

Abstract
Development of DNA aptamer screens that are both simple and informative can increase the success rate of DNA aptamer selection and induce greater adoption. High eIF4e levels contribute to malignancies, thus eIF4e presents itself as a valuable target for DNA aptamer-based inhibition screen. Here, we demonstrate a method for the rapid selection of looped DNA aptamers against eIF4e by combining negative selection and purification in a single step, followed by characterization with high throughput sequencing. The resulting aptamers show functional binding to eIF4e and inhibit translation initiation in biochemical assays. When transfected into cells, eIF4e aptamers cause a dramatic loss of cell proliferation in tumor cells as seen with eIF4e knockdown with antisense oligonucleotides, shRNAs, and siRNAs, hinting at therapeutic possibilities. With the large data set provided by high throughput sequencing, we demonstrate that selection happens in waves and that sequencing data can be used to infer aptamer structure. Lastly, we show that ligation of looped aptamers can enhance their functional effects. These results demonstrate a rapid protocol to screen and optimize aptamers against macromolecules of interest.
Keywords: Aptamer, eIF4e, SELEX, proliferation
Introduction
Aptamers are single-stranded oligonucleotides that form secondary structures that can specifically interact with ligands,1,2 including small molecules,3 glycans,4 and proteins,5 for a wide range of functions including detection of its cognate target6,7 and targeted delivery.8,9 Beyond just purely binding to their targets, DNA aptamers can also inhibit enzymatic activity and binding as well. For example, thrombin-binding aptamers have been shown to inhibit clot formation;5 AS1411 has also been shown to inhibits nucleolin's interaction with arginine methyltransferase 5.10 While a multitude of RNA aptamers and their analogs against various targets have been identified, DNA aptamers are easier to perform a screen with as they are more resistant to nuclease digest and are amenable to standard PCR as opposed to the more tedious amplification processes for RNA and its analogs.
The method of choice for identifying aptamers is called Systematic evolution of ligands by exponential enrichment (SELEX), in which specific and high affinity aptamers are identified through successful rounds of affinity selection and amplification with increasing stringency (typically 8–16 rounds). The initial aptamer library is frequently precleared against the solid phase without the target protein to remove sequences that bind to the solid phase. For DNA aptamers, amplification with PCR after each round of selection is followed with a denaturation and purification step to isolate single stranded aptamers.11 Some groups have also used asymmetric PCR to preferentially generate aptamer strands over antisense complementary strands.12
Following rounds of selection, the resultant library is typically then cloned and sequenced via Sanger sequencing. Recently, a number of groups including ours13,14,15,16,17 have harnessed high-throughput sequencing to the time-consuming cloning stage and dramatically increase the insight into the library sequence space that cannot be adequately covered with Sanger sequencing. While all these studies focused on the discovery of specific motifs, only two analyzed the increase in relative abundance of specific aptamers over time, which is more valuable than taking the end point analysis because some aptamers could dominate earlier on while other sequences appear much later in the selection rounds. Cho et al. 13 analyzed the relative abundance of motifs over three rounds of selection while Schutze et al. 15 analyzed DNA aptamers selected to bind to streptavidin and traced the relative abundance of specific sequences through 10 rounds of selection. Interestingly, Schutze et al. 15 found two differing patterns of enrichment amongst the most abundant sequence clusters after 10 rounds of selection. The first group peaked in relative abundance between selection rounds 5–7 before declining in abundance to give way to the next wave of sequences between selection rounds 8–10. Another piece of data that could be gleaned from tracking the selection of specific aptamer clusters is the relative importance of specific substitutions, insertion, or deletion in binding to the ligand. Although Schutze et al. 15 and Ditzler et al. 16 attempted to analyze the relative importance of particular positions to binding to its cognate target, analyses was complicated by the use of error-prone amplification processes inherent to reverse transcription and Taq polymerase-based PCR.
Cap-dependent translation in eukaryotes is initiated by the 5′ end cap structure (m7GTP) of mRNA binding to eIF4e. eIF4e forms a complex with other initiation factors and the ribosomal 40S subunit, shuttling down the mRNA until the start codon is reached.18 Although eIF4E regulates translation globally, overexpression of eIF4e contributes to tumor malignancy by enabling the increased translation of mRNAs with highly structured, G+C-rich 5′UTRs, typical of proto-oncogenic mRNAs such as VEGF.19 Thus, eIF4e inhibition can potentially be an anticancer strategy.20 Nucleic acid aptamers that can bind and inhibit eIF4e activity can potentially be used to assay for expression of eIF4e, screen for small molecule inhibitors that can mimic its action, even therapeutically to inhibit eIF4e in a specific fashion. Although an RNA aptamer that binds to eIF4e has been identified previously,21 RNA aptamers are long, thus costly to synthesize, and susceptible to nucleases, thus can degrade rapidly once applied, which limits the efficacy of the aptamers over time.
Here, we present a method for the rapid selection of looped DNA aptamers against eIF4e by combining negative selection and purification in a single step, followed by characterization of selection libraries with high throughput sequencing. The resulting aptamers show functional binding to eIF4e and inhibit translation initiation in biochemical assays. When transfected into cells, eIF4e aptamers can result in dramatic loss of cell viability, suggesting that these aptamers could be used for therapeutic ends. With the large data set provided by high throughput sequencing, we show that selection happens in waves and that sequencing data can be used to infer essential bases in aptamers. Lastly, we show that ligation of looped aptamers can enhance their biochemical effects in biochemical assays although not in cell culture experiments.
