

Abstract
As ErbB receptors are expressed in prolactinomas and exhibit downstream effects on prolactin (PRL) production and cell proliferation, we generated transgenic mice using a PRL enhancer/promoter expression system to restrict lactotroph-specific expression of human epidermal growth factor receptor (EGFR) or human EGFR2 (HER2). EGFR or HER2 transgenic mice developed prolactinomas between 13 and 15 months, and confocal immunofluorescence and Western blot analysis confirmed lactotroph-restricted PRL and EGFR or HER2 coexpression. Circulating PRL levels in EGFR and HER2 transgenic mice were increased 5- and 3.8-fold, respectively. Inhibiting EGFR or HER2 signaling with oral lapatinib (100 mg/kg), a dual tyrosine kinase inhibitor for both EGFR and HER2, suppressed circulating PRL by 72% and attenuated tumor PRL expression by 80% and also attenuated downstream tumor EGFR/HER2 signaling. This model demonstrates the role of ErbB receptors underlying prolactinoma tumorigenesis and the feasibility of targeting these receptors for translation to treatment of refractory prolactinomas.
Prolactinomas account for approximately 40% of all pituitary tumors (1). In addition to sellar mass effects, including headache, visual dysfunction, and/or hypopituitarism, patients present with features of excess prolactin (PRL) secretion, including amenorrhea, galactorrhea, and infertility in females and sexual dysfunction in males (2, 3). Dopamine agonists, which suppress PRL synthesis and secretion and tumor growth, are the mainstay therapeutic choice for these commonly encountered tumors (4, 5). However, dopamine agonist resistance and drug intolerance is encountered in approximately 25% of patients without normalization of PRL levels or tumor shrinkage (6). In these patients, transsphenoidal adenometomy may be considered with reported initial remission rates of approximately 75% for microprolactinomas and approximately 34% for macroprolactinomas (7). Surgical outcomes are dependent on tumor size and location, as well as the experience of the surgeon (8). However, up to 50% may recur postoperatively and continue to grow persistently despite antitumor therapy (9, 10). Surgical complications increase with each subsequent resection and include development of new onset hypopituitarism, local tissue damage, cranial nerve injury, as well as enhanced surgical mortality (0.3%–0.5%) and morbidity, especially for larger tumors (>4 cm in diameter) (2). Alternative treatment options are therefore required for tumors resistant to currently available treatments. Anecdotal reports of pharmacotherapy for aggressive and/or resistant prolactinomas include somatostatin analogues, which do not inhibit PRL, and selective estrogen receptor modulators, which may modestly inhibit PRL levels (11, 12). Temozolomide has been shown in small uncontrolled series to inconsistently reduce tumor size, and PRL secretion in aggressive prolactinomas, and effects are not necessarily maintained over time (2, 9, 10).
Human epidermal growth factor receptor (EGFR, ErbB, and HER) family comprises 4 subtypes: EGFR (ErbB1, HER1), p185her2/neu (ErbB2, HER2), ErbB3 (HER3), and ErbB4 (HER4) (13), which regulate cell motility and adhesion, tumor invasion, angiogenesis, and tumor cell proliferation (14). EGFR (14–21) and HER2 (14, 17, 22, 23) are expressed in normal anterior pituitary cells, including lactotrophs. EGFR/HER2 signaling regulates tumor growth and hormone production in experimental lacto-somatotroph tumors and in an experimental Cushing disease model (24–27). Moreover, targeted EGFR/HER2 therapy has also been shown to be effective in 2 dopamine agonist resistant prolactinomas (28).
To directly investigate the role of EGFR/HER2 in lactotroph cell growth and tumorigenesis, we generated transgenic mice expressing lactotroph-targeted human EGFR (hEGFR) or human HER2 (hHER2) transgenes using the PRL promoter/enhancer (29) expression system. Pituitary-specific expression of EGFR or HER2 genes was observed in the transgenic mice. And these mice developed hyperprolactinemia and prolactinomas, which responded to lapatinib, a dual tyrosine kinase inhibitor (TKI), demonstrating the feasibility of targeting EGFR/HER2 for PRL responsiveness in prolactinomas.
