

Abstract
Enterotoxigenic Escherichia coli (ETEC) is a significant cause of morbidity and mortality in the developing world. ETEC-mediated diarrhea is orchestrated by heat-labile toxin (LT) and heat-stable toxins (STp and STh), acting in concert with a repertoire of more than 25 colonization factors (CFs). LT, the major virulence factor, induces fluid secretion after delivery of a monomeric ADP-ribosylase (LTA) and its pentameric carrier B subunit (LTB). A study of ETEC isolates from humans in Brazil reported the existence of natural LT variants. In the present study, analysis of predicted amino acid sequences showed that the LT amino acid polymorphisms are associated with a geographically and temporally diverse set of 192 clinical ETEC strains and identified 12 novel LT variants. Twenty distinct LT amino acid variants were observed in the globally distributed strains, and phylogenetic analysis showed these to be associated with different CF profiles. Notably, the most prevalent LT1 allele variants were correlated with major ETEC lineages expressing CS1 + CS3 or CS2 + CS3, and the most prevalent LT2 allele variants were correlated with major ETEC lineages expressing CS5 + CS6 or CFA/I. LTB allele variants generally exhibited more-stringent amino acid sequence conservation (2 substitutions identified) than LTA allele variants (22 substitutions identified). The functional impact of LT1 and LT2 polymorphisms on virulence was investigated by measuring total-toxin production, secretion, and stability using GM1–enzyme-linked immunosorbent assays (GM1-ELISA) and in silico protein modeling. Our data show that LT2 strains produce 5-fold more toxin than LT1 strains (P < 0.001), which may suggest greater virulence potential for this genetic variant. Our data suggest that functionally distinct LT-CF variants with increased fitness have persisted during the evolution of ETEC and have spread globally.
INTRODUCTION
Infectious diarrheal disease caused by enterotoxigenic Escherichia coli (ETEC) accounts for hundreds of millions of cases each year, mainly in developing countries (1). ETEC strains isolated from humans are capable of colonizing the small intestine through the expression of several colonization factors (CFs) (2). They also secrete two classes of plasmid-encoded enterotoxins, i.e., heat-labile toxin (LT; also termed LT-I) and heat-stable toxin (STh or STp) (1). LT is a member of the AB5 toxin family and is similar to cholera toxin secreted by Vibrio cholerae; these toxins share structural homology and a mechanism of action (3). As with all toxins of the AB5 family, the structure of LT consists of a pentameric ring of receptor-binding B subunits and a single catalytic A subunit. The subunits are encoded by the plasmid-borne genes eltA and eltB and are transcribed as an operon (4). The enzymatically active A subunit consists of a large A1 domain and a short A2 domain. The A1 domain harbors the catalytic function via ADP-ribosylation of stimulatory G proteins, resulting in activation of adenylate cyclase and elevated intracellular cyclic AMP (cAMP) levels (3, 5). The B subunits bind mainly to GM1 ganglioside, but other receptors on intestinal cells have also been identified (6, 7).
LT secretion is initiated by cleavage of the N-terminal signal peptides of subunits A and B followed by sec-dependent transport across the inner membrane to the periplasm (6, 8). In the periplasm, monomers assemble spontaneously or by DsbA disulfide oxidoreductase activity and are then secreted by the general (type II) secretion pathway (GSP) in a pH-dependent manner (9–11). Under classical laboratory culture conditions, individual ETEC isolates differ in their abilities to secrete LT into the medium. Some strains retain LT predominantly in the periplasm or associated with lipopolysaccharide (LPS) in the outer membrane, while other strains secrete as much as 50% of the LT produced into the medium (3, 7, 11, 12). When ETEC attaches to surface intestinal epithelial cells, the mature holotoxin is transferred to the host cell, where it can undergo posttranslational modifications leading to full activation. During this process, the C-terminal A1 domain is released from the A2 domain by proteolytic cleavage, leaving the smaller A2 fragment associated with the B subunit, which is involved in GM1 binding on host cells (6, 13, 14). Subsequently, adenylate cyclase is activated by the A1 domain through ADP-ribosylation of the stimulatory guanine-nucleotide-binding G protein α subunit (Gsα), which leads to increased production of cAMP and deregulation of the cystic fibrosis transmembrane receptor (CFTR) ion channel, resulting in hypersecretion of electrolytes and water into the intestinal lumen, i.e., diarrhea (8).
Several studies of LT-producing ETEC strains—based on genetic, biochemical, and immunological characterization—have shown that LT is a heterogeneous family (6, 8, 15). Two families have been described: LT-I (including the human ETEC reference strain H10407) and the novel family LT-II. The LT-I expressed by ETEC strains isolated from human samples is highly similar to cholera toxin in terms of amino acid sequence, showing 80% sequence homology (6). LT-II (LT-IIa, LT-IIb, and LT-IIc) purified from buffalo stool samples is antigenically distinct from LT-I or cholera toxin (16). Subsequent sequencing analysis has validated such differences, showing high amino acid sequence divergence mainly in the LT-II mature B subunit, which shares only 15 to 16% identity with LT-I and cholera toxin (17). A previous study analyzed the DNA sequences of ETEC LT-I strains isolated from humans in Brazil; 16 LT-I types were identified and were termed LT1 to LT16 (15). These data revealed high levels of polymorphism, mainly in eltA. Since Lasaro et al. analyzed mainly Brazilian strains (15), we were interested in understanding the worldwide distribution of polymorphisms present in the eltAB operon among a geographically and temporally diverse set of clinical ETEC isolates, some of which belong to globally distributed persistent lineages (18). We analyzed the LT-I operons of 192 human ETEC strains isolated from several continents, including Asia, Africa, and Latin America, over 3 decades, both strains belonging to stable lineages and individual isolates with different colonization factor and toxin profiles, in order to evaluate the natural diversity of LT.
MATERIALS AND METHODS
Bacterial strains.
A representative collection of 362 ETEC strains from the University of Gothenburg strain collection (comprising more than 3,500 ETEC strains) were subjected to whole-genome sequencing at the Wellcome Trust Sanger Institute (18); of these, 186 strains were positive for LT and were included in this study. The LT-ETEC strains were collected between 1980 and 2011 from 21 different countries. Strains were isolated from a diverse demographic, including patients younger than the age of 5 years, adults, and travelers and soldiers with acute diarrheal disease; some strains (n = 7) were also isolated from asymptomatic individuals. Six additional LT-expressing strains isolated in cases of diarrhea in Bolivia from 2002 to 2011 were also included in this study. All strains were from anonymous patients and were isolated from stool with informed consent. Permission to use the ETEC strain collection was granted by the Regional Ethical Board of Gothenburg, Sweden (Ethics Committee reference no. 088-10). Strains were characterized as ETEC by the expression of LT and/or ST as determined by GM1–enzyme-linked immunosorbent assays (GM1-ELISA) and inhibition ELISA, respectively, as well as by multiplex PCR. A dot blot assay was used for characterization of CFA/I, CS1 to CS8, CS12, CS14, CS17, CS19, and CS21 (19). BLASTn analysis was used to confirm the presence of CF operons and toxin genes within the genome of each ETEC isolate.
Genomic sequencing and extraction of the eltAB gene.
ETEC strains were grown on horse blood agar plates overnight at 37°C. DNA was isolated from each strain in accordance with the instructions in the Wizard Genomic DNA kit (Promega). The genomic library preparation and DNA sequencing have been described by von Mentzer et al. (18), and genomic extraction of the eltAB gene was performed by nBLAST in this study. GenBank accession number S60731 was used for the eltAB genomic extraction.
LT variant identification and phylogenetic analysis.