Results
Selection of eIF4e binding aptamer
In order to maximize the chance of specific binding and for ease of selection, the biotin-streptavidin interaction was utilized for positive and negative selections as well as isolation of single stranded aptamers (Figure 1a). A 33-mer aptamer library previously utilized in Hoon et al. 17 was synthesized such that the 5′ and 3′ ends of the aptamer would form a loop. Approximately 6 × 1012 unique sequences were amplified tenfold with asymmetric PCR using a high-fidelity DNA polymerase to increase the number of molecules representing each unique sequence. The biotinylated reverse primer allows for the removal of double stranded DNA with streptavidin beads after PCR amplification and also the concurrent removal of streptavidin-binding aptamers (negative selection). The heating and cooling steps within asymmetric PCR would also have allowed single stranded aptamer to form secondary structures, which means the unpurified flowthrough depleted of double stranded PCR products could be utilized for selection straightaway against biotinylated eIF4e immobilized on streptavidin beads. Increasing the number of washes and decreasing the amount of protein-loaded beads and aptamers increased the stringency of selection with each round of selection.
Figure 1.

Selection of DNA aptamers against eIF4e using a rapid selection procedure. (a) A library composed of a looped 33-mer random sequence is amplified tenfold by asymmetric PCR with unmodified forward primer and biotinylated reverse primer before negative selection with streptavidin beads and positive selection with eIF4e beads followed by repeating amplification with asymmetric PCR. (b) The number of unique sequences identified by high throughput sequencing decreases with increasing rounds of selection. Each round generated between 2 and 3 million valid reads.
Following eight rounds of selection, each round of selection was amplified again with specific bar codes and sequenced. After removing poor quality sequencing reads, at least 2 million reads per round was used for further analysis. The number of unique reads decreased with increasing round of selection (Figure 1b), indicating that purifying selection was taking place.
Selection occurs in waves
After sequencing, the sequences from all rounds were grouped into clusters based on sequence similarity, with sequences differing by at most three bases grouped into the same clusters. The most highly represented members of 12 selected clusters without the loop sequences at either end are shown in Figure 2a. Figure 2b shows the relative distribution of these clusters as a function of selection rounds. Based on the relative abundance, these aptamers can be broadly be binned into two classes. One class dominates early on in selection, peaking at rounds 3–6, examples of which are aptamers 1, 3, 4, 7, 9, 10, and 12. The second class begins displacing the first class after round 7, examples of which are aptamers 2, 5, 6, 8, and 11. Schutze et al. 15 also described two waves of selection, suggesting this is a generally applicable phenomenon with DNA aptamer selection. Hence, surveying aptamers at selection end points will invariably lead to omission of classes of aptamers, especially after 10–16 rounds of selection typical of SELEX. Surprisingly, there is no clear motif similarity between the 12 classes examined although several of the aptamer clusters are G-rich (aptamers 5, 6, and 8), suggesting that although G-quadruplexes structures could have formed, most of the aptamers probably do not rely on G-quadruplexes for binding.
Figure 2.
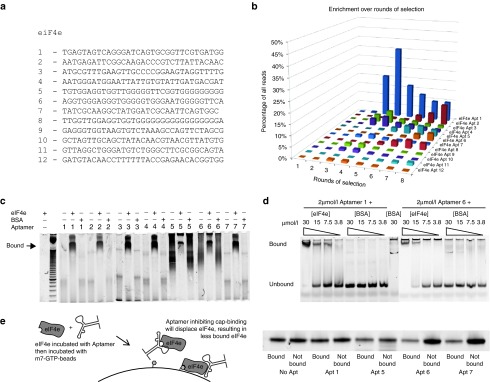
Aptamer clusters are genuine binders of eIF4e. (a) Representative members of the top 12 clusters of aptamers selected. (b) Relative abundance of each aptamer family as a percentage of all reads over increasing rounds of selection. (c,d) 6% polyacrylamide EMSA of the selected aptamers against eIF4e and BSA at 2 µmol/l of aptamer and 15 µmol/l of (c) protein or (d) stated concentration of protein. Aptamers were stained with SyBr Gold after electrophoresis. The images are composite images put together as the aptamers were ran across several polyacrylamide gels. Original gel images can be found in Supplementary Figure S8. (e) 150 nmoles of eIF4e was incubated with 150 nmoles of each aptamer or water then incubated with m7-GTP-Sepharose beads containing ~500 nmoles of m7-GTP. Half the beads and half the supernatant was then run on a polyacrylamide gel and Coomassie stained.