Materials and Methods
Generation of transgenic mice
To generate mice that constitutively express lactotroph-targeted hEGFR or hHER2, we used the rat PRL (rPRL) enhancer/promoter (29). A 3239-bp fragment encoding the 5′-flanking sequence from 17 bp upstream of the first ATG was amplified by PCR. The resulting XbaI (5′-end, natural site) and NheI (3′-end, introduced by PCR) fragment was jointed to the 5′-end of the EGFR or HER2 cDNA (Figure 1A). The jointed fragment was inserted into the Internal Ribosome Entry Site vector carrying Internal Ribosome Entry Site/mCherry cDNA/BDH ployA. Constructs were generated using pGL4 vector (Promega). cDNAs encoding ErbB family (EGFR), and HER2 (N-neu and T-neu), were kindly gifted from Dr Mark Greene (University of Pennsylvania), were subcloned downstream of the rPRL promoter. The hEGFR or hHER2 expression constructs were microinjected into the male pronucleus of fertilized eggs and injected eggs transplanted to pseudopregnant foster mothers following standard procedures, all approved by the Institutional Animal Care and Use Committee.
Figure 1. Generation of hEGFR or hHER2 transgenic mice.

A, hEGFR or hHER2 transgenic mice were generated using the rPRL enhancer/promoter system. Structure of hEGFR or hHER2 expression units were illustrated. B, Whole-cell extracts were prepared using pituitaries from WT, EGFR, and HER2 transgenic mice, and immunoblotting was performed using anti-EGFR, anti-HER2, and antiactin (as a control). To show expression levels of human transgenes and endogenous mouse EGFR and Her2 (observed in WT mice), a short (a, 60 s) and a long (b, 300 s) exposed image are depicted. C, Immunobloting using whole-cell extracts from indicated tissues of the transgenic mice was performed with anti-EGFR, anti-HER2, and antiactin. D, Histochemical staining of pituitary from EGFR/HER2 transgenic mouse using anti-EGFR and anti-HER2 conjugated with Alexa Fluor goat antirabbit 488 (green) and anti-PRL conjugated with Alexa Fluor donkey antimouse 568 (red). Staining was analyzed by confocal microscopy. To confirm colocalization of hEGFR/PRL and hHER2/PRL, merged image is also shown (×63 magnification).
To identify transgenic animals, genomic DNA was isolated from 10- to 14-day offspring using a KAPA Mouse Genotyping kit (KAPABIOSYSTEM) and analyzed by PCR using 2 sets of primers. Primers binding to PRL promoter (PRL-F1 and PRL-F2) and EGFR (EGR-R) or HER2 (HER2-R) cDNA and primers binding to the mCherry sequence (mCherry-forward primer [F] and mCherry-reverse primer [R]) were used with the next sequences: PRL-F, TGCAGATGAGAAAGCAGTGG; EGFR-R, TGCCTTGGCAA ACTTTCTTT; PRL-F2, ATCCTTCCTTTCTGGCCACT; HER2-R, AGGCACGTAGGTAAGCTCCA; mCherry-F, TACGAGGGCACCCAGACCGC; and mCherry R, AAGTTGGTGCCGCGCAGCTT.
Animal husbandry
Animals were housed in accordance with Institutional Animal Care and Use Committee guidelines of the Department of Comparative Medicine, at Cedars-Sinai Medical Center.
Immunoblotting
Whole-cell extracts were prepared from pituitary, brain, lung, kidney, adrenal, and heart. These tissues were lysed in 100-μL radioimmunoprecipitation assay buffer (Sigma-Aldrich) containing protease inhibitor (Roche Molecular Biochemicals) and phosphatase inhibitor cocktails (Sigma-Aldrich). Lysates were centrifuged at 13 000g for 10 minutes at 4°C, and protein concentrations in the resulting whole-cell extracts were determined by bicinchoninic acid protein assay reagent (Thermo Scientific). A total of 50 μg of proteins in the sodium dodecyl sulfate sample buffer (2× Laemmli sample buffer; Life Science) was heated for 5 minutes at 100°C, separated on 4%–12% NuPAGE Bis-Tris gels, and electrotransferred for 1 hour to polyvinylidene difluoride (Invitrogen) and transferred to membranes. Membranes were blocked for 1 hour in 5% nonfat dry milk or 5% BSA in Tris-Buffered Saline and Tween 20 (TBS-T) buffer and incubated overnight with primary antibodies, including anti-pErk1/2 (Cell Signaling Technology), anti-Erk1/2 (Cell Signaling Technology), anti-pserine-threonine protein kinase (Akt) (Cell Signaling Technology), anti-Akt (Cell Signaling Technology), anti-pEGFR (Cell Signaling Technology), anti-EGFR (Cell Signaling Technology), anti-pHER2 (Cell Signaling Technology), and anti-HER2 (Cell Signaling Technology). After washing with TBS-T, membranes were incubated with peroxidase-conjugated secondary antibody (Amersham ECL HRP-Linked; Life Sciences) for 1 hour (5% nonfat dry milk or 5% BSA in TBS-T buffer). Blots were washed, and antibody binding proteins were detected using chemiluminescence detection system (Amersham Biosciences) (Table 1).