Multisequence alignment of 192 amino acid sequences translated from eltAB was performed using ClustalW. A concatenated sequence was constructed for phylogenetic analysis by subtracting the sequences corresponding to the signal peptides of the LTA and LTB subunits. The MEGA program (version 5.2) was used to extract the variables in the translated amino acid sequence of each strain. Sequences were compared to LT variants reported in previous studies: LT1 (15), LT2 (20), and LT3 to LT16 (GenBank accession numbers EU113242 [LT3], EU113243 [LT4], EU113244 [LT5], EU113245 [LT6], EU113246 [LT7], EU113247 [LT8], EU113248 [LT9], EU113249 [LT10], EU113250 [LT11], EU113251 [LT12], EU113252 [LT13], EU113253 [LT14], EU113254 [LT15], and EU113255 [LT16]) (15). Phylogenetic trees were generated in MEGA (version 5.2) using the neighbor-joining algorithm.
GM1-ELISA.
A single-read GM1-ELISA for phenotypic demonstration and quantification of LT produced by a subset of 155 ETEC strains included in the study was adapted from the work of Svennerholm and Wiklund (21) with the following modifications. Briefly, 1 ml of culture was collected from a 5-h culture of an ETEC strain in Luria broth; cells were sonicated in phosphate-buffered saline (PBS) and were then transferred to GM1-coated ELISA plates in triplicate without dilutions. The plates were developed by following the protocol described in reference 21. All samples were tested in triplicate, and mean optical density (OD) values for the detection of LT in each strain were used to extrapolate the amount of LT produced. A subset of 31 strains (16 strains expressing LT1 and 15 strains expressing LT2) was selected for quantitative ELISA, which was performed using a 3-fold dilution series and recombinant cholera toxin subunit B (rCTB) as a reference as described previously (21). In addition, a test to determine the amounts of free B subunits relative to the amounts of the AB holotoxin in the culture was set up. Bacterial extracts from sonicated pellets and supernatant fractions were transferred to two GM1-coated microtiter plates. We used an in-house monoclonal antibody (MAb) for the LTB and CTB subunit (LT39:1:1) in the first plate (19) and another in-house monoclonal antibody specific for the LTA subunit (LT17A) in the second plate. Antibody LT39:1:1 detected both the free B subunit and fully assembled LT holotoxin, while antibody LT17A detected only assembled holotoxin. Quantitative ELISA using 3-fold dilutions was performed as described above. Assays were performed in duplicate, and the ratio of AB5 holotoxin to the total amount of B5 was determined by measurement of the amounts of product obtained in the ELISA targeting anti-CTA and anti-CTB. We assumed that anti-CTA detects the AB5 holotoxin bound through the B5 subunit-mediated binding to the GM1-coated plates, while anti-CTB detects both holotoxin and dissociated B5 subunits, i.e., the total amount of B5 formed.
LT1 and LT2 (AB5) modeling.
The predicted stability of the LT1 holotoxin was compared to that of the LT2 holotoxin using in silico modeling. The region of S190 on LTA was flexible; therefore, the coordinates for that region were missing in all available crystal structures. High-quality homology models of the LT1A-LT1B and LT2A-LT2B pentamers were generated using the MPACK software package (22). The underlying template for the models was a refined (1.95-Å) E. coli crystal structure of LTp (1LTS); the LT2 holotoxin was modeled onto the template and was then compared with LT1. The quality of the models was very high, since the sequence identity with the template was 97%. The LT1 and LT2 (AB5) models were individually soaked in a TIP3 water box. After initial energy minimizations, 2-ns molecular dynamics (MD) simulations were performed with the AMBER software package (23). The last conformation of the 2-ns MD simulation trajectories was used for further analysis and visualization. The LT2A part of this conformation was used to perform a potential protein-protein interface prediction with InterProSurf (24). The interactions of position 75 on LTB in both the LT1 and LT2 models were analyzed separately.
Statistical analysis.
The different LT alleles determined by single-read ELISA were analyzed using one-way analysis of variance (ANOVA). Nonpaired Mann-Whitney U tests were applied to the rest of the data. All tests were performed using GraphPad Prism, version 6.00 for Windows (GraphPad Software, La Jolla, CA). P values of <0.05 were considered statistically significant.
RESULTS
LT-I is highly diverse and encompasses several natural variants.
ETEC disease is a set of overlapping global epidemics of individual ETEC lineages, which have been stable over substantial periods in areas of endemicity (18). To identify genetic variations in LT-I in ETEC lineages and individual isolates, a 1,152-bp nucleotide sequence encompassing the entire eltAB operon was extracted from whole-genome sequences of 192 ETEC isolates collected from different geographic locations spanning 31 years from 1980 to 2011 (18). A total of 192 eltAB operons were successfully extracted. Toxin characterization showed that 90 (46.9%) ETEC strains expressed LT alone as the major virulence factor and 102 isolates expressed LT in combination with either STh or STp. Colonization factor profiles had been determined previously by dot blot assays or PCR and were verified by BLASTn analysis to confirm the presence of toxin and colonization factor operons in each strain. The most common toxin-colonization factor profiles in the collection were LT/STh CS1 + CS3 + CS21 (n = 17) and LT/STh CS5 + CS6 (n = 17), followed by LT CS6 (n = 11) and LT/ST CS19 (n = 11); these represent 4 lineages of closely related ETEC isolates. Seventy-four of the strains were negative for any previously described colonization factor (Table 1).
TABLE 1.
Virulence gene profiles of ETEC strains included in this studya
CF, colonization factor.
To identify any genetic variability harbored within eltA and eltB, the eltAB operons of the 192 clinical ETEC isolates were compared to the previously described LT1 reference allele (15) by using both the concatenated open reading frame encoding the A and B subunits and translated amino acid sequences excluding the signal peptides in order to compare results described previously (15). In total, 44 single nucleotide polymorphisms (SNPs) and 24 amino acid substitutions were found among the 192 LTAB sequences at the nucleotide and amino acid levels, respectively. More polymorphic sites (37 SNPs) were found in the A subunit than in the B subunit (7 SNPs), representing 22 and 2 amino acid substitutions, respectively, compared to the reference LT1 variant. Our collection included 12 novel variants designated LT17 to LT28 and 8 of 16 previously reported LT variants (15). Positions and individual amino acid substitutions are listed in Table 2. Among the 192 human ETEC strains, LT1 and LT2 were the most common natural variants, representing 40.6% and 25.0% of the sequence library, respectively, followed by LT13 at 6.8% and the novel variant LT18 at 6.3%. In total, all novel LT natural variants accounted for 15.1% (n = 29) of our strain collection. No difference in LT variants was found between isolates from the small number of asymptomatic cases, which encompassed 4 variants, LT1, LT20, LT23, and LT8, and the isolates from diarrheal cases.
TABLE 2.
Frequency and characterization of polymorphisms among natural variants of LT detected among ETEC strains analyzed in this study
| No. | LT variant | Alternative designation | No. (%) of ETEC strains (n = 192) | Amino acid substitution(s) in: |
No. of amino acid replacements |
||
|---|---|---|---|---|---|---|---|
| A subunit | B subunit | A subunit | B subunit | ||||
| 1 | LT1 | 78 (40.6) | 0 | 0 | |||
| 2 | LT2 | 48 (25) | S190L, G196D, K213E, S224T | T75A | 4 | 1 | |
| 3 | LT3 | LT3 + R13H | 6 (3.2) | K213E, R235G | R13H | 2 | 1 |
| 4 | LT7 | 2 (1) | P12S, S190L, G196D, K213E, S224T | T75A | 5 | 1 | |
| 5 | LT8 | 7 (3.6) | T203A, K213E | R13H | 2 | 1 | |
| 6 | LT11 | 7 (3.6) | M37I, T193A, K213E, I232 M | 4 | 0 | ||
| 7 | LT12 | LT12 + R18H | 2 (1) | R18H, M37I | 2 | 0 | |
| 8 | LT13 | 13 (6.8) | R18H, M23I | 2 | 0 | ||
| 9 | LT17 | 4 (2.1) | H27N | 1 | 0 | ||
| 10 | LT18 | 12 (6.3) | G196D | 1 | 0 | ||
| 11 | LT19 | 1 (0.5) | S216T | 1 | 0 | ||
| 12 | LT20 | 3 (1.6) | D170N | 1 | 0 | ||
| 13 | LT21 | 1 (0.5) | H27Y | 1 | 0 | ||
| 14 | LT22 | 1 (0.5) | S190L, T193A, G196D, K213E, S224T | T75A | 5 | 1 | |
| 15 | LT23 | 1 (0.5) | I236V | 1 | 0 | ||
| 16 | LT24 | 2 (1) | V103I | 1 | 0 | ||
| 17 | LT25 | 1 (0.5) | P12S | 1 | 0 | ||
| 18 | LT26 | 1 (0.5) | S228L | 1 | 0 | ||
| 19 | LT27 | 1 (0.5) | P12S, E229V | 2 | 0 | ||
| 20 | LT28 | 1 (0.5) | R237Q | 1 | 0 | ||
Eight of the previously reported natural human isolate variants (LT4, LT5, LT6, LT9, LT10, LT14, LT15, and LT16) were not identified. To further verify our results, all LT sequences reported (15) were downloaded from GenBank, and sequences were translated. Some minor differences were discovered; hence, we assigned alternative names to LT3 and LT12, including one additional amino acid substitution in the LT3 sequence at position 13 (R to H) in the B subunit and one in the LT12 sequence at position 18 (R to H) in the A subunit (Table 2). Furthermore, the nucleotide sequence of LT15 in our analysis was translated to an amino acid sequence identical to that of LT2 in the mature A and B subunits.