In order to establish the aptamers could bind eIF4e, electrophoretic mobility shift assay (EMSA) was performed on the seven most highly represented aptamers against eIF4e and BSA. These aptamers were shown to specifically bind to eIF4e as opposed to BSA in an excess of protein (Figure 2c). Two of the seven aptamers were shown to bind in a concentration-dependent manner to eIF4e but not BSA (Figure 2d). Furthermore, an excess of aptamers 1, 5, 6, and 7 affected the migration of eIF4e in a polyacrylamide native gel and can be found colocalized with eIF4e when the gel is stained with SyBr Gold followed by Coomassie (Supplementary Figure S1a). The aptamers were not stained with Coomassie stain and neither were the proteins stained by SyBr Gold. The only surprise was aptamer 2 which did not seem to significantly shift the eIF4e band. To further confirm specificity, aptamers 1 and 6, which demonstrate concentration-dependent shift, did not bind purified eIF4A1 and eIF4B either (Supplementary Figure S1b)
These results demonstrate that the selection protocol yielded genuine binders. Interestingly, aptamers 6 and 7, when prebound to eIF4e, resulted in decreased eIF4e binding onto m7-GTP-beads, while aptamers 1 and 5 did not (Figure 2e), implying that aptamers 6 and 7 bind to and inhibit cap-binding surface of eIF4e while aptamers 1 and 5 do not.
Translation is inhibited by eIF4e aptamers
As eIF4e is a relatively small compact globular protein (25 kDa), it is likely one of the aptamers would be bound to epitopes essential for its function, thus the aptamer could serve to inhibit eIF4e function in cells. To establish if the aptamers could inhibit eIF4e, we used an in vitro transcription and translation kit utilizing cap analogs, a luciferase expressing plasmid and T7 RNA polymerase to generate a mRNA mimic before the insect cell extract, with the translational machinery including eIF4e, is used to convert the mRNA into protein. The relative luciferase activity would thus indicate translation initiation efficiency of eIF4e in the presence of aptamers. Although all selected aptamers bound eIF4e, only three out of the seven inhibited translation at 20 µmol/l (Supplementary Figure S2). This result is borne out in experiments in triplicate and Apt 1, which showed dramatic inhibition of translation previously, demonstrated that this effect is dependent on the concentration of aptamer (Figure 3a). To eliminate the possibility that eIF4e aptamer affected transcription, the total RNA from the TnT reactions was DNase I treated then harvested by Trizol extraction to measure the total RNA produced by the T7-mediated transcription. The RNA content appears to be comparable, which demonstrates that luciferase inhibition was due to translation rather than transcriptional inhibition (Supplementary Figure S3). To establish if the binding of Apt 1 to eIF4e was responsible for translational inhibition, 25 µg/µl of recombinant purified human eIF4e added in the presence of Apt 1 rescued the inhibition partially, probably by acting as a decoy and releasing the inhibition on insect eIF4e, suggesting that eIF4e is indeed the target of Apt 1-mediated inhibition (Figure 3b). Surprisingly, when the same experiment was performed using a rabbit reticulocyte transcription and translation kit, all aptamers surveyed except for Apt 5 and 7 resulted in inhibition of translation at 20 µmol/l (Supplementary Figure S4), suggesting that the binding surfaces of the aptamers are different, hence aptamers 1 and 6 are capable of inhibiting both insect eIF4e and rabbit eIF4e but aptamers 2–4 are only capable of affecting mammalian eIF4e. Thus, we were successful at demonstrating that the selected aptamers bound to eIF4e to inhibit translation in a biochemical system.
Figure 3.

eIF4e aptamers inhibit translation. (a) Insect cell transcription and translation kit using a luciferase template and stated dilution of aptamer or (b) 20 µmol/l of aptamer 1 in the presence of 120 µmol/l BSA or human eIF4e. NS is a random oligonucleotide that can form a hairpin structure. (c) Number of reads from eIF4e Aptamer 1 cluster round 8 that deviated at specific positions from the most abundant member of the cluster. Red letters indicate highly conserved nucleotide, green indicates a position that has relatively more substitutions. (d) MFold prediction of the secondary structure of eIF4e Apt 1 with the associated bases highlighted in red or green. (e) Insect cell transcription and translation assay with eIF4e Apt 2 and eIF4e Apt 1 and mutants with a single modification at specific positions. All error bars reflect standard deviations. *P < 0.05; **P < 0.01; ***P < 0.001; two-tailed Student's t-test. For a, P values refer to comparisons against control/no aptamer.
Sequence determinants of eIF4e inhibition can be deduced from sequence abundance
As high throughput sequencing was used to survey the library space, a large dataset of sequences could be probed. Furthermore, the use of high fidelity polymerases for amplification of the libraries reduced the chance of random mutations being responsible for sequence variation within a cluster. Thus, we hypothesized that the relative abundance of specific sequence substitutions in each position of the consensus could be used to predict how essential the base is to its function. Sequences from cluster 1 were curated and all unique sequences with at least three reads per million in the final round of selection were used to construct the relative abundance of each substitution (Figure 3c). Based on this data, mutations made in the red positions are predicted to negatively impact its binding and hence inhibition because they are poorly represented. Highly represented mutations in green, is conversely predicted to not affect its function. The Apt 1 secondary structures predicted by MFold22 to have the lowest free energy and the relative positions of the mutated residues are shown in Figure 3d. As predicted, changing positions 7, 11, and 21 (Mut 7, 11, and 21) all resulted in a significant increase in luciferase activity which is indicative of reduced binding, while changing position 22 does not seem to result in a big difference in eIF4e mediated inhibition (Figure 3e). The only unexpected result was a substitution at position 29, which did not seem to be affect eIF4e inhibition, despite having no aptamers deviate from the consensus. This may be due to the algorithm for clustering starting its alignment from the 5′ end, thus biasing mismatches towards the 5′ end while throwing out sequences that deviated 4 or more nucleotides from the consensus which may contain deviations on the 3′ end. This result demonstrates the first example of sequencing data being used to infer essential residues for function and hints at the possibility of designing further mutagenesis of nonessential residues to improve function.
