

Abstract
Protein and small-molecule acylation reactions are widespread in nature. Many of the enzymes catalyzing acylation reactions belong to the Gcn5-related N-acetyltransferase (GNAT; PF00583) family, named after the yeast Gcn5 protein. The genome of Salmonella enterica serovar Typhimurium LT2 encodes 26 GNATs, 11 of which have no known physiological role. Here, we provide in vivo and in vitro evidence for the role of the MddA (methionine derivative detoxifier; formerly YncA) GNAT in the detoxification of oxidized forms of methionine, including methionine sulfoximine (MSX) and methionine sulfone (MSO). MSX and MSO inhibited the growth of an S. enterica ΔmddA strain unless glutamine or methionine was present in the medium. We used an in vitro spectrophotometric assay and mass spectrometry to show that MddA acetylated MSX and MSO. An mddA+ strain displayed biphasic growth kinetics in the presence of MSX and glutamine. Deletion of two amino acid transporters (GlnHPQ and MetNIQ) in a ΔmddA strain restored growth in the presence of MSX. Notably, MSO was transported by GlnHPQ but not by MetNIQ. In summary, MddA is the mechanism used by S. enterica to respond to oxidized forms of methionine, which MddA detoxifies by acetyl coenzyme A-dependent acetylation.
INTRODUCTION
The Gcn5-related N-acetyltransferase (GNAT; PF00583) superfamily of proteins (>10,000 members) is present in all domains of life. GNATs transfer the acetyl group from acetyl coenzyme A (acetyl-CoA) to proteins or small molecules (for reviews, see references 1 and 2). The acetylation targets of GNATs include the N termini of proteins (3, 4), aminoglycoside antibiotics (5), glutamate (6), spermidine (7), aminoalkylphosphonic acid (8), dTDP-fucosamine (9), and tRNAs (10). Some of the first bacterial GNATs characterized were the aminoglycoside N-acetyltransferases from Enterococcus faecium (5) and Serratia marcescens (11), demonstrating GNAT-dependent acetylation and inactivation of antibiotics.
GNATs provide protection against a myriad of cellular stressors (5, 11, 12), and it is possible that the diversity of stressors controlled by GNATs correlates with the environment encountered by the cell. Therefore, the relevance of GNAT function to cell physiology varies among organisms, with respect to not only cellular stressors but metabolic pathways as well. For example, Salmonella enterica (13) and Escherichia coli (14) each contain ∼26 GNATs, yet actinomycetes, such as Streptomyces lividans (15), encode up to ∼70 putative GNATs. This suggests that S. lividans occupies a more diverse environment while also maintaining a more complex metabolism.
At present, there is limited to no information available on the cellular processes several putative S. enterica GNATs may affect. Not surprisingly, the signals that trigger the synthesis of GNATs, the transcription factors involved in sensing such signals, and the determinants of GNAT substrate specificity remain unknown.
In S. enterica, MddA (methionine derivative detoxifier A; formerly YncA [STM1590]) is a putative GNAT with no characterized function. Homology searches reveal the presence of MddA-like proteins in Pseudomonas aeruginosa (63% identity) and Acinetobacter baylyi (36% identity) and suggest a role for S. enterica MddA (SeMddA) in controlling the toxic effects of methionine sulfoximine (MSX) and methionine sulfone (MSO) (Fig. 1) (12, 16). The protein structures of SeMddA homologues have been solved in P. aeruginosa (Protein Data Bank [PDB] 2J8R) and A. baylyi (PDB 2JLM), showing these enzymes contain the structural core representative of members of the GNAT family (12, 16, 17). The P. aeruginosa MddA homologue was solved in complex with MSX and showed a conformational change in the active site upon binding to MSX (12).
FIG 1.

Chemical structures of methionine analogues.
MSX is similar in structure to phosphinothricin (PHO) (Fig. 1), and at least some MddA homologues have been incorrectly annotated as PHO acetyltransferases (12, 16), an activity performed by the Bar acetyltransferase (18). The Bar protein is a GNAT of Streptomyces spp. involved in protection against a self-produced toxin, Bialaphos, a natural herbicide consisting of the tripeptide PHO-Ala-Ala (19). The toxin is activated when PHO, a glutamate analogue, is cleaved from the peptide. Bialaphos is a potent herbicide, and plants have been genetically engineered to be resistant by carrying the bar gene (18, 20). PHO and MSX inhibit the activity of glutamine synthetase (GlnA). Notably, MSX does not inhibit glutamine, γ-aminobutyrate, glutamate transaminases, or glutamate decarboxylase (21, 22). GlnA plays an important role in nitrogen metabolism, converting glutamate to glutamine using ATP and ammonium (23, 24).