To assess the genetic relatedness of the LT-I natural variants, a phylogenetic tree was generated (Fig. 1). As reported previously, the LT variants fell into four phylogenetic groups termed groups I to IV (15). To determine the relatedness of both novel and previously described variants, we used amino acid sequences of the 12 novel natural LT variants identified in this study and the translated sequences derived from GenBank. Figure 1 shows that although the LT-I variants fell into 4 major groups, confirming the previous analysis, LT11 branched off from group III, forming a fifth group (group V). Group I included the previously reported LT variants LT1, LT9, LT10, LT12, and LT13 and a majority of the new LT variants (LT17, LT18, LT19, LT20, LT21, LT23, LT24, LT25, LT26, LT27, and LT28). Hence, group I is more diverse than other groups in the current collection and is characterized by several amino acid substitutions along the sequence of the A subunit, compared with the reference sequence (LT1). Group II consisted of previously reported variants LT2, LT7, LT14, LT15, and LT16 and the novel variant LT22. LT2 and LT15 are identical in the mature A and B subunits and are termed LT2 below. The novel allele LT22 differs from LT2 in one additional amino acid substitution at T193A in the A subunit. LT variants belonging to group II therefore encompass several alterations in the amino acid sequences of both the A and B subunits from LT1. Group III comprised the previously reported LT variants LT3, LT5, and LT8, where LT3 and LT8 variants were also identified among the CF-negative strains. In addition, ETEC expressing LT CS1 and LT CS8 only—which are rare combinations—were identified as LT8. The group IV variants found by Lasaro et al. included LT4 and LT6, which were not found in our study. LT4 is identical to porcine LT (LTp) and displays 3 additional amino acid changes in the B subunit from that of LT1 (15, 25). The LT4 variant is commonly found in porcine ETEC strains, and it is thus not surprising that we did not find it in our collection of strains from clinical isolates. Finally, the new group V included only the LT11 variant.
FIG 1.

Phylogenetic analysis of the LT variants. An unrooted phylogenetic tree was used to determine the phylogenetic relatedness of LT variants, including the LT variants reported previously (LT1 to LT16) (15) and the new LT variants found in this study (LT17 to LT28). The tree was constructed by the neighbor-joining method using MEGA, version 5.2.
A majority of LT-ETEC strains that express known colonization factors belong to the two major LT variants LT1 and LT2, which have spread globally.
Since the ETEC isolates in our study were collected over more than 3 decades from remote regions across the world, we were interested in determining if LT variants have evolved over time or show geographic clustering. Therefore, a phylogenetic tree was constructed based on the concatenated LTA and LTB peptides, and metadata were mapped back onto the tree. The overall result of the phylogenetic analysis revealed three distinct clusters, which were designated A, B, and C (Fig. 2). The topology of the tree shows that cluster A contained closely related LT variants belonging to group I. Cluster B included LT variants of groups III, IV, and V, which showed a distant branching, while cluster C included LT variants of group II. Interestingly, no clear relation was found with the country or year of isolation. However, the clusters shared distinct CF profiles. Cluster A is composed of two subclusters, designated A1 and A2. A1 harbored the majority of the isolates, whereas subcluster A2 contained 12 LT18 isolate with CS12 or CS6 + CS21. Cluster A1 harbored strains with diverse CF profiles, including CS1 + CS3 (+ CS21), CS2 + CS3 (+ CS21), CS2 + CS21, CS3 + CS21, CS4 + CS6, CS6 + CS8, CS6 + CS21, CS7, CS17, CS19, and CS21 as well as CF-negative strains. Some of these strains belonged to major lineages of ETEC. Most of these cluster A strains in subclusters A1 and A2 had the LT1 allele, while a minority belonged to LT12, LT13, and LT17 to LT28. Single amino acid substitution variants of LT1, representing novel LT variants, were found mainly in single CF-negative ETEC isolates of cluster A (Fig. 2). Cluster A strains were isolated over 30 years from the Americas, Africa, and Asia. Hence, the LT1 variant of LT is a conserved variant that has persisted in several linages, with different CF profiles that have spread globally over time. Cluster B habored LT3, LT8, and LT11; the first two variants were found in CS1-, CS8-, and CS12-positive isolates, while LT11 was found only in CF-negative strains. The 19 ETEC strains of cluster B were isolated from the Americas and Asia during the period 1983 to 2009. Cluster C harbored lineages including CS5 + CS6-, CS14-, CFA/I + CS21-, CS21-, and CS23-positive isolates, as well as CF-negative strains with the majority expressing LT2 (except for 2 CF-negative isolates that expressed LT7 and LT22). Strains in cluster C were isolated from the Americas, Africa, and Asia over a period of 31 years, suggesting that LT2 has spread globally.
FIG 2.

Phylogenetic analysis of ETEC strains based on LT sequences. A total of 192 LT sequences of 192 human ETEC strains and 16 sequences of LT variants reported previously (15) were used in this analysis. The tree was based on the deduced amino acid sequence of the concatenated LT gene using the neighbor-joining algorithm as implemented in the MEGA program, version 5.2. Branches are colored according to the cluster pattern: red, cluster A; green, cluster B; blue, cluster C. Each strain designation is followed by the toxin profile, CF profile, and year of isolation. Bootstrap values greater than 20% are presented at the nodes of the neighbor-joining tree, indicating the confidence for the clade grouping.
Distribution of polymorphic sites along the LT protein.
The B subunit was much more conserved (only 2 amino acid substitutions) than the A subunit, which exhibited 22 amino acid changes. The A2 domain was slightly more diverse (13 amino acid substitutions) than the A1 domain (9 amino acid changes). Most of the amino acid substitutions in A1 were located between positions 12 and 37 (5 amino acid changes) and between positions 103 and 190 (4 amino acid changes), involving different structural folds within the protein, including an α-helix and β-sheets. Perhaps not surprisingly, no polymorphisms were found in the A1 subunit loop comprising residues 47 to 56, which covers the active site. These residues were also found to be under purifying selection, indicating that they are conserved (see Fig. S1 in the supplemental material). The 13 polymorphic sites of the A2 domain were distributed along the α-helix, which interacts with the B subunit; residues under positive selection were identified, but these changes were not significant (see Fig. S1 in the supplemental material). The R13H and T75A amino acid changes found in the B subunit were located in structures that form a turn and α-helix, respectively.