eIF4e aptamer result in dramatic loss of cell viability in HeLa and HEK293 cells
Since eIF4e knockdown reduces cancer cell proliferation,23 it is possible that eIF4e inhibition with aptamers can affect cell proliferation. Thus, seven aptamers were transfected at 100 nmol/l with lipofectamine 2000 in HEK293 cells and viability measured after 2 days using a redox-based cell viability assay (Figure 4a). As controls, a hairpin-forming single stranded DNA was used (NS seq). Aptamer 6 showed the strongest inhibition, dramatically reducing cell viability to ~3% of negative controls. Furthermore, this inhibition is concentration-dependent (Figure 4b) and is also effective in HeLa cells (Figure 4c and Supplementary Figure S5). This is similar to the decrease in proliferation observed when using shRNAs against eIF4e (Figure 4d).
Figure 4.
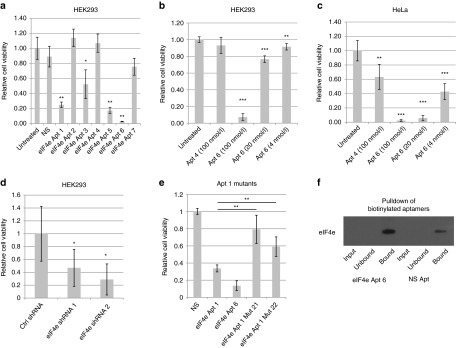
eIF4e aptamers inhibit cell proliferation in HeLa and HEK293 cells. HeLa and HEK293 cells were transfected with varying concentrations of aptamers using Lipofectamine 2000 and 2 days after transfection, cell viability was measured with Cell Titer Blue for 3 hours. (a) HEK293 cells transfected with 100 nmol/l of each aptamer. NS is an oligonucleotide that can form hairpin structures. (b) HEK293 and (c) HeLa cells transfected with varying amount of aptamers. (d) HEK293 cells transfected with shRNAs against eIF4e or a control shRNA against DMPK TA cloned into pCR2.1. (e) HEK293 cells transfected with 100 nmol/l of each aptamer. (f) HEK293 cells were transfected with 100 nmol/l of biotinylated Apt 6 or biotinylated NS. Six hours after transfection, cells were fixed with paraformaldehyde and lysed. Cell lysates were then incubated with Streptavidin Dynabeads. Western blots of the input, unbound, and bound fractions were probed with an eIF4e antibody. **P < 0.01; ***P < 0.001; two-tailed Student's t-test compared to untreated cells. n = 5 for experiments a–e.
To demonstrate that the inhibition observed was through eIF4e, mutants of aptamer 1 previously identified via sequencing were compared to aptamer 1 for their ability to affect cellular proliferation (Figure 4e). Mutation in position 21, which reduced aptamer 1–mediated translational inhibition in insect cell lysates, almost fully negated aptamer 1's function, which suggests that the specific base is crucial to the binding of aptamer to eIF4e. Surprisingly, the mutation at position 22, which did not affect translation in insect cell lysates, also significantly reduced aptamer 1's function albeit not as strongly as mutation at position 21. This may discrepancy may result from the structural differences in eIF4e between insect and mammalian eIF4e that renders the aptamer particular sensitive to a specific mutation. Since the mutations that rendered weaker eIF4e binding, as predicted by sequencing data, also rendered the aptamer ineffective in cell-based proliferation assay, it is likely that the cell proliferation inhibition effect seen with the aptamer is due to eIF4e inhibition. Further verification of specificity can be found in a pulldown assay with lysates derived from cells transfected with a biotinylated aptamer 6 or a control hairpin (Figure 4f). Upon paraformaldehyde fixation, pulldown of the biotinylated aptamer with streptavidin beads also resulted in eIF4e pulldown as determined by Western blot. Although the control nonspecific aptamer could also coprecipitate eIF4e, perhaps due to specific base-pair interaction with mRNAs crosslinked to eIF4e with paraformaldehyde, it is clear that eIF4e aptamer 6 resulted in a much higher amounts of eIF4e than the control aptamer. These results suggest that the action of these aptamers are through eIF4e inhibition.
Ligation enhances inhibitory efficacy of eIF4e aptamers
Bifunctional aptamers, which contain two aptamers targeted to different epitopes on thrombin, have been shown to enhance the activity of inhibitory aptamers.24 Concatamerization of linear aptamers also appear to increase the affinity of HER2 DNA aptamers to HER2.25 As the eIF4e aptamers were designed as loops with blunt ends, they are amenable to ligation. Phosphorylation of aptamers does not seem to alter its ability to inhibit translation at 4 µmol/l concentration (Figure 5a versus 3a). Subsequent ligation of these aptamers resulted in either homodimerization or heterodimerization as demonstrated by a shift in migration in a polyacrylamide gel (Supplementary Figure S6) and both types of dimerization increased the efficacy of the aptamers at inhibiting translation and the effect is not affected by denaturation and refolding of the ligated circular aptamer (Figure 5a). Surprisingly, the same effect could not be seen with eIF4e inhibition in vitro, which in fact seem to reduced the efficacy of the aptamers (Figure 5b).
Figure 5.