MSX is found in the roots and seeds of members of the Connaraceae family of plant species and has been determined to be a toxic component of these plants (25). While this is the only example of naturally occurring MSX, in the late 1940s, MSX was identified as a toxic by-product in bleached flour (26–28). It was produced by addition of nitrogen trichloride, which reacted with wheat proteins in the flour. This had severe effects on individuals who consumed flour treated in this way, and around 1950, this method of flour bleaching was discontinued. Researchers have hypothesized that long-term exposure to MSX in processed foods may account for an increase in neurodegenerative disorders in humans, such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (ALS) (29, 30).
Here, we present in vivo and in vitro evidence that SeMddA is necessary for the survival of S. enterica in the presence of MSX and MSO, that addition of glutamine or methionine prevents this toxicity, and that the SeMddA protein acetylates the nitrogen bound to the alpha carbon of MSX. We report that deletion of two amino acid transporters, MetNIQ and GlnHPQ, fully restores the growth of a ΔmddA strain exposed to MSX. Our data also indicate that the transport of MSO differs from that of MSX, as MetNIQ cannot transport MSO. Taken together, these data demonstrate that SeMddA acetylates MSX and MSO, thereby blocking their toxic effects.
MATERIALS AND METHODS
Culture media and chemicals.
Nutrient broth (NB) (Difco) containing NaCl (85 mM) was used as rich medium. The minimal medium used was no-carbon essential (NCE) minimal medium (31) containing MgSO4 (1 mM), Wolfe's trace minerals (1×) (32), and glycerol (22 mM) as the sole source of carbon and energy. When used, antibiotics were added at the following concentrations: tetracycline, 20 μg ml−1; kanamycin, 50 μg ml−1; chloramphenicol, 20 μg ml−1; and ampicillin, 100 μg ml−1. When added to the medium, the calcium chelator EGTA was present at 10 mM, and X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) was added to a final concentration of 40 μg ml−1. All chemicals were purchased from Sigma-Aldrich unless otherwise noted: kanamycin, ampicillin, NaCl, and HEPES (Fisher Scientific); tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (Soltec Ventures); IPTG (isopropyl β-d-1-thiogalactopyranoside) (IBI Scientific); dithiothreitol (DTT) (Gold BioTechnology); and [1-14C]acetyl-CoA (Moravek).
Bacterial strains.
All the strains are derivatives of S. enterica serovar Typhimurium strain LT2 (unless otherwise specified) and are listed in Table S1 in the supplemental material. Tn10d(tet+) refers to the transposase-defective mini-Tn10 (Tn10Δ16Δ17) (33).
Primers.
All the primers used in this study were synthesized by IDT (Coralville, IA) and are listed in Table S2 in the supplemental material.
Phage transductions.
P22 phage-mediated transduction crosses were performed as described previously (34), using the high-frequency general transducing mutant of bacteriophage P22 HT105/1 int-210 (35, 36). Phage-free, phage-sensitive transductants were isolated on nonselective green indicator plates as described previously (37).
Construction of gene deletions.
Strains carrying in-frame deletions of each gene of interest were constructed following the protocol established for E. coli (38). Briefly, PAGE-purified primers with 36 bp of homology with the 5′ and 3′ ends of the gene of interest (IDT, Coralville, IA) were used to amplify the cat+ or kan+ cassette from template plasmid pKD3 or pKD4, respectively. The PCR products were analyzed on a 1% agarose gel and purified using the Wizard SV Gel and PCR Clean-Up System (Promega) according to the manufacturer's protocol, followed by subsequent construction of the deletion of the gene as described in the original protocol (38). PCR was used to confirm the insertion of the cat+ or kan+ amplicon compared to the wild-type allele size.
All mutations were reconstructed in the appropriate strain background using P22 phage-mediated transduction crosses. Briefly, P22 phage was propagated on a strain containing an allele of interest. P22 lysates were generated from these strains to transduce the original recipient strain to the appropriate antibiotic resistance, yielding a reconstructed strain with the allele of interest (see Table S1 in the supplemental material).
Plasmid construction.
All the plasmids used in this work are listed in Table S1 in the supplemental material. The cloning method using unique BspQ1 restriction sites, as published elsewhere (39), was used to construct all the plasmids in this study unless otherwise stated. DNA sequencing (Georgia Genomics Facility, University of Georgia, Athens, GA) was used to verify all the plasmids constructed in this study. Each gene of interest was amplified from S. enterica serovar Typhimurium LT2 genomic DNA.
Genes of interest (mddA+, glnPQ+, and metNI+) were cloned into the l-(+)-arabinose-inducible vector pBAD24 (40) engineered to contain BspQ1 sites (C. M. VanDrisse and J. C. Escalante-Semerena, unpublished data). The resulting plasmids (pMDD8, pGLN2, and pMETN1) were used in complementation studies.
