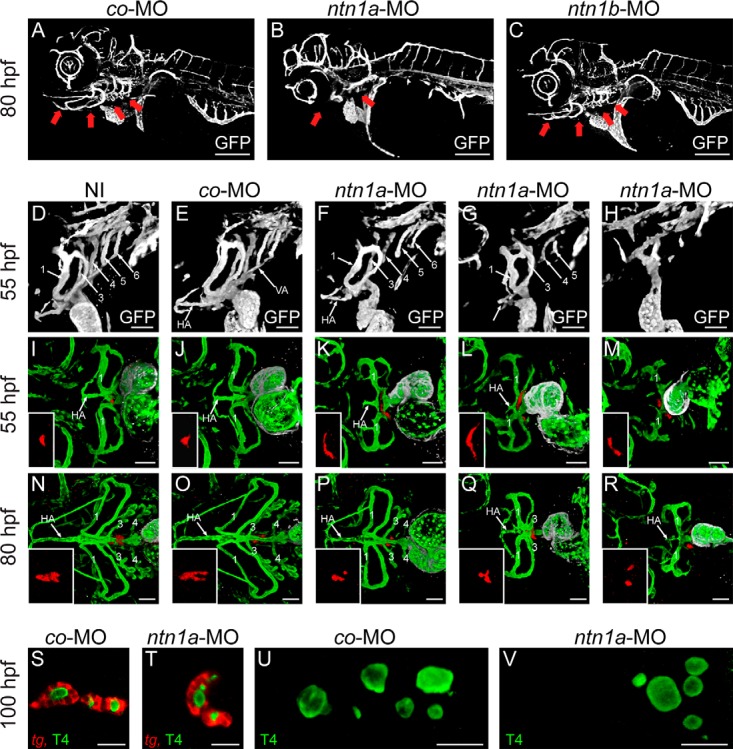
A–C, GFP immunofluorescence staining of 80-hpf transgenic tg(kdrl:EGFP) embryos injected with co-MO, ntn1a-MO, or ntn1b-MO. Pharyngeal vascular development was defective in ntn1a-deficient embryos (see panel B) but not in embryos injected with ntn1b-MO (see panel C). Note that defective formation of AAs and associated vessels (red arrows in panels A–C) occurred despite an overall normal vascular development in ntn1a-morphants. 3D reconstructions of confocal images are shown, lateral view, anterior is to the left. D–H, GFP immunofluorescence staining of 55-hpf transgenic tg(kdrl:EGFP) embryos. NI embryos and co-MO-injected embryos had formed bilateral pairs of five AAs (indicated as numbers 1, 3, 4, 5, and 6). Formation of the HA is evident, and a paired ventral aorta (VA) has formed connecting the heart outflow tract via branchial AAs 3–6 to the lateral dorsal aorta. In contrast, ntn1a-morphants displayed defective pharyngeal vessel morphogenesis, ranging from limited and incomplete AA formation to an almost complete absence of branchial AAs (see panels F–H). Defects in VA and HA formation correlated with the severity of AA abnormalities in ntn1a-morphants (see panels F–H). 3D reconstructions of confocal images are shown, lateral view, anterior is to the left. I–R, Confocal microscopy of transgenic tg(tg:mCherry; kdrl:EGFP) embryos after immunofluorescence staining for mCherry (thyroid, red), GFP (endothelium/endocardium, green), and cardiac troponin T (myocardium, white). Thyroid anomalies including ectopic lateral thyroid expansion were exclusively found in ntn1a-morphants displaying defects in pharyngeal vascular development, particularly defects in HA morphogenesis. 3D reconstructions of confocal images are shown, ventral view, anterior is to the left. S–V, Despite defects in size, shape, and position of thyroid tissue, follicle formation and functional maturation of thyroid tissue appeared unaffected in ntn1a-morphants. Panels S and T show confocal sections of embryos (ventral view, anterior to the left) after double staining for tg mRNA and colloidal T4. Panels U and V show confocal projections of embryos (ventral view, anterior to the left) after colloidal T4 staining. Scale bar, 200 μm (A–C), 50 μm (D–R), 20 μm (S–V).