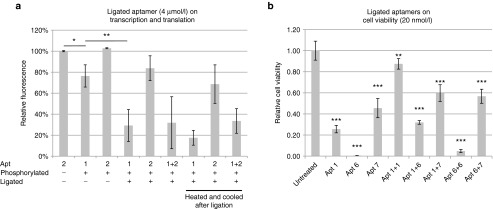
Ligation of aptamers enhances activity. (a) Aptamers phosphorylated with T4 DNA ligase and ligated at 4 µmol/l applied to the insect cell TnT kit and luciferase levels measured 4 hours after application. Aptamers were heated and cooled before ligation, two samples were heated to 85 °C after ligation and cooled again instead of directly being applied to the TnT mix. (b) HeLa cells were transfected with aptamers ligated or aptamers alone at 20 nmol/l with lipofectamine 2000 and measured 2 days after transfection with Cell Titer Blue.
Discussion
DNA aptamers serve a different niche when compared to antibodies in the therapeutic and diagnostic space, with key advantages being the ease of synthesis and modification and the ability to be delivered into the cells at relatively high efficiency with current transfection and gene delivery technologies. This is counterbalanced by the lack of chemical diversity on the backbone of the aptamers, which limits the types of interactions possible with ligands and consequently results in lower affinity as compared to antibodies. However, the development of modified26 or unnatural27 bases in aptamer libraries has partially ameliorated the problem and it is likely that aptamers can make up for weaker contact interactions with favorable secondary structures that fit better. One of the key challenges towards greater adoption of aptamer technology is the development of a rapid selection protocol to build a database of aptamers against diverse ligands to facilitate technology uptake. To this end, we have developed a DNA selection protocol which does not require extensive cleanup between selections and allows researchers to perform up to four cycles of selection a day.
The other limiting factor, prohibitively high costs of characterizing libraries obtained from a screen, has recently been solved with the graduation from Sanger sequencing to high throughput sequencing with the associated huge dataset. As discussed in the introduction, several groups, including ours, have already managed to utilize high throughput data to identify sequence motifs for binding. However, no study has thus far used deviation from a consensus as a mapping tool for essential bases because few studies utilize high fidelity enzymes in library amplification, which confounds inference that can be drawn as deviation from consensus could be just a PCR fidelity issue. Another inference that may be drawn from the high throughput sequencing dataset is the binding characteristics of the aptamer to its target. We noticed that selection in our study happened in waves, with the first wave dominating when the number of washes was small and the second wave when the number of washes increased. Based on this, it may be possible to classify the first group of aptamers as having low dissociation constants and strong binding and the second waves as having low off-rates and weaker binding. Future kinetic studies may be able to substantiate this hypothesis and shed light on the nature of the sequences. Knowing the relative dissociation constants and off-rates can aid in designing conjugates for targeting or inhibition.
The aptamers discovered are also interesting because there is no sequence or structural similarity to an eIF4e-binding inhibitory RNA aptamer.21 Moreover, no obvious consensus motif could be obtained from alignment of all clusters. The diversity of secondary structures of the clusters predicted by MFold (Supplementary Figure S7) also suggests that the clusters we obtained were not binding to eIF4e in the same fashion, suggesting different epitopes. Furthermore, ligation of aptamer clusters seem to enhance their activity in a test tube, suggesting that they bound to different epitopes and worked in a cooperative fashion to increase binding of the target. Thus, it may be generally applicable to identify multiple aptamers targeting different epitopes, each with moderate dissociation constant, and construct multimers that possess the low dissociation constant that will fulfill clinical needs.
Here, we present a selection protocol that has the following advantages. It is rapid, requiring only two steps between rounds of selection to regenerate and produce functional single stranded DNA aptamers while performing negative selection simultaneously. It also uses high throughput sequencing data to characterize evolution of aptamer populations while producing data that predicts functional importance of specific bases in each aptamer cluster. Due to the design, aptamer sequences selected are amenable to conjugation via ligation, which produces multifunctional aptamers that are functionally better than the monomers. This protocol is thus capable of enhancing the odds of identifying functional specific aptamers. Together with other rapid screening protocols for aptamers, this protocol will hopefully increase the uptake and utilization of aptamer-based reagents by simplifying aptamer selection.
Material and methods
Reagents. All chemical reagents and oligonucleotides unless otherwise stated were obtained from Sigma-Aldrich, Singapore. All antibodies used unless otherwise stated were obtained from Abcam, Hong Kong, China.