Plasmid pTEV5 (41), which directs the synthesis of the protein with a cleavable N-terminal hexahistidine tag, was modified to contain BspQ1 restriction sites. The resulting plasmid was named pTEV16 (VanDrisse and Escalante-Semerena, unpublished) and used for overexpression of wild-type SeMddA (SeMddAWT, encoded by pMDD7).
Site-directed mutagenesis was performed using primers designed using PrimerX (available at http://www.bioinformatics.org/primerx/) to mutate the predicted catalytic residue of SeMddAWT, glutamate (E82), to a noncatalytic glutamine residue (Q82) to construct a catalytic variant (SeMddAE82Q) in both the complementation (pMDD11) and overexpression (pMDD10) vectors.
Growth behavior analyses.
Starter cultures were grown overnight at 37°C with shaking in nutrient broth containing the appropriate drug marker and used to inoculate fresh medium (1% [vol/vol]) in a volume of 200 μl per well of a 96-well plate with appropriate antibiotics. Strains containing plasmids were induced with varying concentrations of l-(+)-arabinose, as described in the figure legends. Additional chemicals, such as glutamine, methionine, MSX, MSO, or PHO, were added at the concentrations indicated in the figures and figure legends. The plates were incubated at 37°C with shaking for 20 to 48 h in a microplate reader (Bio-Tek Instruments). Growth curves were determined in triplicate in three independent experiments, with a representative growth curve shown. The data were analyzed using Prism v6 (GraphPad) analytical software. Error bars represent standard deviations.
Overproduction and purification of the SeMddAWT and SeMddAE82Q proteins.
Vectors encoding SeMddAWT (pMDD7) and SeMddAE82Q (pMDD10) were transformed into E. coli C41(λDE3). Overnight cultures of the transformants were subcultured (1:100 [vol/vol] [inoculum/medium]) in 1 liter of LB containing ampicillin (100 μg ml−1). The cultures were grown at 37°C with shaking to an optical density at 600 nm (OD600) of 0.6, induced with IPTG (1 mM), and shaken overnight at ∼28°C. Cells were harvested by centrifugation at 6,000 × g for 15 min at 4°C. The collected cell paste was resuspended in binding buffer A (HEPES buffer [50 mM, pH 7.5] containing NaCl [500 mM] and imidazole [20 mM]) plus lysozyme (1 mg ml−1), DNase I (25 μg ml−1), and the protease inhibitor phenylmethanesulfonyl fluoride (PMSF; 0.5 mM). The cells were lysed by sonication for 1 min (2 s; 50% duty) for 2 rounds on ice using a 550 Sonic Dismembrator (Fisher Scientific) at setting 4. Clarified cell lysates were obtained after centrifugation for 45 min at 4°C and 43,667 × g, followed by filtration of the supernatant through a 0.45-μm filter (Millipore). Samples were loaded onto a 2-ml HisPur Ni-nitrilotriacetic acid (NTA) resin column (Thermo Scientific) at 4°C, preequilibrated with binding buffer. The Ni+ column was washed first with buffer B (HEPES buffer [50 mM, pH 7.5] with NaCl [500 mM]) that contained 40 mM imidazole to remove nonspecifically bound proteins. Following that, His6-tagged SeMddA proteins were eluted in the same buffer system containing 500 mM imidazole.
To cleave the tag, His6-TEV protease (rTEV) was purified as described previously (42), and cleavage of tagged SeMddA proteins was performed as follows: rTEV was added to the eluted protein supplemented with DTT (1 mM) at a 1:100 protease/tagged-protein ratio, and the mixture was incubated at room temperature for 3 h. Proteins were dialyzed at 4°C in buffer C (HEPES buffer [50 mM, pH 7.5] containing NaCl [500 mM], TCEP [0.5 mM], and EDTA [0.5 mM]). The dialyzed, cleaved protein was reloaded onto the column and eluted using a 40 mM imidazole wash step, followed by an imidazole wash (500 mM, which allowed separation of the untagged [SeMddAWT or SeMddAE82Q] from the tagged [His6-TEV protease] protein). SeMddAWT and SeMddAE82Q eluted from the column during the wash step. The proteins were stored in HEPES buffer (50 mM, pH 7.2) containing NaCl (100 mM), TCEP (0.5 mM), and glycerol (10% [vol/vol]); drop frozen in liquid nitrogen; and stored at −80°C. Both proteins were purified to 99% homogeneity, as determined using Total Lab v2005 software (Nonlinear Dynamics).
Analytical gel filtration.