To analyze the potential impact of the amino acid substitutions, we modeled the LT1AB5 and LT2AB5 (Fig. 3a) complexes based on the crystal structure 1LTS. The model complexes were refined during a 2-ns MD simulation in an explicit water box. During the 2-ns simulation, the LTB domain pentamers were compact and stable (Fig. 3b). At the same time, the LTA domains started to change their positions relative to the LTB pentamers. This flexibility was expected, since the A domains were anchored to the LTB pentamers only via the C terminus of the A domain. Here S or T at position 224 (on LT1 or LT2, respectively) contacted and anchored the A domain to only one monomer (Fig. 3c and d). On the other hand, position S228, further down the pentamer cavity, contacted several changing monomers. Residue K or E at position 213 on the A domain was solvent exposed and was not near the LTB pentamer. It did not contribute to AB5 complex stabilization. On the LTB pentamer, residue T or A at position 75 did not contribute to complex stability either, since it contacted only neighboring residues on the same monomer. In the case of LT2, this residue contacted only neighboring backbone atoms on the helix. Most probably, the T75A variant is neutral and has no structural or functional effects on LTB. Using the LT2A model, we predicted potential protein-protein interface residues (Fig. 3). These potential interface patches are shown as brown surface patches in Fig. 3a. Interestingly, variable positions L190, D196, E213, and T224 were part of, or very close to, potential interface regions. The contact partner around T224 is obviously the LTB pentamer. However, the nature of the other interfaces is not clear at present.
FIG 3.

Structural analysis of the LT1 and LT2 variants. (a) The model of LT2 (AB5) is shown as a ribbon diagram, with select residues and regions represented by spheres and surface patches, respectively. The model was generated using the crystal structure 1LTS as the template. The last conformation of a 2-ns MD simulation of the model is shown. The A and B subunits are represented by light blue and gray ribbons. Red spheres represent the A75 atoms on LT2B, and blue spheres represent the atoms of L190, D196, E213, and T224. Brown patches represent LT2A surface-exposed portions of residues that are predicted to be in protein-protein interface regions (Tyr24, Ser28, His45, Phe49, Asp50, Arg51, Gly52, Thr53, Gln54, Met55, Asn56, Gly69, Val71, Ser81, Leu82, Ser83, Leu84, Arg85, Ser86, His88, Leu89, Ala90, Gln92, Ser93, Ile94, Ser96, Gly97, Tyr98, Ser99, Thr100, Tyr102, Asn114, Val115, Asn116, Asp117, Val121, Tyr122, Ser123, Pro124, His125, Pro126, Tyr127, Glu128, Gln129, Glu130, Trp145, Tyr146, Arg147, Asn149, Phe150, Gly151, Val152, Ile153, Asp154, Glu155, Arg156, Leu157, His158, Ile173, Pro175, Ala176, Glu177, Asp178, Tyr180, Arg181, Arg193, Glu194, Glu195, Pro196, Trp197, Ile198, His199, His200, Ala201, Asn207, Leu208, Ser209, Asn223, Leu224, Ile227, Tyr228, Arg230, Glu231, Lys237, Arg238, Ile240, Phe241, Tyr244, Gln245, Tyr251, Asn252, Arg255, Glu257, and Leu258). (b) Structural fit of the LT1B (green ribbon) and LT2B (gray ribbon) pentamer models. The T or A residue at position 75 on the LTB subunit is indicated by red spheres. (c) LT1A contacts LT1B at position S224 at the inner top rim of the pentamer (blue spheres and bonds). Residue T75 (red spheres) on the LTB subunit makes only intramonomer contacts. Atoms in contact range (d < 3.5 Å) are shown in yellow spheres, and the side chain bonds of these residues are also shown in yellow. Residue K213 is solvent exposed. (d) Same as panel c but only for LT2.
LT2-expressing strains produce significantly more LT than strains that express LT1.
The amino acid sequence differences in the various LT variants could have an impact on the stability and/or folding of the toxin itself and could therefore impair production and secretion (6). To examine this, we performed a single-read ELISA to assess total LT assembly by ETEC strains expressing different variants. A total of 155 ETEC strains were included in this analysis, representing 80.7% of the strains used in this study. As a preliminary test, bacterial cell lysates were analyzed by GM1-ELISA, and OD450 (optical density at 450 nm) values were normalized to bacterial numbers (an OD600 of 0.8 corresponds to 109 bacteria). Strains were categorized as high, medium, or low LT producers. The amounts of LT produced were high for LT2- and LT21-expressing strains (OD450, >0.5), medium for LT11 and LT13 (OD450, 0.5 to 0.25), and low for LT1 and LT18 (OD450, <0.25) (Fig. 4).
FIG 4.

Differences in the amount of LT among strains expressing different LT variants as determined using GM1-ELISA. A total of 155 ETEC strains expressing 16 of 20 LT variants were tested for LT production. The number of strains expressing the respective LT variant is given above each box plot. The OD values came from the one-read ELISA. Results shown are averages of the OD values of triplicate samples. ODs of 0 to 0.25, 0.25 to 0.5, and >0.5 were considered to indicate small, medium, and large amounts of LT produced by ETEC strains, respectively. Statistical analysis was performed using ANOVA with Turkey's multiple-comparison posttest by using Prism, version 6.00. For the statistical analysis, LT22, LT23, LT25, LT26, LT27, and LT28 were excluded due to the low numbers of samples. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
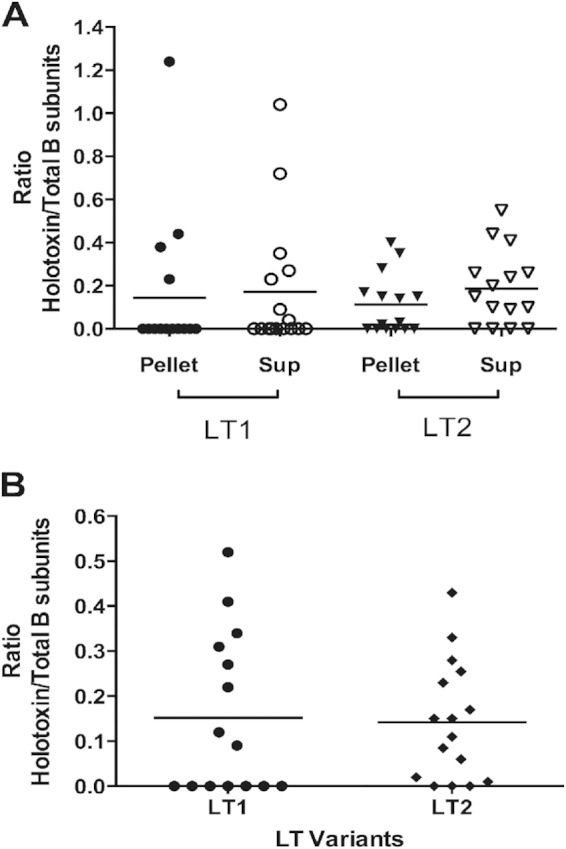
More-detailed analyses of LT production and secretion by LT1 and LT2 strains were performed using quantitative GM1-ELISA. These analyses revealed that LT2 strains produced 5-fold more LT than LT1 strains (30.77 ng/ml versus 6.53 ng/ml) (P < 0.001). Similar results were obtained using the pellet and supernatant fractions (Fig. 5A and B). In the pellet fraction, LT2 ETEC produced 9-fold more LT than LT1 strains (P < 0.001), and in the supernatant fraction, LT2 ETEC produced 3-fold more LT than LT1 strains (P < 0.05). Next, the ability to secrete LT was analyzed as a percentage of the formed toxin found in the supernatant and was calculated from the toxin in the supernatant divided by total production in both the pellet and the supernatant multiplied by 100. When the secretion percentage was determined, almost equal values were found (50.29% for LT1 and 50.91% for LT2), and no statistical difference was found (Fig. 5C). Thus, secretion rates are similar for strains expressing LT2 and LT1.
FIG 5.

Production and secretion of LT by ETEC strains expressing LT1 and LT2 variants as determined by quantitative GM1-ELISA. (A) Total production in LT1 and LT2 strains. (B) Comparison of LT production in LT1 and LT2 strains using the sonicated bacterial (Pellet) and supernatant (Sup) fractions. (C) Percentage of LT secretion. Results are means from duplicate independent tests of each strain. The statistical analysis was performed by the Mann-Whitney test using Prism, version 6.00. Horizontal lines indicate median values. *, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant.