Confocal microscopy of double transgenic tg(tg:mCherry;kdrl:EGFP) embryos, expressing mCherry in thyroid and EGFP in endothelial cells, showed that aberrant lateral thyroid expansion occurred predominantly in ntn1a-morphants displaying abnormal HA morphologies (Figure 4, I–M, and Figure 5). Confocal analyses of 80-hpf embryos showed that lack of a normal AP expansion of thyroid tissue in ntn1a-morphants was correlated with an overall poorly developed hypobranchial vasculature including the defective AA and HA formation (Figure 4, N–R). When counting the number of thyroid cells (cells double positive for mCherry and DAPI) in 80 hpf tg(tg:mCherry) embryos, no differences were detected between MO-controls and ntn1a-morphants (Figure 6). At 100 hpf, however, thyroid tissue of ntn1a-morphants contained significantly fewer thyroid cells than MO-controls (Figure 6). On the contrary, the functional maturation of thyroid tissue appeared largely unaffected in ntn1a-morphants as judged by their capacity to form functional follicles producing the thyroid hormone T4 (Figure 4, S–V).
Figure 5. Three-dimensional reconstruction of confocal images of the heart OFT region of 55 hpf tg(tg:mCherry;kdrl:EGFP) embryos expressing mCherry (red) in thyroid cells and EGFP (green) in endothelial cells and endocardium.
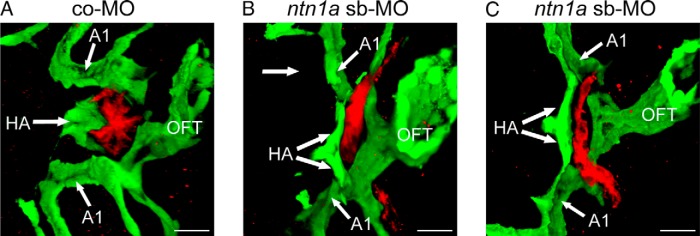
A, In embryos injected with a co-MO, the endothelial cells of the HA embrace the thyroid primordium, which is present as a compact ovoid midline structure located rostral to the heart OFT. B and C, In contrast, embryos injected with a ntn1a-MO displayed irregular thyroid morphologies. Uni- or biaterally expanding thyroid tissue was predominantly observed along the course of an abnormally bifurcated HA. AA1, aortic arch artery 1. Scale, 20 μm.
Figure 6. Determination of thyroid cell number in tg(tg:mCherry) embryos injected with a ctrl-MO and ntn1a sb-MO.
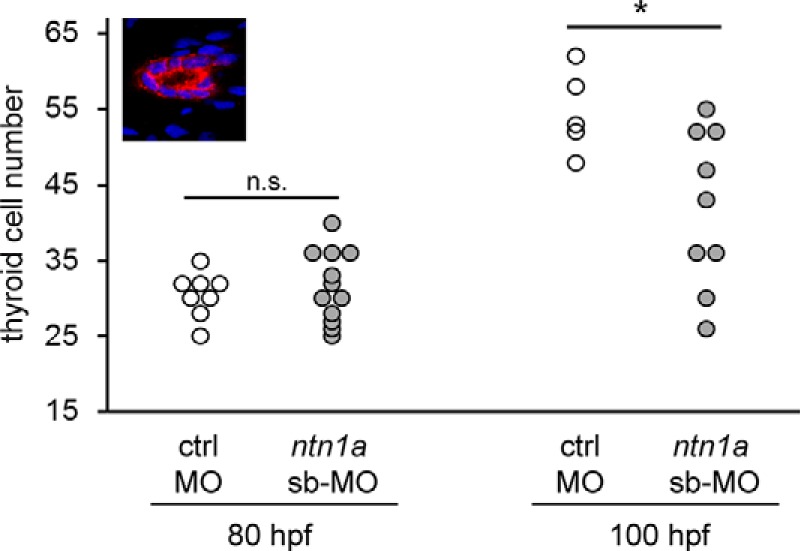
Embryos were fixed at 80 and 100 hpf, and thyroid cells were labeled by immunofluorescence using an anti-mCherry antibody. Counterstaining with DAPI was used to label cell nuclei. Confocal z-stacks (1 μm sections) comprising the whole thyroid tissue were acquired, and the number of all cells double positive for mCherry and DAPI was measured for each embryo (see insert for a single confocal section from a control thyroid). N denotes the number of embryos analyzed. Asterisk denotes the significant differences between treatment means. *, P < .05 (unpaired t test). Ctrl, control; n.s., not significant.