Experiments were performed at 4°C. Per run, a 500-μl sample volume of 100 μg of SeMddAWT or SeMddAE82Q protein was injected onto a Superdex 200 HR 10/30 gel filtration column (GE Healthcare) attached to an ÄKTA purifier fast protein liquid chromatography (FPLC) system that was equilibrated with buffer D (HEPES [50 mM, pH 7.4] and NaCl [100 mM]). A calibration standard containing a mixture of molecular masses ranging from 1.35 to 670 kDa (Bio-Rad) was used to generate a standard curve to determine the molecular mass. The standards mixtures contained vitamin B12 (1.35 kDa), equine myoglobin (17 kDa), chicken ovalbumin (44 kDa), bovine gamma globulin (158 kDa), and thyroglobulin (670 kDa). The negative control used was injection of the protein storage buffer, with no peak in absorbance observed. A flow rate of 0.5 ml min−1 was used to develop the column, and elution peak analysis was performed using UNICORN v4.11 software (GE Healthcare Life Sciences). Data were graphed and analyzed using Prism v6 (GraphPad) analytical software. Linear regression analyses of the standard curves yielded r2 values of 0.98.
Thin-layer chromatography.
Reaction mixtures included HEPES buffer (50 mM, pH 7.0), containing TCEP (1 mM), [1-14C]acetyl-CoA (20 μM), substrate (0.5 mM), and SeMddAWT or SeMddAE82Q (1 μg). The reaction mixtures were incubated at 37°C for 30 min, spotted onto a polyester-backed silica gel plate (Whatman Ltd.), and developed in a chamber preequilibrated with a mobile phase of n-butanol, acetic acid, and distilled H2O (dH2O) (3:1:1). Thin-layer chromatography (TLC) plates were incubated for 3 to 4 h, dried, and developed with a phosphor screen overnight. The resulting phosphorimage was detected using a Typhoon Trio+ Variable Mode Imager (GE Healthcare) with ImageQuant v5.2 software.
SeMddA in vitro activity assays.
A SpectraMax Plus 384 microplate spectrophotometer (Molecular Devices) equipped with SoftMax Pro v4 software was used for data acquisition. Assays were performed at 30°C in 100-μl volumes in 96-well microplates using a continuous spectrophotometric assay that employed 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) (Ellman's reagent) as a reporter of free sulfhydryl groups at 412 nm (43, 44). Reaction mixtures contained HEPES buffer (50 mM, pH 7.2), DTNB (0.3 mM), acetyl-CoA, SeMddA, and MSX or MSO. Reactions were initiated by the addition of SeMddA enzyme. A control containing enzyme but lacking acetyl-CoA was used to correct for background. A secondary negative control containing MddAE82Q (96 nM), acetyl-CoA, and MSX was also used. In the above-mentioned controls, absorbance measurements were not higher than the background control. Data were acquired every 10 s over a 2-min period.
To determine kinetic parameters when MSX or MSO was the substrate, SeMddA was present at 96 nM, acetyl-CoA was used at a saturating concentration (1 mM), and MSX or MSO concentrations were varied from 0.010 to 2 mM. In assays where the acetyl-CoA concentration was varied (from 0.050 to 2 mM), MSX was present at a saturating concentration (1 mM). SeMddA enzyme was present in the reaction mixture at 96 nM. Kinetic parameters were determined using Prism v6 (GraphPad) analytical software. The molar extinction coefficient used for the concentration of the TNB2− anion was 14,150 M−1 cm−1 (45).
Synthesis and identification of acetyl-methionine sulfoximine and acetyl-methionine sulfone.
To confirm the location of acetylation of MSX and MSO, acetyl-MSX (Ac-MSX) and acetyl-MSO (Ac-MSO) were generated enzymatically. The reaction components included acetyl-CoA (1 mM), MSX or MSO (0.5 mM), SeMddAWT (20 μg), and ammonium bicarbonate (20 mM) in a reaction volume of 500 μl. A no-enzyme control reaction was also performed. The reaction mixtures were incubated at 37°C for 2 h. SeMddAWT was removed from the reaction mixture by filtration using Amicon Ultra centrifugal filters (Millipore) with a molecular weight cutoff of 3,000 (3K), according to the manufacturer's protocol. Samples were concentrated in a Vacufuge plus speed vacuum (Eppendorf) at 30°C and resuspended in 50% acetonitrile, 50% dH2O, with 1% formic acid. The identities of acetyl-MSX and acetyl-MSO were confirmed by mass spectrometry (MS) (Protein and Mass Spectrometry Facility, UGA). Electrospray ionization (ESI)-MS was performed after sample dilution in acetonitrile and run on an Esquire 3000 Plus (Bruker) Ion Trap Mass Spectrometer at 0.3 ml/h.
Isolation of a Tn10d(tet+) insertion in an mddA1::cat+ strain by transposon mutagenesis.