Protein expression, purification, and biotinylation. Rossetta pLysS competent bacteria was transformed with the pET11d expression plasmid containing the full-length eIF4e clone. The cells were grown in LB medium at 37 °C to OD600 ~0.6 and eIF4e induced with 1 mmol/l IPTG for 3 hours at 37 °C. Cells were resuspended in 50 mmol/l Tris pH 8.0, 10% sucrose, and sonicated. The sonicated sample was centrifuged for 10 minutes at 17,000g at 4 °C and resuspended in Tris/Triton buffer (50 mmol/l Tris pH 8.0, 2 mmol/l EDTA, 100 mmol/l NaCL, 0.5% Triton X-100). The sample was then centrifuged at 25,000g for 15 minutes at 4 °C and resuspended in Tris/Triton buffer. After recentrifugation, the remaining pellet was solubilized in 6 mol/l guanidinium hydrochloride, 50 mmol/l Hepes-KOH pH 7.6, 5 mmol/l DTT. The protein concentration of the sample was then adjusted to 1 mg/ml. The denatured protein was refolded via a 1/10 dilution into refolding buffer consisting of 20 mmol/l Hepes-KOH, 100 mmol/l KCl, and 1 mmol/l DTT. The refolded protein was concentrated and desalted using a Amersham PD10 column into refolding buffer. The eIF4e protein sample was run over a monoQ column and eluted with a 1 mol/l KCl gradient. eIF4e eluted as a sharp peak at a ~0.3 mol/l KCl. eIF4e protein was then biotinylated with EDC biotinylation kit (ThermoScientific-Pierce) and washed thrice with wash buffer with a 10 KDa MW centrifugal ultrafiltration cartridge (centricon) (EMD-Millipore) to remove unbound biotin. 80 µg of the resultant protein was conjugated to 40 µl of MyOne-Streptavidin Dynabeads (Life Technologies, Singapore) over 2 days at 4 °C on shaker. For eIF4A1 and eIF4B, full length human eIF4A1 and truncated eIF4B (aa 1–327) was cloned into pET28a with an N-terminal His-tag and a biotinylation signal sequence. The constructs were transformed into T7 Express cells (New England Biolabs, Singapore) and induced with 0.3 mmol/l IPTG overnight at 16 °C. The cells were then lysed in lysis buffer (10 mmol/l HEPES pH 8, 500 mmol/l NaCl) by sonication and His-tag proteins eluted with 500 mmol/l imidazole after washes with lysis buffer, 50 mmol/l imidazole and 100 mmol/l imidazole. The pure protein was then quantified by Bradford.
Aptamer selection. The aptamer library (5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-(N)33-AGATCGGAAGAGCTC-3′) was synthesized by Integrated DNA Technologies (Coralville, IA) and PAGE purified, exactly the same library as described in Hoon et al. 17 Approximately 6 × 1012 molecules (10 pmoles) out of a theoretical diversity of 7 × 1019 unique sequences was PCR amplified assymetrically with KOD polymerase (EMD-Millipore, Singapore), 100 pmoles of the L primer (5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′) and 10 pmoles of the R primer (5′-(biotin)AGACGTGTGCTCTTCCGATCT-3′) as per manufacturer's instructions to amplify each unique sequence approximately tenfold. PCR annealing was performed at 57 °C for 3 cycles (linear amplification of negative strand) then 65 °C for 18 cycles (linear amplification of aptamer strand). Subsequent amplification of the aptamers retained by each round of selection by asymmetric PCR was performed with a similar protocol with 57 °C for 20 cycles then 65 °C for 15 cycles. The first 20 cycles is to allow for exponential amplification of the retained aptamer and the next 15 cycles for linear amplification of the positive aptamer strand. The resultant amplified library was then passed through 10 µl of MyOne Streptavidin Dynabeads to remove double stranded PCR products and Streptavidin-binding aptamers to produce single stranded nonstreptavidin-binding DNA aptamers. 3 µl of beads conjugated with biotinylated eIF4e was used for each aptamer selection thereafter, decreasing to 2 µl and 1 µl for rounds 7 and 8 respectively. The entire unbound library was used for round 1 selection. 50% of previous round's amplified unbound aptamer library used for the second round, 25% for the third, 25% for the fourth round, 20% for the fifth round, and 10% in subsequent rounds. Three washes in PBST for 5 minutes was done in the first round, 4 washes in the second, 5 washes in the third, 7 washes in the fourth, 10 washes in the fifth, and 12 washes in subsequent rounds. The beads were mixed in 50 µl PCR reactions and amplified as described earlier.
For validation, aptamers were ordered as single stranded DNA oligonucleotides with 10 nucleotide overhangs on either end for the ends to form a stem loop structure (i.e., 5′-CTTCCGATCT-(N)33-AGATCGGAAG-3′).
High throughput sequencing. Aptamer libraries were prepared by amplification of 1 µl of recovered and amplified selection with Primer GX1 (Illumina, Singapore) and reverse primer with a unique barcoded sequence from ScriptSeq RNAseq barcode primers (Epicenter, Madison, WI) using KOD polymerase. Barcoded libraries were pooled at roughly equimolar ratios based on band intensity on SyBr Gold stained polyacrylamide gel and the pooled sample was size selected by PAGE purification (6% TBE PAGE). The pooled size-selected sample was then sequenced on a Genome Analyzer IIx for 36 cycles following manufacturer's protocols. The image analysis and base calling were done using Illumina's GA Pipeline. Adapters were trimmed with Biopieces remove_adapter script and remaining sequences were aligned and clustered according to sequence similarity.
Electrophoretic mobility shift assay. 20 pmoles of each aptamer was heated to 95 °C and cooled in 10 µl of PBS on ice followed by the addition of 0.5 µl protein at the appropriate dilution. The mixture is then incubated for 5 minutes on ice before being loaded into a 10% polyacrylamide gel in Tris-borate buffer and subjected to electrophoresis. The gel is then stained thereafter with 1x SyBr Gold (Life Technologies) before visualization on a UV transilluminator.