Discussion
The etiology of CHTD is one of the remaining enigmas in the pathophysiology of thyroid diseases (46). CHTD is a sporadic condition that has a discordance rate of 92% between monozygotic twins (4) and a significant association with congenital heart defects (2, 47). This prompted us to consider de novo germline genetic events (ie, de novo CNVs and/or point mutations) as the underlying cause. In this pilot study, we used a combination of targeted candidate gene sequencing and high-density CNV analysis to assess the role of rare alleles with major effects in CHD and CHTD. We found rare variants in all three patients, and we selected netrin-1 as the biologically most plausible contributory factor for functional studies in zebrafish. This study also underlines the value of combining phenotype and genotype profiling with zebrafish functional studies to uncover new pathogenic genes from a small cohort of patients (11, 48).
In patient 3 we found three potentially relevant rare structural variants: an 47, XYY karyotype; an atypical 22q11 deletion; and an 17q deletion.
First, standard karyotyping revealed a 47, XYY chromosome constitution that was corroborated by high-resolution karyotyping. An association between congenital hypothyroidism and sex chromosome aneuploidy (ie, especially in 47, XXY) has been previously reported (49). Thorwarth et al (20) also reported a wide Y duplication in a patient with thyroid hypoplasia and a VSD.
The 22q11 deletion in patient 3 overlaps with deletions associated with DGS in which thyroid dysfunction is a variable component of the phenotype. However, genotype-phenotype correlations have been restricted to classic deletions as defined by the TUPLE1 probe (50). The TUPLE1 standard probe did not detect the deletion in our patient even though the clinical picture was typical for DGS. According to studies in mice, deletion of Tbx1 fully replicates the thyroid phenotype seen in DGS (51), yet this gene was not deleted in patient 3. A role for Crkl in anteroposterior patterning of the pharyngeal apparatus has been described; this process requires the cooperation with Tbx1 and local retinoic acid signaling (52). Of note, even if up to 50% of classical DGS present with thyroid hypoplasia (53), thyroid ectopy is not reported in DGS, and in a series of atypical DGS (ie, CRKL deletion), no patients were reported to have hypothyroidism (36). Therefore, CRKL deletion is associated with neither thyroid ectopy nor hypothyroidism, and we did not select CRKL for further validation with functional assays.
More importantly, patient 3 carries a de novo 12-kb deletion on chromosome 17p13.1 within the NTN1 gene, which is predicted pathogenic because two consecutive exons are deleted. NTN1 is of great interest in light of findings that place this gene in the Sonic hedgehog signaling pathway and implicate it as a physical and functional interactor of the Down syndrome cell adhesion molecule, a gene implicated in the cardiac phenotype of Down syndrome patients (54–56). NTN1 is crucial for normal brain development but recent studies have implicated NTN1 signaling also in the regulation of nonneuronal cell migration and survival and vascular development as well as epithelial cell adhesion and migration during lung and pancreas morphogenesis (57, 58). Similar to lungs and pancreas, the thyroid is derived from foregut endoderm, but a role of netrin-1 action during thyroid development has not yet been reported.