To identify mutations that allow the growth of an mddA1::cat+ strain in the presence of MSX, a mutagenesis screen using Tn10d(tet+) transposons was utilized. To obtain a phage pool lacking the mddA gene, a transposition experiment was performed by transducing a P22 phage stock carrying a pool of Tn10d(tet+) transposons inserted throughout the S. enterica genome into an mddA1::cat+ strain containing a plasmid carrying a transposase (JE18543). This was done to prevent the recovery of growth due to repair of the mddA1::cat+ deletion. The transduction reaction mixtures were plated on NB plates containing tetracycline (20 μg ml−1) to select for transposon insertion. An estimated total of 61,308 colonies were pooled, resulting in ∼13.5-fold coverage of the S. enterica genome.
A P22 lysate grown on this pool of strains was used to transduce an mddA1::cat+ strain (JE18333) to tetracycline resistance (20 μg ml−1) on NB plates, followed by replica printing onto NCE minimal medium plates containing glycerol (22 mM) and MSX (10 μM). Colonies arising on the selection plates were freed of phage, patched onto NB plates, incubated for 4 to 6 h at 37°C, and replica printed to several selection plates, including NB with tetracycline (20 μg ml−1), NB with chloramphenicol (20 μg ml−1), and NCE minimal medium with MSX (10 μM). Strains that grew under all of these conditions were freed of phage, and P22 lysates were generated. The resulting phage lysate was used as a donor in crosses with the parental mddA1::cat+ strain (JE18333). Transductions were plated on NB with tetracycline (selecting for the transposon) and, after 24 h of growth at 37°C, replica printed to plates containing MSX (10 μM) to confirm the phenotype.
The location of the insertion on the chromosome was determined in the reconstructed strain by sequencing the DNA flanking the Tn10d(tet+) element using a PCR-based protocol. A DNA product was amplified with degenerate primers and primers derived from the Tn10d(tet+) insertion sequences as reported previously (46) and used as a template for sequencing reactions. DNA sequencing was performed using BigDye Terminator v3.1 protocols (Applied Biosystems), and the reactions were analyzed at the University of Georgia Genomics Facility.
RESULTS
MSX and MSO inhibit growth of an mddA1::cat+ strain.
We examined the ability of an S. enterica mddA1::cat+ strain (JE18333) to grow in the presence of MSX or MSO (Fig. 2). In the absence of MSX or MSO, no growth differences were observed between the mddA+ and mddA1::cat+ strains in either rich or minimal medium (data not shown). Addition of MSX (10 μM) caused complete growth inhibition of the mddA1::cat+ strain in minimal medium (Fig. 2A, diamonds). The same concentration of MSX did not have any effect on the growth of the mddA+ strain (Fig. 2A, inverted triangles). The observed effects were different when strains were grown in rich medium. Under these conditions, MSX partially inhibited growth of the mddA1::cat+ strain, but only when the MSX concentration was at least 50 μM (Fig. 2B, triangles).
FIG 2.

MSX and MSO inhibit growth of an mddA::cat+ strain. The growth of S. enterica mddA+ and mddA1::cat+ strains was examined in the presence of MSX (10 μM or 50 μM) or MSO (50 μM or 100 μM), as indicated. NCE minimal medium with glycerol (22 mM) or nutrient broth was used in these experiments. Growth curves were determined using a microplate reader (Bio-Tek Instruments) as described in Materials and Methods. The following strains were analyzed: JE10079 (mddA+), JE18333 (mddA1::cat+), JE18961 (mddA1::cat+/pMDD8 mddA+), and JE19029 (mddA1::cat+/pmDD11 mddA+ [encoding SeMddAE82Q]). The error bars represent standard deviations.
Addition of MSO (50 μM) also negatively affected the growth of the mddA1::cat+ strain relative to that of the mddA+ strain in minimal medium, but the effect was less severe than the effect caused by MSX (Fig. 2C, triangles). A short delay in the onset of exponential growth was observed for the mddA1::cat+ strain in rich medium containing 200 μM MSO (Fig. 2D, triangles).
SeMddA activity blocks the negative effects of MSX and MSO.
We performed in vivo experiments to determine whether SeMddA played a role in circumventing the toxic effects of MSX and MSO. For this purpose, plasmid pMDD8 (mddA+) was introduced into the mddA1::cat+ strain (JE18333), yielding strain JE18961. As a control, an inactive variant of SeMddA in which the predicted catalytic residue E82 was changed to Q82 (SeMddAE82Q) was constructed by site-directed mutagenesis. The presence of plasmid pMDD11 encoding the SeMddAE82Q variant did not restore growth of the mddA1::cat+ strain in the presence of MSX (Fig. 2A, circles). However, the wild-type mddA+ allele in trans (pMDD8) supported the growth of the mddA1::cat+ strain in the presence of MSX or MSO (Fig. 2A, squares, and D, inverted triangles). It should be noted that at higher levels of inducer, i.e., ≥250 μM l-(+)-arabinose, an mddA1::cat+ strain synthesizing the SeMddAE82Q variant grew in medium containing MSX (10 μM), albeit with an extended lag phase, indicating that SeMddAE82Q retained some catalytic activity (see Fig. S1 in the supplemental material).