Table 1.
Antibody Table
| Peptide/Protein Target | Antigen Sequence (if known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| EGFR | NA | EGF Receptor (D38B1) XP rabbit mAb | Cell Signaling Technology, catalog #4267 | Rabbit, monoclonal | 1:1000 for WB, 1:200 for IF |
| pEGFR | NA | Phospho-EGF receptor (Tyr1068) antibody | Cell Signaling Technology, catalog number 2234 | Rabbit, polyclonal | 1:1000 for WB |
| HER2 | NA | HER2/ErbB2 (D8F12) XP rabbit mAb | Cell Signaling Technology, catalog number 2234 | Rabbit, monoclonal | 1:1000 for WB, 1:200 for IF |
| pHER2 | NA | Phospho-HER2/ErbB2 (Tyr877) antibody | Cell Signaling Technology, catalog number 2241 | Rabbit, polyclonal | 1:1000 for WB |
| PRL | NA | Prolactin(B-8), sc-271758 | Santa Cruz Biotechnology, Inc, catalog number sc-271758 | Mouse, monoclonal | 1:1000 for WB, 1:200 for IF |
| Erk1/2 | NA | p44/42 MAPK (Erl1/2) (137F5) rabbit mAb | Cell Signaling Technology, catalog number 4695 | Rabbit, monoclonal | 1:1000 for WB |
| pErk1/2 | NA | Phospho-p44/42 MAPK (Erl1/2) (Thr202/Tyr204) antibody | Cell Signaling Technology, catalog number 9101 | Rabbit, polyclonal | 1:1000 for WB, 1:200 for IF |
| Akt | NA | Akt(pan)(11E7) rabbit mAb | Cell Signaling Technology, catalog number 4685 | Rabbit, monoclonal | 1:1000 for WB |
| pAkt | NA | Phospho-Akt (Ser473) (D9E) XP rabbit mAb | Cell Signaling Technology, catalog number 4060 | Rabbit, monoclonal | 1:1000 for WB, 1:200 for IF |
| Actin | NA | Monoclonal antiactin antibody produced in mouse | Sigma-Aldrich, A4700 | Mouse, monoclonal | 1:5000 for WB |
Hormone assays
Serum PRL and TSH levels were analyzed by RIAs using reagents purchased from MP Biomedicals.
Immunohistochemical staining
Tumor specimens or pituitary glands were fixed in 10% formalin and embedded in paraffin. After deparaffinization and antigen retrieval, slides were blocked in 10% goat serum in 1% BSA-PBS, incubated overnight with rabbit polyclonal anti-EGFR (Cell Signaling Technology) and mouse monoclonal anti-PRL (Santa Cruz Biotechnology, Inc). After washing, samples were incubated with Alexa Fluor goat antirabbit 488 (Heavy + Light chains, 1:500 dilution; Invitrogen) and Alexa Fluor donkey antimouse 568 (Invitrogen) for 2 hours at room temperature, then mounted with Prolong Gold antifade reagent (Invitrogen). Confocal microscope images were obtained using True Confocal Scanner confocal scanner (Leica Microsystems) in a dual emission mode to separate autofluorescence from specific staining.
Lapatinib treatment
Female mice with PRL levels more than 40 ng/mL were divided into 2 groups for lapatinib treatment and controls (n = 14 per group). Mice were treated with lapatinib (100 mg/kg) or vehicle (0.5% methylcellose, 0.5% Tween 80/PBS; 100 μL) by oral gavage daily for 3 weeks. Blood (200 μL) was collected twice for hormone assessment (before treatment) by retro-orbital bleeding under isoflurane inhalational anesthesia via a nose cone connected to a rodent anesthesia machine (Kent Scientific Corp). On the last treatment day (d 21), mice were euthanized within 3 hours of drug administration, cardiac blood was collected with 18-gauge syringes, and pituitary glands or tumors were harvested. Fragments of each pituitary or tumor were fixed in formalin and embedded in paraffin for immunohistochemical staining and preserved in liquid nitrogen for subsequent protein extraction.
Statistics
Results are expressed as mean ± SEM. Differences were assessed by one-way ANOVA following the Scheffe's F test. P< .05 was considered significant.