Although the clinical significance of the detected variants at the NTN1 locus remains to be determined, our functional studies in zebrafish embryos provide significant evidence linking netrin-1 action with pharyngeal vessel and thyroid morphogenesis. First, we detected dynamic expression of zebrafish homologs of NTN1 in the pharyngeal region, although neither ntn1a or ntn1b was expressed in the developing thyroid itself. Instead, ntn1a was expressed in pharyngeal tissue surrounding the developing AAs, and defective AA formation was one hallmark of the ntn1a loss-of-function phenotype. Although defective thyroid morphogenesis became apparent at 55 hpf, after thyroid separation from the pharyngeal floor, perturbed AA formation was already evident at earlier stages. Another key observation was that the presence of irregularly shaped and ectopically located thyroid tissue was strictly associated with aberrant morphogenesis of pharyngeal vessels (eg, the HA) that are important for guiding late thyroid relocalization (23). Together these observations suggest that abnormal pharyngeal vessel formation might be the primary effect of ntn1a deficiency, whereas the thyroid anomalies most likely represent a secondary response to the lack of a proper guidance function exerted by dysplastic pharyngeal vessels. In this regard, our data reinforce the concept that embryonic blood vessels play a critical role in thyroid organogenesis (24, 59) and that vascular anomalies may account for certain cases of CHTD. Consistently, there are clinical observations of vascular malformations (eg, hypoplasia or agenesis of thyroid arteries) in some cases of ectopic thyroids in humans (60, 61). Ntn1a-deficient zebrafish embryos also displayed cardiac laterality defects, but neither abnormal heart jogging nor heart looping was correlated with the presence of thyroid anomalies.
Two other inherited variants were found. First, the NKX2.5 p.R25C variant identified in patient 1 was previously found in patients with both CHD and CHTD (11). In addition, functional assays suggested that the R25C mutant exhibits impaired binding and transactivation properties (11, 62). Because this variant was also found in the father of patient 1 as well as at low frequency in some control series with a minor allele frequency of 0.003 on dbSNP, we postulate that it constitutes a reduced penetrance risk allele. Second, we found a deletion in SYT17 in patient 2, again inherited from an unaffected father. Given that SYT17 deletion was inherited from the healthy father, we did not select this gene for further validation with functional assays (48).
Our study implicates known pathways of thyroid and heart development and replicates previous results suggesting a possible contributory role for the NKX2.5 p.R25C variant. The incomplete penetrance of this variant in our patient 1 is compatible with either genetic or environmental modifiers. These observations suggest a complex mode of inheritance in CHTD and CHD, which are now genetically traceable using modern high-resolution platforms (63).
Based on the high yield of rare variants identified in this pilot analysis, future studies in patients with CHTD and CHD are warranted, and these studies should include a comprehensive analysis of protein-coding mutations and structural genomic variation. High-resolution chip platforms and whole genome sequencing in trios with affected CHTD/CHD children will yield insight into complex inheritance patterns affecting interacting pathways of embryonic development of the thyroid and heart.
Acknowledgments
We thank the patients and their parents for their cooperation. We also thank Drs C. Deal and G. Van Vliet (Centre Hospitalier Universitaire Sainte-Justine, University of Montréal) for their continuous support during this project. In addition, we thank Fabien Magne for technical assistance. We also thank C. Stacher-Hörndli (University of Utah Medical Center, Salt Lake City, Utah) and G. Peng (Fudan University, Shanghai, China) for their generosity with morpholino antisense oligonucleotide reagents, V. Janssens (Université Libre de Bruxelles, Brussels, Belgium) for technical assistance in the morpholinos injections, J.-M. Vanderwinden, and F. Bollet-Quivogne (Light Microscopy Facility, Université Libre de Bruxelles) for technical assistance in the confocal microscopy.