High levels of MSX are inhibitory to wild-type S. enterica.
An increase in the lag phase of a culture of the mddA+ strain (JE10079) was seen at higher levels of MSX (20 to 50 μM). However, the strain eventually grew at the same rate observed in medium devoid of MSX and reached the same optical density as a culture not exposed to MSX (Fig. 3A). The effect of MSX in rich medium was notably different, as the mddA+ strain showed no inhibition when the MSX concentration was 500 μM (data not shown), and only partial inhibition of growth occurred at 1 mM (Fig. 3B, triangles). In sharp contrast, as stated previously, no growth of the mddA1::cat+ strain was observed in the presence of as little as 10 μM MSX in minimal medium (Fig. 3C); however, in rich medium, growth of the mddA1::cat+ strain was abolished only at a concentration of 1 mM MSX (Fig. 3D, inverted triangles).
FIG 3.

The effect of increasing MSX concentration on mddA+ and mddA::cat+ strains is detrimental to growth. The growth of S. enterica mddA+ and mddA1::cat+ strains was examined with increasing concentrations of MSX, as indicated. The minimal medium used was NCE with glycerol (22 mM), and the rich medium used was nutrient broth. Growth curves were determined using a microplate reader (Bio-Tek Instruments) as described in Materials and Methods. The following strains were analyzed: JE10079 (mddA+) and JE18333 (mddA1::cat+). The error bars represent standard deviations.
High concentrations (e.g., 200 μM) of other methionine derivatives, such as methionine sulfoxide and buthionine sulfoximine (BSX), did not affect the growth rate of either the mddA+ or the mddA1::cat+ strain in minimal medium (doubling times [h]: with methionine sulfoxide, 1.4 and 1.5, respectively; with BSX, 1.6 and 1.7, respectively).
SeMddA does not block the inhibitory effects of PHO.
Addition of PHO (100 μM) was inhibitory to S. enterica growth; however, the mddA and mddA1::cat+ strains were equally affected, indicating that the deleterious effect of PHO could not be blocked by SeMddA (Fig. 4). At present, it is unclear whether PHO is acetylated in S. enterica, and if so, which acetyltransferase catalyzes the reaction.
FIG 4.
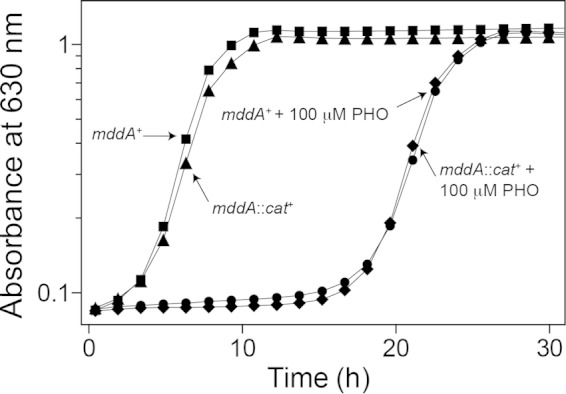
SeMddA does not prevent growth inhibition by PHO. Growth of the S. enterica mddA+ and mddA1::cat+ strains was examined with or without PHO (100 μM). NCE minimal medium supplemented with glycerol (22 mM) was used in these experiments. Growth curves were determined using a microplate reader (Bio-Tek Instruments) as described in Materials and Methods. The following strains were analyzed: JE10079 (mddA+) and JE18333 (mddA1::cat+).
Inhibition of an S. enterica mddA1::cat+ strain by MSX or MSO is alleviated by the addition of glutamine or methionine.
MSX and MSO are glutamate analogues that inhibit glutamine synthetase (GlnA). GlnA function is essential in nitrogen metabolism, and MSX has been shown to bind tightly to the enzyme, causing irreversible inhibition of activity (47, 48). This inhibition is partially resolved by the addition of the product, glutamine, bypassing the requirement for GlnA (49). When glutamine (200 μM) was present in the culture medium, exposure of an mddA1::cat+ strain to MSX (10 μM) resulted in a modest increase in the growth yield, plateauing at an OD630 of ∼0.2 (Fig. 5A). The addition of higher concentrations of glutamine (i.e., 500 and 1,000 μM) correlated with higher growth yields (Fig. 5A), suggesting that saturation of the GlnA active site with glutamine outcompeted binding of the inhibitor.
FIG 5.