LT1 and LT2 toxin variants are equally stable.
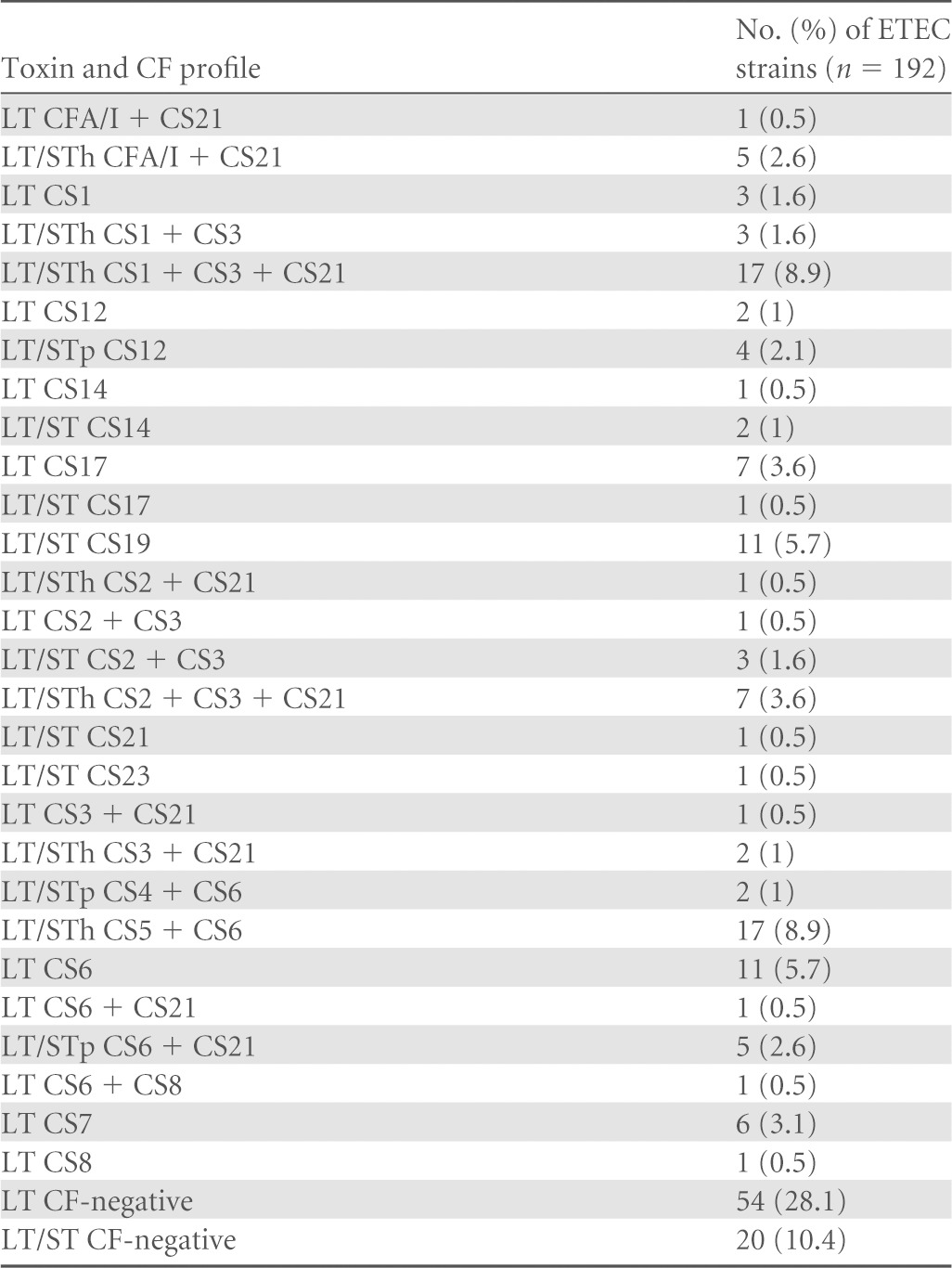
Once the LTA and LTB subunits reach the periplasm, they assemble into the holotoxin. This formed holotoxin is remarkably stable; however, changes in the LT amino acid sequence could influence absolute stability (6). To determine whether LT1 and LT2 have differences in their stability, we measured the amount of LTA and full folded LTB subunits in each isolate by GM1-ELISA. The ELISA was performed on 16 LT1 and 15 LT2 strains using two different monoclonal antibodies: one targeting the LTA subunit specifically, which detects the intact LT holotoxin (when bound to GM1 via the B5 subunit), and a second targeting the total B subunit (which can detect both holotoxin and free B5 subunits bound to GM1 but without the A subunit). A ratio between the amounts of LTAB and LTB was calculated to infer LT stability. When the amounts of stable LT expressed by LT1 and LT2 strains were compared, the ratios were slightly different in the pellet and supernatant fractions (Fig. 6A). In the pellet, the ratios for the LT1 and LT2 holotoxins were 0.14 and 0.11, respectively. In the supernatant, LT2 strains reached a ratio of 0.19 and LT1 strains reached a ratio of 0.17. However, no statistical differences were found in the analysis of the amino acid differences between the two major LT variants; LT1 and LT2 did not affect the assembly of LT holotoxins in the periplasm or the stability of secreted, fully folded LT (Fig. 6B).
FIG 6.
LT stability analysis in ETEC strains expressing LT1 and LT2. (A) Proportion of stable LT in the sonicated bacteria (Pellet) and supernatants (Sup). (B) Total ratio of stable LT in both the pellet and supernatants. The ratio of stability was calculated as (nanograms of LT holotoxin)/(nanograms of the B subunit). The amount of LT holotoxin was determined with a MAb against the A subunit, and the amount of the B subunit was determined with a MAb against the B subunit. Horizontal lines indicate mean values. The statistical analysis was performed by the Mann-Whitney U test using Prism, version 6.00. No statistical differences were found.
DISCUSSION
ETEC strains are characterized by the production of one or both of LT and ST. However, ETEC is a highly diverse pathogen with regard to serotype, toxin type, and CF expression (1, 26). We were initially interested in determining whether LT evolves into more-virulent variants over time and whether we could follow this in a set of strains isolated over 3 decades. The recent discovery of several natural variants in the LT sequence among ETEC strains isolated in Brazil (15), as well as the emergence of novel variants of the related cholera toxin (27), prompted us to evaluate the diversity of LT (LT-I) in human ETEC strains on a global scale. We identified novel LT variants and could show that both known and novel LT variants are associated with specific ETEC CF profiles, but no association with time was found. In fact, we have recently reported that globally distributed ETEC lineages with stable toxin-CF profiles have been stable over substantial periods in areas of endemicity (18).
In our 192 human ETEC strains isolated from children and adults globally over 31 years, 20 natural variants of the translated LT were identified. This finding, together with previous studies (8, 28), demonstrated a remarkable genetic diversity of LT in ETEC strains with worldwide distribution. Lasaro et al. (15) reported 16 LT variants in a set of 51 Brazilian ETEC strains, and we found 8 of these 16 LT variants, as well as 12 additional novel variants, among ETEC strains isolated from areas of endemicity all over the world. These findings support previous results showing the heterogeneity and variability within the LT sequence and demonstrate even greater LT diversity than was previously known. Some LT variants identified (15) were not found in this study, suggesting that some variants may be nonclinical isolates, or they could have a local impact or circulate in specific geographic locations. This may also suggest that certain mutations occur temporarily but do not persist in the population of human ETEC strains. In support of this, most of the novel LT variants identified in our study were found only in a single strain, while the more common LT variants, such as LT1, LT2, LT11, LT13, and LT18, seem to be associated with ETEC lineages that are widespread and not only circulate among continents but also persist over time.