This work was supported by grants from the Canadian Institutes of Health Research (to J.D. and G.A.); by the Fonds de Recherche du Québec-Santé (to J.D. and G.A.); by Grant GMHD79045 from the Heart and Stroke Foundation of Canada (to G.A.); by the Girafonds/Fondation du Centre Hospitalier Universitaire Sainte-Justine (to J.D.); by the European Society for Pediatric Endocrinology (ESPE Research Unit grant, to J.D. and S.C.); by Grant FRSM 3_4598_12 from the Belgian Fonds de la Recherche Scientifique Medicale (to S.C.); by Grant ARC AUWB-2012-12/17-ULB3 from the Action de Recherche Concertée de la Communauté Française de Belgique (to S.C.); by the Fonds d'Encouragement à la Recherche; and grants from the Belgian National Fund for Scientific Research (FNRS). R.O. is an FNRS Postdoctoral Researcher, I.V. is an FNRS Research Fellow, A.T. is an FNRS Short-Term Foreign Postdoctoral Fellow, and S.C. is an FNRS Senior Research Associate.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AA
- arch artery
- ASD
- atrial septal defect
- CHD
- congenital heart disease
- CHTD
- congenital hypothyroidism caused by thyroid dysgenesis
- CNV
- copy number variant
- 3D
- three dimensional
- DAPI
- 4′,6′-diamino-2-phenylindole
- DGS
- DiGeorge syndrome
- DIG
- digoxigenin
- EGFP
- enhanced GFP
- FISH
- fluorescent in-situ hybridization
- GFP
- green fluorescent protein
- HA
- hypobranchial artery
- hpf
- hours postfertilization
- MO
- morpholino antisense oligonucleotide
- NI
- noninjected
- NTN1
- netrin-1
- OFT
- outflow tract
- PDA
- patent ductus arteriosus
- PFA
- phosphate-buffered paraformaldehyde
- sb-MO
- splice-blocking MO
- SNP
- single-nucleotide polymorphism
- SYT17
- synaptotagmin-17
- tb-MO
- translation-blocking MO
- VSD
- ventricular septal defect
- WIF
- whole-mount immunofluorescence
- WISH
- whole-mount in situ hybridization.
References
- 1. Deladoey J, Belanger N, Van Vliet G. Random variability in congenital hypothyroidism from thyroid dysgenesis over 16 years in Quebec. J Clin Endocrinol Metab. 2007;92(8):3158–3161. [DOI] [PubMed] [Google Scholar]
- 2. Devos H, Rodd C, Gagne N, Laframboise R, Van Vliet G. A search for the possible molecular mechanisms of thyroid dysgenesis: sex ratios and associated malformations. J Clin Endocrinol Metab. 1999;84(7):2502–2506. [DOI] [PubMed] [Google Scholar]
- 3. Castanet M, Polak M, Bonaiti-Pellie C, Lyonnet S, Czernichow P, Leger J. Nineteen years of national screening for congenital hypothyroidism: familial cases with thyroid dysgenesis suggest the involvement of genetic factors. J Clin Endocrinol Metab. 2001;86(5):2009–2014. [DOI] [PubMed] [Google Scholar]
- 4. Perry R, Heinrichs C, Bourdoux P, et al. Discordance of monozygotic twins for thyroid dysgenesis: implications for screening and for molecular pathophysiology. J Clin Endocrinol Metab. 2002;87(9):4072–4077. [DOI] [PubMed] [Google Scholar]
- 5. Calcagni G, Digilio MC, Sarkozy A, Dallapiccola B, Marino B. Familial recurrence of congenital heart disease: an overview and review of the literature. Eur J Pediatr. 2007;166(2):111–116. [DOI] [PubMed] [Google Scholar]
- 6. Manning N, Archer N. A study to determine the incidence of structural congenital heart disease in monochorionic twins. Prenat Diagn. 2006;26(11):1062–1064. [DOI] [PubMed] [Google Scholar]
- 7. Clifton-Bligh RJ, Wentworth JM, Heinz P, et al. Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat Genet. 1998;19(4):399–401. [DOI] [PubMed] [Google Scholar]