Results
Generation of EGFR or HER2 transgenic mice using the rPRL enhancer/promoter system
To investigate the role of hEGFR or hHER2 in prolactinoma pathogenesis, we generated female transgenic mice using the rPRL enhancer/promoter (Figure 1A) that has previously shown lactotroph-targeted expression (29). hEGFR expression in the pituitary was detected in 4-month-old mice by immunoblotting (Figure 1B). Transgenic hEGFR expression was 13-fold more abundant than endogenous EGFR in wild-type (WT) mice and was seen to be restricted to transgenic pituitary glands (Figure 1C). Similarly, pituitary-specific hHER2 expression observed in HER2 transgenic mice was enhanced approximately 9-fold compared with endogenous HER2 levels and was restricted to transgenic pituitary glands. Endogenous expression of mouse pituitary HER3 and HER4 in both transgenic mice were both not detectable by Western blotting. These results indicate that lactotroph-targeted EGFR or HER2 expression is controlled by the rPRL enhancer/promoter system in these transgenic mice. To confirm pituitary cell colocalization of EGFR and PRL, confocal immunofluorescence was performed on the EGFR transgenic mouse pituitary (Figure 1D). At age 13–15 months, enlarged pituitaries, or adenomas, were observed in 35% (13 of 37) euthanized female transgenic mice (Figure 2, A–D). Five of 20 EGFR transgenic mice developed pituitary tumors, and 1 had an enlarged pituitary gland (3 × 2 mm). Seven (4 T-neu and 3 N-neu) of 17 HER2 (9 T-neu and 8 N-neu) transgenic mice developed tumors, whereas only 1 WT mouse had an enlarged pituitary gland (3 × 2 mm) (Table 2). Circulating PRL levels in EGFR or HER2 transgenic mice were significantly increased compared with WT, respectively (110 ± 20 vs 22 ± 6 ng/mL; P = .0025 and 84 ± 18 vs 22 ± 6 ng/mL; P = .0087) (Figure 3A). In summary, circulating PRL levels in both EGFR and HER2 transgenic mice were 4.4-fold increased compared with WT (98 ± 14 vs 22 ± 6 ng/mL; P = .0029). Using confocal immunofluorescence, pituitary PRL expression in transgenic mice was observed to be higher than in WT pituitary glands, consistent with results of the Western blotting (Figure 3B).
Figure 2. Prolactinoma development in EGFR or HER2 transgenic mice.
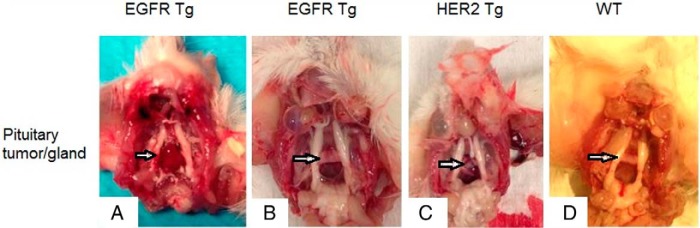
A, Prolactinoma in EGFR transgenic mouse (female, 13 mo), 4 × 5 mm. B. Hyperplastic pituitary gland in EGFR transgenic mouse (female, 14 mo), 3 × 2 mm. C, Prolactinoma in HER2 (T-neu) transgenic mouse (female, 13 mo), 3 × 4 mm. D, Pituitary gland in WT mouse (female, 13 mo), 2 × 1 mm.
Table 2.
Pituitary Tumorigenesis in Female ErbB Transgenic Mice, Age 11–13 Months
| WT | EGFR | HER2 | |
|---|---|---|---|
| Total (n) | 12 | 20 | 17 (9 T-neu and 8 N-neu) |
| % tumor penetrance or enlarged pituitary | 8% | 30% | 41% (4 T-neu and 3 N-neu) |
| Circulating PRL levels (ng/mL) | 22 ± 6 | 110 ± 20a | 84 ± 18a |
P < .05 vs WT.
Figure 3. PRL expression in EGFR or HER2 transgenic mice.

A, Serum PRL levels in EGFR or HER2 transgenic mice were analyzed using indicated mice by RIA (**, P < .01). B. Histochemical staining using pituitaries (×100 magnification) from female EGFR transgenic mouse (B1), HER2 (T-neu) female transgenic mouse (B2), and WT mouse (B3). All mice were 13 months old.