Lasaro et al. (15) also reported that the 16 LT variants were distributed in 4 different clusters. In agreement with their findings, our phylogenetic analysis, including the 16 previously reported LT variants and the 12 new LT variants identified in this study, showed a similar topology. However, we found that the LT11 variant formed a fifth group. This was even more obvious when the phylogenetic tree was based on the SNPs, where LT11 clearly branched off from the rest of the LT variants (see Fig. S2 in the supplemental material). We also observed that 11 of the 12 newly identified variants in this study belonged to group I—centering around LT1—and that most novel LT variants were closely related to LT1. The 12th novel variant fell into the divergent group II formed by LT2. While group I was rooted in LT1, group II was rooted in the LT2 allele, suggesting that LT1 and LT2 could be ancestors for their respective groups. Hence, groups I and II are the most important groups in human ETEC strains, since they encompass the majority of LT variants and strains with a widespread distribution, as well as being found in strains isolated over the entire study duration of 31 years.
We could not find a strong association between specific LT variants and the geographic distribution or year of isolation among the strains analyzed in this study, suggesting that similar polymorphisms in the LT gene could be present in different regions of the world and at different time points (Fig. 2). In contrast, we found a strong relation between the presence of specific LT variants and the CF profile. For instance, CS1, CS2, and CS3 were expressed only in LT1 strains, while CS5 + CS6 and CFA/I expression was associated with LT2-expressing strains. This finding suggests that there is a link between the acquisition of the LT gene and a particular colonization factor by means of lateral transfer of chromosome- and plasmid-borne genes. Our results are in agreement with previous observations showing that ETEC strains expressing the same virulence profile (toxin-CF) fall into the same clonal groups regardless of the place of isolation (18, 29–34). These data also suggest that a possible clonal expansion of ETEC strains expressing the LT variant ancestors LT1 and LT2 could have occurred by means of human migration and travel. In fact, we show that two clusters, A and C, make up the majority of the ETEC strains (Fig. 2). Cluster A is a highly diverse group that includes a large number of LT variants (group I) with a broad range of colonization factor profiles. Also, this cluster is the most polymorphic due to the high number of single amino acid substitutions among the LT sequences. However, the LT sequences of cluster A are all rooted in the LT1 variant, and strains expressing LT1 also express colonization factors such as CS1, CS2, CS4, CS17, and CS19, which were previously reported to belong to the CFA/I family with similar genetic and biochemical features (35–37). However, the strains that express variants related to LT1 were more often colonization factor negative and were present only in a single or few strains. It was reported that the presence of separated clusters is a consequence of recent genomic changes, suggesting that these related LT variants could have emerged and again disappeared recently, while strains with LT1 retain their colonization factors and are persistent virulent strains (33). The second largest cluster, cluster C, contains strains that express CFA/I, as well as CS5 + CS6. Fewer related LT variants are found within this group, but most derivatives from the ancestral variant LT2 were, again, CF negative.
Clusters A and C represent two divergent and prevalent populations of LT-ETEC strains. This suggests that since the majority of the colonization factors and toxin are normally encoded on plasmids, the different LT variants have been acquired together with certain colonization factors on the same plasmid or a compatible coplasmid(s) (31, 38, 39). Although further analyses are required to demonstrate whether LT and colonization factors are physically located on the same plasmid, our data suggest that the alleles of both toxins and CFs are conserved within lineages and hence might have been acquired simultaneously by one ancestor strain at one point and then spread clonally. A previous report indicated that around 130 million years ago, before V. cholerae and E. coli diverged as species, LT genes were acquired by horizontal transfer (40). Also, it has been known that the LT sequence is flanked by insertion sequence (IS) elements, similar to those found next to genes encoding fimbriae, suggesting a general mechanism for the transmission of virulence-related genes (41, 42). Our data, together with the findings that ETEC strains with the same toxin-CF profile often are genetically related, suggest that LT acquisition is not due solely to horizontal gene transfer but rather is also due to lateral gene transfer.
When studying the natural diversity of LT, we observed more polymorphisms in the A subunit than in the B subunit, where only 2 amino acid substitutions were identified (in contrast to 22 changes in the A subunit). A previous report (43) found that single mutations in the A subunit (K63, D53, K7, K104, K97, and K114) and the double mutation K7 and K97 caused a considerable decrease in the proportion of fully assembled molecules of LT. However, in our study, the four mutations identified in the LT2 A subunit apparently did not affect the assembly of the LT molecule, suggesting that these polymorphic sites are not involved in the formation of the AB5 complex. This is supported by the fact that these variants are present in clinical isolates from patients with diarrhea and hence are expected to express a virulent LT toxin. On the other hand, we identified a considerable number of polymorphic places in the A2 helix domain of the A subunit. This structure is located near the B pentamer and continues into the pore of the B subunit, creating points of hydrophobic interactions between A and B subunits. Here we found that S224T (LT2, LT7, and LT22) and S228L (LT26) in LTA are located in close proximity to A2-B interaction residues, i.e., close to T75A in LTB; such polymorphism could possibly affect the positioning of the A subunit during holotoxin assembly (44, 45). However, our in silico protein modeling does not suggest that the T75A substitution in LTB would affect the stability of the holotoxin. Based on our results, however, we cannot determine whether export to the periplasm or efficiency of assembly is affected by the amino acid substitutions. A previous study reported that deletion of the last 14 residues of the LTA subunit could dramatically affect holotoxin assembly but also that deletion of the last 4 amino acids could be critical for the stability of the toxin (46). We found that LT28 (n = 1) and LT23 (n = 1) have an amino acid change at residues in that critical region. These strains displayed very low levels of LT production, which may be related to a deficiency in holotoxin assembly due to a reduction in LTA-LTB interaction. However, the effect of polymorphism in this region needs to be studied in more detail.
The LTB subunit was more conserved than the LTA subunit, possibly reflecting host specificity, since the B subunit binds to host receptors. LTB binds to GM1, glycoproteins, and glycolipids, as well as to carbohydrate epitopes of the ABO blood group system (47), and specific amino acid substitutions can interfere with binding (6, 48, 49). For instance, amino acid changes at residues 46, 47, and 57 have been reported to diminish binding affinity, since they were located close to the binding pocket (25, 26). Additional mutations in the LTB sequence have been described before in LTp (isolated from pigs), and these polymorphisms resulted in reduced binding to human GM1 and blood sugars (8, 48). In this study, such mutations were not identified, We found amino acid changes at residues 13 (LT3 and LT8) and residue 75 (LT2) among high-LT-producing strains, which are not involved in direct binding to GM1, although residue 13 is close to a proposed binding site. A histidine at residue 13 was found in strains that clustered in group B, which are the closest relatives to porcine variants that do not bind to human epithelial cells; the effect of this alteration should hence be determined in more detail. However, our findings in general corroborated that all strains expressed human LT with intact binding specificity to human host receptors.
With regard to secretion, it has been shown that LT release is basically dependent on the LTB5 unit (6). In our strains, we observed that secretion capacity was not affected by the differences in the amino acid sequences between the LT1 and LT2 variants, since the average LT secretion levels of both LT1 and LT2 remained constant around 50%. These data support the finding that polymorphism detected in the B subunit does not have a biological and functional impact on LT, which was corroborated by the protein modeling.
Importantly, we found a significant difference in LT production among the different LT variants, and especially between LT1 and LT2. A previous study indicated that LT1 and LT2 strains showed no significant difference with regard to binding affinity in the GM1 ganglioside assays (15). Furthermore, no differences were found in cAMP production using purified and trypsin-activated purified LT1 and LT2 (28), supporting the notion that these two major toxin variants are equally virulent. However, mice infected with LT2-producing ETEC strains displayed a highly effective protective anti-LT antibody response to subsequent infections with LT-producing strains (28). These data corroborate our observation that strains expressing LT2 produce more toxin than strains expressing LT1 under laboratory conditions. However, whether this is the case in the human small intestine remains to be investigated.