- 8. Macchia PE, Lapi P, Krude H, et al. PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat Genet. 1998;19(1):83–86. [DOI] [PubMed] [Google Scholar]
- 9. Castanet M, Sura-Trueba S, Chauty A, et al. Linkage and mutational analysis of familial thyroid dysgenesis demonstrate genetic heterogeneity implicating novel genes. Eur J Hum Genet. 2005;13(2):232–239. [DOI] [PubMed] [Google Scholar]
- 10. Biben C, Weber R, Kesteven S, et al. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2–5. Circ Res. 2000;87(10):888–895. [DOI] [PubMed] [Google Scholar]
- 11. Dentice M, Cordeddu V, Rosica A, et al. Missense mutation in the transcription factor NKX2–5: a novel molecular event in the pathogenesis of thyroid dysgenesis. J Clin Endocrinol Metab. 2006;91(4):1428–1433. [DOI] [PubMed] [Google Scholar]
- 12. Martinez Barbera JP, Clements M, Thomas P, et al. The homeobox gene Hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development. 2000;127(11):2433–2445. [DOI] [PubMed] [Google Scholar]
- 13. Abu-Khudir R, Paquette J, Lefort A, et al. Transcriptome, methylome and genomic variations analysis of ectopic thyroid glands. PLoS One. 2010;5(10):e13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deladoey J, Vassart G, Van Vliet G. Possible non-mendelian mechanisms of thyroid dysgenesis. Endocr Dev. 2007;10:29–42. [DOI] [PubMed] [Google Scholar]
- 15. Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36(9):949–951. [DOI] [PubMed] [Google Scholar]
- 16. Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sebat J, Lakshmi B, Malhotra D, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hitz MP, Lemieux-Perreault LP, Marshall C, et al. Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet. 2012;8(9):e1002903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uccellatore F, Sava L, Giuffrida D, et al. Cytogenetic analysis in congenital hypothyroidism. J Endocrinol Invest. 1990;13(7):605–607. [DOI] [PubMed] [Google Scholar]
- 20. Thorwarth A, Mueller I, Biebermann H, et al. Screening chromosomal aberrations by array comparative genomic hybridization in 80 patients with congenital hypothyroidism and thyroid dysgenesis. J Clin Endocrinol Metab. 2010;95(7):3446–3452. [DOI] [PubMed] [Google Scholar]
- 21. Reller MD, Strickland MJ, Riehle-Colarusso T, Mahle WT, Correa A. Prevalence of congenital heart defects in metropolitan Atlanta, 1998–2005. J Pediatr. 2008;153(6):807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Best K, Draper E, Kurinczuk J, et al. PPO.17. Is congenital heart disease on the increase in the UK? A register-based study. Arch Dis Child Fetal Neonatal Ed. 2014;99(suppl 1):A155. [Google Scholar]
- 23. Opitz R, Maquet E, Huisken J, et al. Transgenic zebrafish illuminate the dynamics of thyroid morphogenesis and its relationship to cardiovascular development. Dev Biol. 2012;372(2):203–216. [DOI] [PubMed] [Google Scholar]
- 24. Alt B, Elsalini OA, Schrumpf P, et al. Arteries define the position of the thyroid gland during its developmental relocalisation. Development. 2006;133(19):3797–3804. [DOI] [PubMed] [Google Scholar]
- 25. Samuels ME, Gallo-Payet N, Schwartzentruber J, et al. Bioinactive ACTH causing glucocorticoid deficiency. J Clin Endocrinol Metab. 2013;98(2):736–742. [DOI] [PubMed] [Google Scholar]
- 26. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203(3):253–310. [DOI] [PubMed] [Google Scholar]