Inhibitory effects of lapatinib on EGFR or HER2-induced prolactinomas
To test the responsiveness of prolactinomas induced by EGFR or HER2 expression to lapatinib, a dual TKI for both EGFR and HER2 (see reference 41 below) mice with circulating PRL levels more than 40 ng/mL was divided into treatment and control groups (n = 14 per group) and treated with lapatinib (100 mg/kg) or vehicle (0.5% methylcellose, 0.5% Tween 80/PBS; 100 μL) by oral gavage daily for 3 weeks, respectively. After 3 weeks of treatment, circulating PRL levels in EGFR transgenic mice receiving lapatinib treatment were significantly decreased compared with pretreatment levels (158 ± 86 vs 59 ± 48 ng/mL; P = .0055) (Figure 4A). In contrast, PRL levels in control mice were unchanged after vehicle treatment (142 ± 73 vs 131 ± 67 ng/mL; P = .127) (Figure 4A). In HER2 transgenic mice, lapatinib attenuated circulating PRL levels significantly (103 ± 79 vs 59 ± 39 ng/mL; P = .04) (Figure 4B), whereas PRL levels in control group were not changed (90 ± 67 vs 86 ± 54 ng/mL; P = .59) (Figure 4B). According to one-way ANOVA, no significant differences on magnitude of effects were observed between EGFR and HER2 transgenics. Circulating TSH levels were also analyzed and unchanged in EGFR or HER2 transgenic mice after lapatinib treatment, respectively (174 ± 48 vs 179 ± 83 ng/mL; P = .885 and 175 ± 83 vs 164 ± 71 ng/mL; P = .666), indicating the inhibitory effect of lapatinib specifically on PRL production. Notably, mice body weights were not altered after oral lapatinib treatment.
Figure 4. Effect of lapatinib on PRL levels in EGFR or HER2 transgenic mice.

Circulating PRL and TSH (as a control) levels in EGFR or HER2 transgenic mice before and after treatment of oral lapatinib (100 mg daily for 3 wk). Same number (7 in each group) of mice were also treated with vehicle as a control. A, PRL levels before and after treatment in EGFR transgenic mice. B, PRL levels before and after treatment in HER2 transgenic mice. C, TSH levels pre- and postlapatinib in EGFR transgenic mice. D, TSH levels pre- and postlapatinib in EGFR transgenic mice *, P < .01; #, P > .05.
p-Akt and p-Erk were reduced by lapatinib treatment
To investigate downstream signaling effects of lapatinib in these transgenic animals, immunoblot analysis was performed on pituitary tumor extracts derived from EGFR/HER2 transgenic mice. Four mice from each group were selected based on tumor size (>3 × 2 mm), and PRL levels in selected mice are shown above the immunoblotting results (Figure 5A). Compared with 4 mice treated with vehicle, lapatinib attenuated tumor PRL expression by 74% in 4 EGFR transgenic mice (Figure 5A). Furthermore, expression of EGFR signal transducing molecules p-Akt and p-Erk was also suppressed in tumors treated with oral lapatinib by 53% and 74%, respectively. Total Akt and total Erk levels were unchanged (Figure 5A). Similar results were also seen in HER2 transgenic mice (Figure 5B). Lapatinib attenuated tumor PRL, p-Akt, and p-Erk expression by 86%, 60%, and 71%, compared with vehicle, respectively, whereas total Akt and total Erk levels were not changed (Figure 5B). Confocal immunofluorescence results confirmed PRL, p-Akt and p-Erk suppression after lapatinib treatment (Figure 5C).
Figure 5. Effects of lapatinib on EGFR or HER2 downstream signaling.

Levels of PRL, p-Akt, p-Erk and t-Erk expression were analyzed by immunoblotting using pituitaries derived from EGFR (A) or HER2 (B) transgenic mice treated with oral lapatinib or vehicle (100 mg daily for 3 wk). Four mice were selected based on matched tumor size (>3 × 2 mm), and effectiveness of lapatinib or vehicle on PRL level in each individual mouse is depicted above the blot. C, Histochemical staining of PRL, p-Erk, and p-Akt in pituitary glands derived from EGFR transgenic mice treated with lapatinib or vehicle were analyzed (×20 magnification).
Discussion
Mechanisms underlying prolactinoma pathogenesis are largely unclear mainly due to the unavailability of human prolactinoma cell lines, reproducible animal models, and cellular heterogeneity. Here, we report that lactotroph-targeted transgenic hEGFR/HER2 expression, induced prolactinomas between 13 and 15 months of age. Moreover, inhibiting EGFR/HER2 activity with lapatinib, a dual TKI for both EGFR and HER2, leads to suppression of hyperprolactinemia and MAPK and Akt pathways.