In summary, ETEC strains that express either the LT1 or LT2 variant express the most prevalent colonization factors associated with the occurrence of diarrheal disease worldwide (2, 50), and major lineages expressing specific colonization factor profiles are linked to the two variants. Although LT2 strains express significantly larger amounts of LT than LT1 strains, both LT1 and LT2 ETEC strains are frequently and repeatedly identified in cases of severe diarrhea worldwide and over time, supporting their virulence and successful dissemination.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Swedish Research Council grant K2012-56X-22029-01-3, VINNOVA grant 2011-03491, and a grant from Groschinsky's Foundation to Å.S. and by Swedish Foundation for Strategic Research (SSF) grant SB12-0072 to A.-M.S. and Å.S. The project was performed as part of the UMSA-IBMB Diarrheal Disease Project supported by the Swedish Agency for Research Economic Cooperation (SIDA) (to A.-M.S. and Å.S.). E.J. acknowledges financial support from the Swedish Institute and the International Science Programme (ISP). We also acknowledge RO1 NIAID AI0094001 funding to T.S.
We acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing high-performance computing resources that have contributed to the research results reported in this paper (http://www.tacc.utexas.edu).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02050-14.
REFERENCES
- 1.Qadri F, Svennerholm AM, Faruque AS, Sack RB. 2005. Enterotoxigenic Escherichia coli in developing countries: epidemiology, microbiology, clinical features, treatment, and prevention. Clin Microbiol Rev 18:465–483. doi: 10.1128/CMR.18.3.465-483.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaastra W, Svennerholm AM. 1996. Colonization factors of human enterotoxigenic Escherichia coli (ETEC). Trends Microbiol 4:444–452. doi: 10.1016/0966-842X(96)10068-8. [DOI] [PubMed] [Google Scholar]
- 3.Sánchez J, Holmgren J. 2005. Virulence factors, pathogenesis and vaccine protection in cholera and ETEC diarrhea. Curr Opin Immunol 17:388–398. doi: 10.1016/j.coi.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 4.Dallas WS, Gill DM, Falkow S. 1979. Cistrons encoding Escherichia coli heat-labile toxin. J Bacteriol 139:850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sears CL, Kaper JB. 1996. Enteric bacterial toxins: mechanisms of action and linkage to intestinal secretion. Microbiol Rev 60:167–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mudrak B, Kuehn MJ. 2010. Heat-labile enterotoxin: beyond GM1 binding. Toxins 2:1445–1470. doi: 10.3390/toxins2061445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmgren J, Svennerholm AM, Lindblad M. 1983. Receptor-like glycocompounds in human milk that inhibit classical and El Tor Vibrio cholerae cell adherence (hemagglutination). Infect Immun 39:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodrigues JF, Mathias-Santos C, Sbrogio-Almeida ME, Amorim JH, Cabrera-Crespo J, Balan A, Ferreira LC. 2011. Functional diversity of heat-labile toxins (LT) produced by enterotoxigenic Escherichia coli: differential enzymatic and immunological activities of LT1 (hLT) AND LT4 (pLT). J Biol Chem 286:5222–5233. doi: 10.1074/jbc.M110.173682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wülfing C, Rappuoli R. 1997. Efficient production of heat-labile enterotoxin mutant proteins by overexpression of dsbA in a degP-deficient Escherichia coli strain. Arch Microbiol 167:280–283. doi: 10.1007/s002030050444. [DOI] [PubMed] [Google Scholar]
- 10.Tauschek M, Gorrell RJ, Strugnell RA, Robins-Browne RM. 2002. Identification of a protein secretory pathway for the secretion of heat-labile enterotoxin by an enterotoxigenic strain of Escherichia coli. Proc Natl Acad Sci U S A 99:7066–7071. doi: 10.1073/pnas.092152899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzales L, Ali ZB, Nygren E, Wang Z, Karlsson S, Zhu B, Quiding-Järbrink M, Sjöling Å. 2013. Alkaline pH is a signal for optimal production and secretion of the heat labile toxin, LT in enterotoxigenic Escherichia coli (ETEC). PLoS One 8:e74069. doi: 10.1371/journal.pone.0074069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lasaro MAS, Rodrigues JF, Mathias-Santos C, Guth BEC, Régua-Mangia A, Piantino Ferreira AJ, Takagi M, Cabrera-Crespo J, Sbrogio-Almeida ME, de Souza Ferreira LC. 2006. Production and release of heat-labile toxin by wild-type human-derived enterotoxigenic Escherichia coli. FEMS Immunol Med Microbiol 48:123–131. doi: 10.1111/j.1574-695X.2006.00134.x. [DOI] [PubMed] [Google Scholar]
- 13.Grant CC, Messer RJ, Cieplak W. 1994. Role of trypsin-like cleavage at arginine 192 in the enzymatic and cytotonic activities of Escherichia coli heat-labile enterotoxin. Infect Immun 62:4270–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleckenstein JM, Hardwidge PR, Munson GP, Rasko DA, Sommerfelt H, Steinsland H. 2010. Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes Infect 12:89–98. doi: 10.1016/j.micinf.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lasaro MA, Rodrigues JF, Mathias-Santos C, Guth BE, Balan A, Sbrogio-Almeida ME, Ferreira LC. 2008. Genetic diversity of heat-labile toxin expressed by enterotoxigenic Escherichia coli strains isolated from humans. J Bacteriol 190:2400–2410. doi: 10.1128/JB.00988-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casey TA, Connell TD, Holmes RK, Whipp SC. 2012. Evaluation of heat-labile enterotoxins type IIa and type IIb in the pathogenicity of enterotoxigenic Escherichia coli for neonatal pigs. Vet Microbiol 159:83–89. doi: 10.1016/j.vetmic.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 17.Jobling MG, Holmes RK. 2012. Type II heat-labile enterotoxins from 50 diverse Escherichia coli isolates belong almost exclusively to the LT-IIc family and may be prophage encoded. PLoS One 7:e29898. doi: 10.1371/journal.pone.0029898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.von Mentzer A, Connor TR, Wieler LH, Semmler T, Iguchi A, Thomson NR, Rasko DA, Joffré E, Corander J, Pickard D, Wiklund G, Svennerholm A-M, Sjöling Å, Dougan G. 10 November 2014. Identification of enterotoxigenic Escherichia coli (ETEC) clades with significant long-term global distribution. Nat Genet doi: 10.1038/ng.3145. [DOI] [PubMed] [Google Scholar]
- 19.Sjöling Å Wiklund G, Savarino SJ, Cohen DI, Svennerholm AM. 2007. Comparative analyses of phenotypic and genotypic methods for detection of enterotoxigenic Escherichia coli toxins and colonization factors. J Clin Microbiol 45:3295–3301. doi: 10.1128/JCM.00471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imamura S, Kido N, Kato M, Kawase H, Miyama A, Tsuji T. 1997. A unique DNA sequence of human enterotoxigenic Escherichia coli enterotoxin encoded by chromosomal DNA. FEMS Microbiol Lett 146:241–245. doi: 10.1111/j.1574-6968.1997.tb10200.x. [DOI] [PubMed] [Google Scholar]