- 27. Jin SW, Herzog W, Santoro MM, et al. A transgene-assisted genetic screen identifies essential regulators of vascular development in vertebrate embryos. Dev Biol. 2007;307(1):29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Suli A, Mortimer N, Shepherd I, Chien CB. Netrin/DCC signaling controls contralateral dendrites of octavolateralis efferent neurons. J Neurosci. 2006;26(51):13328–13337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stacher Horndli C, Chien CB. Sonic hedgehog is indirectly required for intraretinal axon pathfinding by regulating chemokine expression in the optic stalk. Development. 2012;139(14):2604–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilson BD, Ii M, Park KW, et al. Netrins promote developmental and therapeutic angiogenesis. Science. 2006;313(5787):640–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang C, Gao J, Zhang H, Sun L, Peng G. Robo2-slit and Dcc-netrin1 coordinate neuron axonal pathfinding within the embryonic axon tracts. J Neurosci. 2012;32(36):12589–12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yelon D, Horne SA, Stainier DY. Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev Biol. 1999;214(1):23–37. [DOI] [PubMed] [Google Scholar]
- 33. Thompson MA, Ransom DG, Pratt SJ, et al. The cloche and spadetail genes differentially affect hematopoiesis and vasculogenesis. Dev Biol. 1998;197(2):248–269. [DOI] [PubMed] [Google Scholar]
- 34. Opitz R, Maquet E, Zoenen M, Dadhich R, Costagliola S. TSH receptor function is required for normal thyroid differentiation in zebrafish. Mol Endocrinol. 2011;25(9):1579–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Carre A, Castanet M, Sura-Trueba S, et al. Polymorphic length of FOXE1 alanine stretch: evidence for genetic susceptibility to thyroid dysgenesis. Hum Genet. 2007;122(5):467–476. [DOI] [PubMed] [Google Scholar]
- 36. Verhagen JM, Diderich KE, Oudesluijs G, et al. Phenotypic variability of atypical 22q11.2 deletions not including TBX1. Am J Med Genet Part A. 2012;158A(10):2412–2420. [DOI] [PubMed] [Google Scholar]
- 37. Lai Wing Sun K, Correia JP, Kennedy TE. Netrins: versatile extracellular cues with diverse functions. Development. 2011;138(11):2153–2169. [DOI] [PubMed] [Google Scholar]
- 38. Cirulli V, Yebra M. Netrins: beyond the brain. Nat Rev Mol Cell Biol. 2007;8(4):296–306. [DOI] [PubMed] [Google Scholar]
- 39. Levy-Strumpf N, Culotti JG. Netrins and Wnts function redundantly to regulate antero-posterior and dorso-ventral guidance in C. elegans. PLoS Genet. 2014;10(6):e1004381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lauderdale JD, Davis NM, Kuwada JY. Axon tracts correlate with netrin-1a expression in the zebrafish embryo. Mol Cell Neurosci. 1997;9(4):293–313. [DOI] [PubMed] [Google Scholar]
- 41. Strahle U, Fischer N, Blader P. Expression and regulation of a netrin homologue in the zebrafish embryo. Mech Dev. 1997;62(2):147–160. [DOI] [PubMed] [Google Scholar]
- 42. Lu X, Le Noble F, Yuan L, et al. The netrin receptor UNC5B mediates guidance events controlling morphogenesis of the vascular system. Nature. 2004;432(7014):179–186. [DOI] [PubMed] [Google Scholar]
- 43. Rohr KB, Concha ML. Expression of nk2.1a during early development of the thyroid gland in zebrafish. Mech Dev. 2000;95(1–2):267–270. [DOI] [PubMed] [Google Scholar]
- 44. Bakkers J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc Res. 2011;91(2):279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trinh LA, Stainier DY. Cardiac development. Methods Cell Biol. 2004;76:455–473. [DOI] [PubMed] [Google Scholar]
- 46. Vassart G, Dumont JE. Thyroid dysgenesis: multigenic or epigenetic. or both? Endocrinology. 2005;146(12):5035–5037. [DOI] [PubMed] [Google Scholar]