Receptor-specific ligands, including EGF, heparin-binding EGF, TGF-α, and heregulins, bind extracellular domains of individual ErbB receptors and induce receptor homo- or heterodimerization and activation of downstream signaling (30, 31), which regulate processes for tumor initiation and progression. Specifically, ErbB2 possesses an active tyrosine kinase domain, and no ligand has been identified. ErbB receptor overexpression in transgenic mice driven by tissue-specific promoters induced lung and breast cancers (32–34). Expression of ErbB receptors and ligands has been reported in normal anterior pituitary cells, inducing PRL secretion in lactotrophs (35–38). EGF and heregulin also regulate PRL gene expression and induce lactotroph differentiation (26, 27, 39), further suggesting a role for ErbB receptors in lactotroph tumor development.
To identify mechanisms underlying prolactinoma tumorigenesis, we generated transgenic mice expressing EGFR or HER2 driven by the tissue-specific rPRL enhancer/promoter (29). As shown in Figure 1, we successfully generated these transgenic mice with this system. Because we used the natural rPRL promoter and enhancer for EGFR or HER2 expression, their expression levels were not very high compared with other reports using strong promoters, such as the cytomegalovirus promoter (40, 41). Indeed, we could not detect mCherry fluorescence regulated by IRIS in the transcripts. Levels of hEGFR and HER2 expression seem to be more physiological in younger (before 13 mo) transgenic mice, and enlarged pituitary glands or prolactinomas were observed at 13–15 months of age in 35% of transgenic mice. Circulating PRL levels were significantly increased (>40 ng/mL) with hEGFR or HER2 expression, supporting the notion that EGFR or HER2 expression is a factor contributing to lactotroph tumorigenesis. However, lactotrophs expressing EGFR or HER2 did not develop pituitary tumors at earlier stages, indicating that EGFR or HER2 activation appears not to be the sole requirement for full transformation of a normal lactotroph and other changes (ie, a second hit) are required to invoke transformation.
Prolactinomas resistant to dopamine agonist therapy and surgery are a continuing challenge for endocrinologists. Lapatinib, a TKI that targets both EGFR and HER2, is used in combination for advanced HER2-posivtive breast cancer (42) and inhibits receptor signaling by preventing autophosphorylation and subsequent MAPK-Erk signaling (43). MAPK-Erk is also activated in PRL and growth hormone (GH)-secreting GH4C1 and GH3 cells treated with either heregulin or EGF (42). In AtT20 cells, overexpression of EGFR led to activation of MAPK-Erk signaling, which was inhibited by gefitinib, an EGFR TKI (24). Furthermore, overexpressed HER2 leading to activation of MAPK-Erk signaling in GH3 cells was inhibited by both gefitinib and lapatinib (27).
The results shown here indicate that lapatinib suppressed serum PRL levels and attenuated tumor PRL expression in EGFR- and HER2-driven prolactinomas, supporting the feasibility of ErbB-targeted therapy for prolactinomas.
Acknowledgments
We thank Dr Song-Guang Ren and Roy Heltsley for technical assistance; Kolja Wawrowsky at the Confocal Core of Cedars-Sinai Medical Center; and Dr Mark Greene and Dr Qiang Wang for kindly providing us EGFR and HER2 cDNA.
This work was supported by National Institutes of Health Grants CA07597 (to S.M.), T32DK007770 (to T.A.), and K23DK085148 (to O.C.) and by the Doris Factor Molecular Endocrinology Laboratory.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Akt
- serine-threonine protein kinase
- EGFR
- epidermal growth factor receptor
- ErbB
- epidermal growth factor receptor
- F
- forward primer
- GH
- growth hormone
- hEGFR
- human EGFR
- HER
- epidermal growth factor receptor
- hHER2
- human HER2
- neu
- epidermal growth factor receptor 2
- PRL
- prolactin
- R
- reverse primer
- rPRL
- rat PRL
- TBS-T
- Tris-Buffered Saline and Tween 20
- TKI
- tyrosine kinase inhibitor
- WT
- wild type.