Translation assay. The assay was performed with TnT T7 Insect Cell Extract protein expression kit (Promega, Madison, WI) and rabbit reticulocyte TnT protein expression kit (Promega). The kits generate capped mRNA with cap analogs and T7 RNA polymerase, before the insect or rabbit cell components translate the mRNA. 0.2 µl of Luciferase ICE T7 Control DNA (provided in the kit) was mixed with 4 µl of TnT T7 ICE Master Mix and 1 µl of aptamer/protein mix at the appropriate concentrations. The aptamer would have been heated to 95 °C for 5 minutes in distilled water and cooled on ice immediately prior to addition. The reactions are mixed and incubated at 30 °C for 5 hours and 2 µl of sample is used for measurement of luciferase activity after addition of 20 µl of Luciferase Assay Reagent (Promega) in a monochromator plate reader (Tecan Safire 2). To prepare ligated aptamers, aptamers were phosphorylated with T4 polynucleotide kinase (New England Biolabs) at 37 °C for 20 minutes and heated to 95 °C then cooled on ice before ligated using T4 DNA ligase (New England Biolabs) at 10 µmol/l each for 1 hour at room temperature.
Cell culture and transfection. HEK293 and HeLa cells were cultured in DMEM High Glucose (Life Technologies) supplemented with 10% FBS and antibiotics and incubated at 37 °C in 5% CO2. Transfection of cells were carried out with using Lipofectamine 2000 (Life Technologies) as per manufacturer's instruction in 24-well plates or 96-well plates. For shRNAs, eIF4e-specific shRNAs were Taq amplified with the U6 forward primer (5′-gatcgggcccgtcgacaaggtcgggcaggaagagggcct-3′) and reverse primers containing the eIF4e shRNAs containing guide strands previously used in Schwarzer et al.28 (shRNA 1:5′-aaaaataatcacagccaggcattaactcttgat taatgcctggctgtgactacggtgtttcgtcctttccacaa-3′ and shRNA2:5′-aaaaaggttacttgaggccagaagttctcttgaaacttctggcctcaagtgatcggtgtttcgtcctttccacaa-3′). The resultant product was TA cloned into pCR2.1 via TOPO-TA cloning (Life Technologies). The control shRNA is DMPKsh13, which has been previously used in Seow et al. 29 The shRNAs were transfected with Lipofectamine 2000 as per manufacturer's instructions. For the pulldown experiments, biotinylated aptamers were purchased from Sigma-Aldrich as oligonucleotides with a 5′-biotin. Three wells of a 24-well plate was transfected with 100 nmol/l of each aptamer as per manufacturer's instruction. Six hours after transfection, cells were washed thrice with PBS and fixed for 10 minutes with 4% paraformaldehyde in PBS. Cells were then wash thrice again with PBS to remove residual paraformaldehyde and lysed with 300 µl RIPA lysis buffer. 100 µl of the buffer was incubated with 10 µl of Streptavidin-C1 Dynabeads. Unbound fraction was collected (100 µl) and the remaining beads washed five times with PBST and resuspended in 20 µl of RIPA buffer. 10 µl of each sample was then run on a polyacrylamide gel and a Western blot against eIF4e using anti-eIF4e (Abcam, ab1126) primary and antirabbit HRP secondary (Promega) was performed.
Statistics. All experiments, unless otherwise stated, were all performed in triplicates. All error bars used in this report are standard deviations. Statistical significance was determined by two-tailed Student's t-test assuming unequal variance unless otherwise stated.
SUPPLEMENTARY MATERIAL Figure S1. Aptamer native polyacrylamide mobility shift assay. Figure S2. Insect cell transcription and translation with aptamer. Figure S3. Inhibition due to translation not transcription. Figure S4. Rabbit Reticulocyte transcription and translation. Figure S5. Inhibition of HeLa proliferation. Figure S6. Aptamer ligation on polyacrylamide. Figure S7. MFold prediction of structures for eIF4e Apt 2, 6, and 7. Figure S8. Original gel images for Figure 2c,d.
Acknowledgments
The authors thank Sydney Brenner and Sir David Lane for discussions and advice. The research was supported by the Agency of Science, Technology and Research (A*STAR), Singapore (JCO 11/03/EG/07/05). Funding for open access charge: A*STAR. A patent has been filed based on the findings in this study. The authors declare no conflict of interest.