Glutamine and methionine counteract the deleterious effects of MSX and MSO on growth in the absence of MddA. The growth of S. enterica mddA+ and mddA1::cat+ strains was examined in the presence of MSX (10 μM) and MSO (50 μM), with the addition of glutamine (200 μM, 500 μM, or 1,000 μM) or methionine (500 μM) to the medium. NCE minimal medium supplemented with glycerol (22 mM) was used in these experiments. Growth curves were determined using a microplate reader (Bio-Tek Instruments) as described in Materials and Methods. The following strains were analyzed: JE10079 (mddA+) and JE18333 (mddA1::cat+). The error bars represent standard deviations.
Growth of an mddA1::cat+ strain exposed to MSO (50 μM) was restored to wild-type levels with higher levels of glutamine (i.e., 500 and 1,000 μM) in the medium (Fig. 5C). Addition of glutamine to an mddA1::cat+ strain exposed to either MSX or MSO in rich medium allowed the strain to grow at a rate similar to that of the mddA+ strain, and it reached a growth yield similar to wild-type levels (see Fig. S2A and B in the supplemental material). It is clear that MSX and MSO inhibit GlnA activity (47, 50–54); however, it is less clear whether these compounds inhibit other cellular processes that could account for the observed growth phenotypes.
Previous work showed that the addition of methionine to wild-type S. enterica exposed to MSX restored growth (for wild-type cultures within 8 h), possibly alleviating toxicity by competing with the uptake of MSX (49). The growth behavior of mddA+ and mddA1::cat+ strains in medium containing methionine (500 μM) and MSX (10 μM) or MSO (50 μM) was investigated (Fig. 5B and D). Addition of methionine improved the growth of an mddA1::cat+ strain exposed to MSX, but not to wild-type levels. Methionine also decreased MSX toxicity in the mddA+ strain, resulting in cultures reaching stationary phase ∼10 h sooner than cultures growing in the presence of MSX and in the absence of methionine (Fig. 5B). When MSO was used in lieu of MSX, growth of the mddA1::cat+ strain was restored to wild-type levels when methionine was added to the culture medium (Fig. 5D).
SeMddA is a dimer in solution.
SeMddAWT is a 516-residue, 19.2-kDa protein. SeMddAWT and the SeMddAE82Q catalytic variant were isolated to 99% homogeneity using Ni+ affinity chromatography (see Materials and Methods) (Fig. 6A). To determine the oligomeric state of the proteins in solution, FPLC gel filtration analysis was performed using commercially available molecular mass standards. Under the conditions tested, SeMddAWT and SeMddAE82Q eluted ∼30 min after injection. The retention time was consistent with that of a protein whose mass was approximately 40 kDa, compared to the elution times of molecular mass standards. Since the calculated molecular mass of SeMddAWT was approximately 19 kDa, we inferred that SeMddAWT was a dimer in solution (Fig. 6B). The oligomeric state of SeMddA was consistent with those of MddA homologues from P. aeruginosa (PA4866) and A. baylyi (ACIAD1637) (12, 16).
FIG 6.
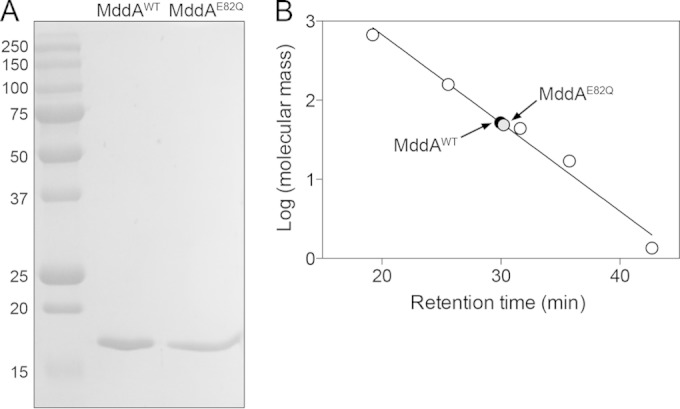
SeMddA is a dimer in solution. (A) SeMddAWT and the catalytic variant SeMddAE82Q (∼19 kDa) were purified using a two-step nickel affinity purification. An SDS-PAGE gel shows the molecular mass standards (kDa) (left lane), purified SeMddAWT protein, and purified SeMddAE82Q. Both proteins were purified to >99% homogeneity. (B) The molecular masses of SeMddAWT and SeMddAE82Q in solution were estimated by gel filtration chromatography as described in Materials and Methods. The molecular mass standards (open circles) were thyroglobulin (bovine; 670 kDa), gamma globulin (bovine; 158 kDa), ovalbumin (chicken; 44 kDa), myoglobin (horse; 17 kDa), and vitamin B12 (1.35 kDa).
SeMddAWT acetylates toxic methionine derivatives.