References
- 1. Melmed S. Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest. 2003;112(11):1603–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gillam MP, Molitch ME, Lombardi G, Colao A. Advances in the treatment of prolactinomas. Endocr Rev. 2006;27(5):485–534. [DOI] [PubMed] [Google Scholar]
- 3. Melmed S. Pathogenesis of pituitary tumors. Nat Rev Endocrinol. 2011;7(5):257–266. [DOI] [PubMed] [Google Scholar]
- 4. Casanueva FF, Molitch ME, Schlechte JA, et al. Guidelines of the Pituitary Society for the diagnosis and management of prolactinomas. Clin Endocrinol (Oxf). 2006;65(2):265–273. [DOI] [PubMed] [Google Scholar]
- 5. Mancini T, Casanueva FF, Giustina A. Hyperprolactinemia and prolactinomas. Endocrinol Metab Clin North Am. 2008;37(1):67–99, viii. [DOI] [PubMed] [Google Scholar]
- 6. Olafsdottir A, Schlechte J. Management of resistant prolactinomas. Nat Clin Pract Endocrinol Metab. 2006;2(10):552–561. [DOI] [PubMed] [Google Scholar]
- 7. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96(2):273–288. [DOI] [PubMed] [Google Scholar]
- 8. Jane JA, Jr, Thapar K, Kaptain GJ, Maartens N, Laws ER., Jr Pituitary surgery: transsphenoidal approach. Neurosurgery. 2002;51(2):435–442; discussion 442–444. [PubMed] [Google Scholar]
- 9. Losa M, Mazza E, Terreni MR, et al. Salvage therapy with temozolomide in patients with aggressive or metastatic pituitary adenomas: experience in six cases. Eur J Endocrinol. 2010;163(6):843–851. [DOI] [PubMed] [Google Scholar]
- 10. Raverot G, Sturm N, de Fraipont F, et al. Temozolomide treatment in aggressive pituitary tumors and pituitary carcinomas: a French multicenter experience. J Clin Endocrinol Metab. 2010;95(10):4592–4599. [DOI] [PubMed] [Google Scholar]
- 11. Bronstein MD, Knoepfelmacher M, Liberman B, Marino R, Jr, Germek OA, Schally AV. Absence of suppressive effect of somatostatin on prolactin levels in patients with hyperprolactinemia. Horm Metab Res. 1987;19(6):271–274. [DOI] [PubMed] [Google Scholar]
- 12. Lamberts SW, Verleun T, Oosterom R. Effect of tamoxifen administration on prolactin release by invasive prolactin-secreting pituitary adenomas. Neuroendocrinology. 1982;34(5):339–342. [DOI] [PubMed] [Google Scholar]
- 13. Zhang H, Berezov A, Wang Q, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117(8):2051–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herbst RS, Fukuoka M, Baselga J. Gefitinib–a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4(12):956–965. [DOI] [PubMed] [Google Scholar]
- 15. Chaidarun SS, Eggo MC, Sheppard MC, Stewart PM. Expression of epidermal growth factor (EGF), its receptor, and related oncoprotein (erbB-2) in human pituitary tumors and response to EGF in vitro. Endocrinology. 1994;135(5):2012–2021. [DOI] [PubMed] [Google Scholar]
- 16. Cooper O, Vlotides G, Fukuoka H, Greene MI, Melmed S. Expression and function of ErbB receptors and ligands in the pituitary. Endocr Relat Cancer. 2011;18(6):R197–R211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ezzat S, Zheng L, Smyth HS, Asa SL. The c-erbB-2/neu proto-oncogene in human pituitary tumours. Clin Endocrinol (Oxf). 1997;46(5):599–606. [DOI] [PubMed] [Google Scholar]
- 18. Halper J, Parnell PG, Carter BJ, Ren P, Scheithauer BW. Presence of growth factors in human pituitary. Lab Invest. 1992;66(5):639–645. [PubMed] [Google Scholar]
- 19. Kontogeorgos G, Stefaneanu L, Kovacs K, Cheng Z. Localization of epidermal growth factor (EGF) and epidermal growth factor receptor (EGFr) in human pituitary adenomas and nontumorous pituitaries: an immunocytochemical study. Endocr Pathol. 1996;7(1):63–70. [DOI] [PubMed] [Google Scholar]
- 20. Peillon F, Le Dafniet M, Garnier P, et al. Receptors and neurohormones in human pituitary adenomas. Horm Res. 1989;31(1–2):13–18. [DOI] [PubMed] [Google Scholar]
- 21. Theodoropoulou M, Arzberger T, Gruebler Y, et al. Expression of epidermal growth factor receptor in neoplastic pituitary cells: evidence for a role in corticotropinoma cells. J Endocrinol. 2004;183(2):385–394. [DOI] [PubMed] [Google Scholar]
- 22. Nose-Alberti V, Mesquita MI, Martin LC, Kayath MJ. Adrenocorticotropin-producing pituitary carcinoma with expression of c-erbB-2 and high PCNA index: a comparative study with pituitary adenomas and normal pituitary tissues. Endocr Pathol. 1998;9(1):53–62. [DOI] [PubMed] [Google Scholar]