- 21.Svennerholm AM, Wiklund G. 1983. Rapid GM1-enzyme-linked immunosorbent assay with visual reading for identification of Escherichia coli heat-labile enterotoxin. J Clin Microbiol 17:596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Savidge TC, Urvil P, Oezguen N, Ali K, Choudhury A, Acharya V, Pinchuk I, Torres AG, English RD, Wiktorowicz JE, Loeffelholz M, Kumar R, Shi L, Nie W, Braun W, Herman B, Hausladen A, Feng H, Stamler JS, Pothoulakis C. 2011. Host S-nitrosylation inhibits clostridial small molecule-activated glucosylating toxins. Nat Med 17:1136–1141. doi: 10.1038/nm.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. 2005. The Amber biomolecular simulation programs. J Comput Chem 26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Negi SS, Schein CH, Oezguen N, Power TD, Braun W. 2007. InterProSurf: a web server for predicting interacting sites on protein surfaces. Bioinformatics 23:3397–3399. doi: 10.1093/bioinformatics/btm474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang C, Rausch D, Zhang W. 2009. Little heterogeneity among genes encoding heat-labile and heat-stable toxins of enterotoxigenic Escherichia coli strains isolated from diarrheal pigs. Appl Environ Microbiol 75:6402–6405. doi: 10.1128/AEM.00952-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolf MK. 1997. Occurrence, distribution, and associations of O and H serogroups, colonization factor antigens, and toxins of enterotoxigenic Escherichia coli. Clin Microbiol Rev 10:569–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Son MS, Megli CJ, Kovacikova G, Qadri F, Taylor RK. 2011. Characterization of Vibrio cholerae O1 El Tor biotype variant clinical isolates from Bangladesh and Haiti, including a molecular genetic analysis of virulence genes. J Clin Microbiol 49:3739–3749. doi: 10.1128/JCM.01286-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lasaro MA, Mathias-Santos C, Rodrigues JF, Ferreira LC. 2009. Functional and immunological characterization of a natural polymorphic variant of a heat-labile toxin (LT-I) produced by enterotoxigenic Escherichia coli (ETEC). FEMS Immunol Med Microbiol 55:93–99. doi: 10.1111/j.1574-695X.2008.00506.x. [DOI] [PubMed] [Google Scholar]
- 29.Pacheco AB, Guth BE, Soares KC, Nishimura L, de Almeida DF, Ferreira LC. 1997. Random amplification of polymorphic DNA reveals serotype-specific clonal clusters among enterotoxigenic Escherichia coli strains isolated from humans. J Clin Microbiol 35:1521–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pacheco ABF, Soares KC, de Almeida DF, Viboud GI, Binsztein N, Ferreira LCS. 1998. Clonal nature of enterotoxigenic Escherichia coli serotype O6:H16 revealed by randomly amplified polymorphic DNA analysis. J Clin Microbiol 36:2099–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pacheco ABF, Ferreira LCS, Pichel MG, Almeida DF, Binsztein N, Viboud GI. 2001. Beyond serotypes and virulence-associated factors: detection of genetic diversity among O153:H45 CFA/I heat-stable enterotoxigenic Escherichia coli strains. J Clin Microbiol 39:4500–4505. doi: 10.1128/JCM.39.12.4500-4505.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sommerfelt H, Steinsland H, Grewal HMS, Viboud GI, Bhandari N, Gaastra W, Svennerholm A-M, Bhan MK. 1996. Colonization factors of enterotoxigenic Escherichia coli isolated from children in north India. J Infect Dis 174:768–776. doi: 10.1093/infdis/174.4.768. [DOI] [PubMed] [Google Scholar]
- 33.Steinsland H, Valentiner-Branth P, Aaby P, Molbak K, Sommerfelt H. 2004. Clonal relatedness of enterotoxigenic Escherichia coli strains isolated from a cohort of young children in Guinea-Bissau. J Clin Microbiol 42:3100–3107. doi: 10.1128/JCM.42.7.3100-3107.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Del Canto F, Valenzuela P, Cantero L, Bronstein J, Blanco JE, Blanco J, Prado V, Levine M, Nataro J, Sommerfelt H, Vidal R. 2011. Distribution of classical and nonclassical virulence genes in enterotoxigenic Escherichia coli isolates from Chilean children and tRNA gene screening for putative insertion sites for genomic islands. J Clin Microbiol 49:3198–3203. doi: 10.1128/JCM.02473-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anantha RP, McVeigh AL, Lee LH, Agnew MK, Cassels FJ, Scott DA, Whittam TS, Savarino SJ. 2004. Evolutionary and functional relationships of colonization factor antigen I and other class 5 adhesive fimbriae of enterotoxigenic Escherichia coli. Infect Immun 72:7190–7201. doi: 10.1128/IAI.72.12.7190-7201.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nada RA, Shaheen HI, Khalil SB, Mansour A, El-Sayed N, Touni I, Weiner M, Armstrong AW, Klena JD. 2011. Discovery and phylogenetic analysis of novel members of class b enterotoxigenic Escherichia coli adhesive fimbriae. J Clin Microbiol 49:1403–1410. doi: 10.1128/JCM.02006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chattopadhyay S, Tchesnokova V, McVeigh A, Kisiela DI, Dori K, Navarro A, Sokurenko EV, Savarino SJ. 2012. Adaptive evolution of class 5 fimbrial genes in enterotoxigenic Escherichia coli and its functional consequences. J Biol Chem 287:6150–6158. doi: 10.1074/jbc.M111.303735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasko DA, Rosovitz MJ, Myers GS, Mongodin EF, Fricke WF, Gajer P, Crabtree J, Sebaihia M, Thomson NR, Chaudhuri R, Henderson IR, Sperandio V, Ravel J. 2008. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol 190:6881–6893. doi: 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shepard SM, Danzeisen JL, Isaacson RE, Seemann T, Achtman M, Johnson TJ. 2012. Genome sequences and phylogenetic analysis of K88- and F18-positive porcine enterotoxigenic Escherichia coli. J Bacteriol 194:395–405. doi: 10.1128/JB.06225-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamamoto T, Gojobori T, Yokota T. 1987. Evolutionary origin of pathogenic determinants in enterotoxigenic Escherichia coli and Vibrio cholerae O1. J Bacteriol 169:1352–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murphy GL, Dallas WS. 1991. Analysis of two genes encoding heat-labile toxins and located on a single Ent plasmid from Escherichia coli. Gene 103:37–43. doi: 10.1016/0378-1119(91)90388-R. [DOI] [PubMed] [Google Scholar]
- 42.Schlör S, Riedl S, Blaß J, Reidl J. 2000. Genetic rearrangements of the regions adjacent to genes encoding heat-labile enterotoxins (eltAB) of enterotoxigenic Escherichia coli strains. Appl Environ Microbiol 66:352–358. doi: 10.1128/AEM.66.1.352-358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magagnoli C, Manetti R, Fontana MR, Giannelli V, Giuliani MM, Rappuoli R, Pizza M. 1996. Mutations in the A subunit affect yield, stability, and protease sensitivity of nontoxic derivatives of heat-labile enterotoxin. Infect Immun 64:5434–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan E, O'Neal CJ, Mitchell DD, Robien MA, Zhang Z, Pickens JC, Tan XJ, Korotkov K, Roach C, Krumm B, Verlinde CL, Merritt EA, Hol WG. 2004. Structural biology and structure-based inhibitor design of cholera toxin and heat-labile enterotoxin. Int J Med Microbiol 294:217–223. doi: 10.1016/j.ijmm.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 45.van den Akker F, Pizza M, Rappuoli R, Hol WGJ. 1997. Crystal structure of a non-toxic mutant of heat-labile enterotoxin, which is a potent mucosal adjuvant. Protein Sci 6:2650–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Streatfield SJ, Sandkvist M, Sixma TK, Bagdasarian M, Hol WG, Hirst TR. 1992. Intermolecular interactions between the A and B subunits of heat-labile enterotoxin from Escherichia coli promote holotoxin assembly and stability in vivo. Proc Natl Acad Sci U S A 89:12140–12144. doi: 10.1073/pnas.89.24.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holmner A, Askarieh G, Okvist M, Krengel U. 2007. Blood group antigen recognition by Escherichia coli heat-labile enterotoxin. J Mol Biol 371:754–764. doi: 10.1016/j.jmb.2007.05.064. [DOI] [PubMed] [Google Scholar]
- 48.Mudrak B, Rodriguez DL, Kuehn MJ. 2009. Residues of heat-labile enterotoxin involved in bacterial cell surface binding. J Bacteriol 191:2917–2925. doi: 10.1128/JB.01622-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bäckström M, Shahabi V, Johansson S, Teneberg S, Kjellberg A, Miller-Podraza H, Holmgren J, Lebens M. 1997. Structural basis for differential receptor binding of cholera and Escherichia coli heat-labile toxins: influence of heterologous amino acid substitutions in the cholera B-subunit. Mol Microbiol 24:489–497. doi: 10.1046/j.1365-2958.1997.3541721.x. [DOI] [PubMed] [Google Scholar]
- 50.Svennerholm A-M, Lundgren A. 2012. Recent progress toward an enterotoxigenic Escherichia coli vaccine. Expert Rev Vaccines 11:495–507. doi: 10.1586/erv.12.12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.