- 47. Medda E, Olivieri A, Stazi MA, et al. Risk factors for congenital hypothyroidism: results of a population case-control study (1997–2003). Eur J Endocrinol. 2005;153(6):765–773. [DOI] [PubMed] [Google Scholar]
- 48. Diez-Roux G, Banfi S, Sultan M, et al. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 2011;9(1):e1000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sarri C, Cote GB, Mengreli C, Lambadaridis I, Pantelakis S. Hypothyroidism and sex chromosomes. J Med Genet. 1988;25(4):247–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Choi JH, Shin YL, Kim GH, et al. Endocrine manifestations of chromosome 22q11.2 microdeletion syndrome. Horm Res. 2005;63(6):294–299. [DOI] [PubMed] [Google Scholar]
- 51. Fagman H, Liao J, Westerlund J, Andersson L, Morrow BE, Nilsson M. The 22q11 deletion syndrome candidate gene Tbx1 determines thyroid size and positioning. Hum Mol Genet. 2007;16(3):276–285. [DOI] [PubMed] [Google Scholar]
- 52. Guris DL, Duester G, Papaioannou VE, Imamoto A. Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev Cell. 2006;10(1):81–92. [DOI] [PubMed] [Google Scholar]
- 53. Stagi S, Lapi E, Gambineri E, et al. Thyroid function and morphology in subjects with microdeletion of chromosome 22q11 (del(22)(q11)). Clin Endocrinol (Oxf). 2010;72(6):839–844. [DOI] [PubMed] [Google Scholar]
- 54. Andrews GL, Tanglao S, Farmer WT, et al. Dscam guides embryonic axons by Netrin-dependent and -independent functions. Development. 2008;135(23):3839–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ahmed RP, Haider KH, Shujia J, Afzal MR, Ashraf M. Sonic Hedgehog gene delivery to the rodent heart promotes angiogenesis via iNOS/netrin-1/PKC pathway. PLoS One. 2010;5(1):e8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fagman H, Grande M, Gritli-Linde A, Nilsson M. Genetic deletion of sonic hedgehog causes hemiagenesis and ectopic development of the thyroid in mouse. Am J Pathol. 2004;164(5):1865–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu Y, Stein E, Oliver T, et al. Novel role for netrins in regulating epithelial behavior during lung branching morphogenesis. Curr Biol. 2004;14(10):897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yebra M, Montgomery AM, Diaferia GR, et al. Recognition of the neural chemoattractant Netrin-1 by integrins α6β4 and α3β1 regulates epithelial cell adhesion and migration. Dev Cell. 2003;5(5):695–707. [DOI] [PubMed] [Google Scholar]
- 59. Fagman H, Andersson L, Nilsson M. The developing mouse thyroid: embryonic vessel contacts and parenchymal growth pattern during specification, budding, migration, and lobulation. Dev Dyn. 2006;235(2):444–455. [DOI] [PubMed] [Google Scholar]
- 60. Declerck S, Casselman JW, Depondt M, Vandevoorde P. Lingual thyroid imaging. J Belge Radiol. 1993;76(4):241–242. [PubMed] [Google Scholar]
- 61. Banna M, Lasjaunias P. The arteries of the lingual thyroid: angiographic findings and anatomic variations. AJNR Am J Neuroradiol. 1990;11(4):730–732. [PMC free article] [PubMed] [Google Scholar]
- 62. Kasahara H, Lee B, Schott JJ, et al. Loss of function and inhibitory effects of human CSX/NKX2.5 homeoprotein mutations associated with congenital heart disease. J Clin Invest. 2000;106(2):299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zaidi S, Choi M, Wakimoto H, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498(7453):220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]