- 23. Roncaroli F, Nosé V, Scheithauer BW, et al. Gonadotropic pituitary carcinoma: HER-2/neu expression and gene amplification. Report of two cases. J Neurosurg. 2003;99(2):402–408. [DOI] [PubMed] [Google Scholar]
- 24. Fukuoka H, Cooper O, Ben-Shlomo A, et al. EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J Clin Invest. 2011;121(12):4712–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fukuoka H, Cooper O, Mizutani J, et al. HER2/ErbB2 receptor signaling in rat and human prolactinoma cells: strategy for targeted prolactinoma therapy. Mol Endocrinol. 2011;25(1):92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vlotides G, Cooper O, Chen YH, Ren SG, Greenman Y, Melmed S. Heregulin regulates prolactinoma gene expression. Cancer Res. 2009;69(10):4209–4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vlotides G, Siegel E, Donangelo I, Gutman S, Ren SG, Melmed S. Rat prolactinoma cell growth regulation by epidermal growth factor receptor ligands. Cancer Res. 2008;68(15):6377–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cooper O, Mamelak A, Bannykh S, et al. Prolactinoma ErbB receptor expression and targeted therapy for aggressive tumors. Endocrine. 2014;46(2):318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crenshaw EB, 3rd, Kalla K, Simmons DM, Swanson LW, Rosenfeld MG. Cell-specific expression of the prolactin gene in transgenic mice is controlled by synergistic interactions between promoter and enhancer elements. Genes Dev. 1989;3(7):959–972. [DOI] [PubMed] [Google Scholar]
- 30. Borg A, Tandon AK, Sigurdsson H, et al. HER-2/neu amplification predicts poor survival in node-positive breast cancer. Cancer Res. 1990;50(14):4332–4337. [PubMed] [Google Scholar]
- 31. Nagy P, Jenei A, Damjanovich S, Jovin TM, Szölôsi J. Complexity of signal transduction mediated by ErbB2: clues to the potential of receptor-targeted cancer therapy. Pathol Oncol Res. 1999;5(4):255–271. [DOI] [PubMed] [Google Scholar]
- 32. Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation. 2007;75(9):770–787. [DOI] [PubMed] [Google Scholar]
- 33. Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arteaga CL. EGF receptor mutations in lung cancer: from humans to mice and maybe back to humans. Cancer Cell. 2006;9(6):421–423. [DOI] [PubMed] [Google Scholar]
- 35. Armstrong J, Childs GV. Changes in expression of epidermal growth factor receptors by anterior pituitary cells during the estrous cycle: cyclic expression by gonadotropes. Endocrinology. 1997;138(5):1903–1908. [DOI] [PubMed] [Google Scholar]
- 36. Mouihate A, Lestage J. Epidermal growth factor: a potential paracrine and autocrine system within the pituitary. Neuroreport. 1995;6(10):1401–1404. [PubMed] [Google Scholar]
- 37. Murdoch GH, Potter E, Nicolaisen AK, Evans RM, Rosenfeld MG. Epidermal growth factor rapidly stimulates prolactin gene transcription. Nature. 1982;300(5888):192–194. [DOI] [PubMed] [Google Scholar]
- 38. Zhao WJ. Expression and localization of neuregulin-1 (Nrg1) and ErbB2/ErbB4 receptors in main endocrine organs of the rhesus monkey. Int J Endocrinol Metab. 2013;11(3):162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Felix R, Meza U, Cota G. Induction of classical lactotropes by epidermal growth factor in rat pituitary cell cultures. Endocrinology. 1995;136(3):939–946. [DOI] [PubMed] [Google Scholar]
- 40. Fang K, Chen MH. Transfection of anti-sense complementary DNA of human epidermal-growth-factor receptor attenuates the proliferation of human non-small-cell-lung-cancer cells. Int J Cancer. 1999;81(3):471–478. [DOI] [PubMed] [Google Scholar]
- 41. Lanteri M, Ollier L, Giordanengo V, Lefebvre JC. Designing a HER2/neu promoter to drive α1,3galactosyltransferase expression for targeted anti-αGal antibody-mediated tumor cell killing. Breast Cancer Res. 2005;7(4):R487–R494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cameron DA, Stein S. Drug insight: intracellular inhibitors of HER2–clinical development of lapatinib in breast cancer. Nat Clin Pract Oncol. 2008;5(9):512–520. [DOI] [PubMed] [Google Scholar]
- 43. Nelson MH, Dolder CR. Lapatinib: a novel dual tyrosine kinase inhibitor with activity in solid tumors. Ann Pharmacother. 2006;40(2):261–269. [DOI] [PubMed] [Google Scholar]