Supplementary Material
References
- Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Huizenga DE, Szostak JW. A DNA aptamer that binds adenosine and ATP. Biochemistry. 1995;34:656–665. doi: 10.1021/bi00002a033. [DOI] [PubMed] [Google Scholar]
- Wen AQ, Yang QW, Li JC, Lv FL, Zhong Q, Chen CY. A novel lipopolysaccharide-antagonizing aptamer protects mice against endotoxemia. Biochem Biophys Res Commun. 2009;382:140–144. doi: 10.1016/j.bbrc.2009.02.152. [DOI] [PubMed] [Google Scholar]
- Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature. 1992;355:564–566. doi: 10.1038/355564a0. [DOI] [PubMed] [Google Scholar]
- Stojanovic MN, de Prada P, Landry DW. Aptamer-based folding fluorescent sensor for cocaine. J Am Chem Soc. 2001;123:4928–4931. doi: 10.1021/ja0038171. [DOI] [PubMed] [Google Scholar]
- Gronewold TM, Glass S, Quandt E, Famulok M. Monitoring complex formation in the blood-coagulation cascade using aptamer-coated SAW sensors. Biosens Bioelectron. 2005;20:2044–2052. doi: 10.1016/j.bios.2004.09.007. [DOI] [PubMed] [Google Scholar]
- McNamara JO, 2nd, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E.et al. (2006Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras Nat Biotechnol 241005–1015. [DOI] [PubMed] [Google Scholar]
- Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moreno AF.et al. (2008Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers Proc Natl Acad Sci USA 1052586–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Girvan AC, Casson LK, Pierce WM, Jr, Qian M, Thomas SD.et al. (2007AS1411 alters the localization of a complex containing protein arginine methyltransferase 5 and nucleolin Cancer Res 6710491–10500. [DOI] [PubMed] [Google Scholar]
- Marimuthu C, Tang TH, Tominaga J, Tan SC, Gopinath SC. Single-stranded DNA (ssDNA) production in DNA aptamer generation. Analyst. 2012;137:1307–1315. doi: 10.1039/c2an15905h. [DOI] [PubMed] [Google Scholar]
- Boiziau C, Dausse E, Yurchenko L, Toulmé JJ. DNA aptamers selected against the HIV-1 trans-activation-responsive RNA element form RNA-DNA kissing complexes. J Biol Chem. 1999;274:12730–12737. doi: 10.1074/jbc.274.18.12730. [DOI] [PubMed] [Google Scholar]
- Cho M, Xiao Y, Nie J, Stewart R, Csordas AT, Oh SS.et al. (2010Quantitative selection of DNA aptamers through microfluidic selection and high-throughput sequencing Proc Natl Acad Sci USA 10715373–15378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupakuwana GV, Crill JE, 2nd, McPike MP, Borer PN. Acyclic identification of aptamers for human alpha-thrombin using over-represented libraries and deep sequencing. PLoS ONE. 2011;6:e19395. doi: 10.1371/journal.pone.0019395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütze T, Wilhelm B, Greiner N, Braun H, Peter F, Mörl M.et al. (2011Probing the SELEX process with next-generation sequencing PLoS ONE 6e29604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditzler MA, Lange MJ, Bose D, Bottoms CA, Virkler KF, Sawyer AW.et al. (2013High-throughput sequence analysis reveals structural diversity and improved potency among RNA inhibitors of HIV reverse transcriptase Nucleic Acids Res 411873–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoon S, Zhou B, Janda KD, Brenner S, Scolnick J. Aptamer selection by high-throughput sequencing and informatic analysis. BioTechniques. 2011;51:413–416. doi: 10.2144/000113786. [DOI] [PubMed] [Google Scholar]
- Montanaro L, Pandolfi PP. Initiation of mRNA translation in oncogenesis: the role of eIF4E. Cell Cycle. 2004;3:1387–1389. doi: 10.4161/cc.3.11.1251. [DOI] [PubMed] [Google Scholar]
- Graff JR, Zimmer SG. Translational control and metastatic progression: enhanced activity of the mRNA cap-binding protein eIF-4E selectively enhances translation of metastasis-related mRNAs. Clin Exp Metastasis. 2003;20:265–273. doi: 10.1023/a:1022943419011. [DOI] [PubMed] [Google Scholar]
- Blagden SP, Willis AE. The biological and therapeutic relevance of mRNA translation in cancer. Nat Rev Clin Oncol. 2011;8:280–291. doi: 10.1038/nrclinonc.2011.16. [DOI] [PubMed] [Google Scholar]
- Mochizuki K, Oguro A, Ohtsu T, Sonenberg N, Nakamura Y. High affinity RNA for mammalian initiation factor 4E interferes with mRNA-cap binding and inhibits translation. RNA. 2005;11:77–89. doi: 10.1261/rna.7108205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni A, Akcakanat A, Singh G, Luyimbazi D, Zheng Y, Kim D.et al. (2008eIF4E knockdown decreases breast cancer cell growth without activating Akt signaling Mol Cancer Ther 71782–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giusto DA, King GC. Construction, stability, and activity of multivalent circular anticoagulant aptamers. J Biol Chem. 2004;279:46483–46489. doi: 10.1074/jbc.M408037200. [DOI] [PubMed] [Google Scholar]
- Mahlknecht G, Maron R, Mancini M, Schechter B, Sela M, Yarden Y. Aptamer to ErbB-2/HER2 enhances degradation of the target and inhibits tumorigenic growth. Proc Natl Acad Sci USA. 2013;110:8170–8175. doi: 10.1073/pnas.1302594110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham JA, Johnson R, Toole JJ. The application of a modified nucleotide in aptamer selection: novel thrombin aptamers containing 5-(1-pentynyl)-2'-deoxyuridine. Nucleic Acids Res. 1994;22:2817–2822. doi: 10.1093/nar/22.14.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimoto M, Yamashige R, Matsunaga K, Yokoyama S, Hirao I. Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat Biotechnol. 2013;31:453–457. doi: 10.1038/nbt.2556. [DOI] [PubMed] [Google Scholar]
- Schwarzer A, Holtmann H, Brugman M, Meyer J, Schauerte C, Zuber J.et al. (2014Hyperactivation of mTORC1 and mTORC2 by multiple oncogenic events causes addiction to eIF4E-dependent mRNA translation in T-cell leukemia Oncogene (epub ahead of print) [DOI] [PubMed]
- Seow Y, Sibley CR, Wood MJ. Artificial mirtron-mediated gene knockdown: functional DMPK silencing in mammalian cells. RNA. 2012;18:1328–1337. doi: 10.1261/rna.030601.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.