TLC was used to identify the substrate(s) of SeMddAWT. An in vitro activity assay was used to monitor the SeMddA-dependent transfer of the 14C-labeled acetyl moiety from [1-14C]acetyl-CoA to putative substrates that were analogous in structure to MSX (see Materials and Methods). Phosphorimaging showed SeMddAWT-dependent acetylation of MSX, MSO, methionine sulfoxide, BSX, methionine, and glutamine (see Fig. S3 in the supplemental material). Transfer of the radiolabel was not seen when PHO, glutamate, or arginine was used as a substrate or when SeMddAE82Q was used as the negative control (see Fig. S3 in the supplemental material).
To examine the specificity of the SeMddAWT enzyme for the substrates identified by TLC, a spectrophotometric assay using DTNB was used to determine the kinetic parameters of SeMddAWT (see Materials and Methods) (Table 1). No acetyltransferase activity was detected, under the conditions tested, when PHO, methionine sulfoxide, BSX, methionine, or glutamine was used as the substrate (data not shown). Activity of SeMddAWT was seen for MSO {apparent Km [Km(app)], 230 ± 56 μM} and MSX [Km(app), 576 ± 89 μM]. Inhibition of SeMddAWT activity was observed at MSO concentrations of >1.5 mM, while no inhibition was seen when MSX was tested (<2 mM). Also, concentrations of acetyl-CoA of >750 μM were inhibitory to SeMddAWT.
TABLE 1.
Kinetic parameters of S. enterica MddAWTa
| Substrate | Km(app) (mM) | kcat(app) (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|---|
| MSX | 576 ± 89 | 35 ± 2 | 6 × 104 |
| MSO | 230 ± 56 | 31 ± 2 | 1 × 104 |
| Acetyl-CoA | 156 ± 41 | 23 ± 1 | 2 × 104 |
The apparent kinetic parameters of the SeMddAWT protein were determined using a spectrophotometric assay described in Materials and Methods. The catalytic variant SeMddAE82Q was used as a negative control.
The SeMddAE82Q catalytic variant (96 nM; negative control) displayed no activity at saturating substrate concentrations (MSX and acetyl-CoA, 1 mM each), and appreciable activity of SeMddAE82Q was not observed until a 50-fold-higher protein concentration (5 μM) was reached. Although the activity of SeMddAE82Q was substantially decreased in vitro, the residual activity observed may account for the growth of an mddA1::cat+ strain containing a plasmid encoding SeMddAE82Q in medium with MSX (10 μM) at high levels of induction [0.25 to 1 mM l-(+)-arabinose] (see Fig. S1 in the supplemental material).
Location of acetylation of MSX and MSO.
To confirm the location of acetylation of MSX and MSO, Ac-MSX and Ac-MSO were generated enzymatically (see Materials and Methods), and the structures were resolved by mass spectrometry (Protein and Mass Spectrometry Facility, UGA). Signals for the predicted masses of acetylated MSX (m/z 221) and acetylated MSO (m/z 222) were observed using ESI-MS (data not shown). Liquid chromatography-tandem MS (LC–MS-MS) of the Ac-MSX signal resulted in a strong peak at m/z 142 (data not shown), indicative of the location of the acetyl group on the nitrogen bound to the alpha carbon.
S. enterica cannot use MSX or acetyl-MSX as a source of methionine.
To try to understand the fate of Ac-MSX, the ability of the compound to be utilized by S. enterica as a methionine source was tested. We used a methionine auxotroph that could acetylate MSX (i.e., strain JE6583 [metE mddA+]). Strain JE6583 was grown in minimal medium that lacked methionine (negative control) or that contained methionine (positive control; 100 μM), MSX (100 μM), or methionine and MSX (100 μM each). Growth was observed only when methionine was present (Fig. 7). These results indicated that neither MSX nor its acetylated form was used by strain JE6583 to generate methionine, or if generated, it was not to levels high enough to satisfy the methionine requirement for growth.
FIG 7.
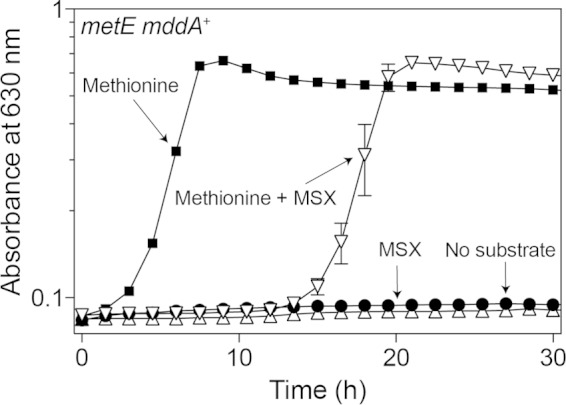
Neither MSX nor acetyl-MSX permits growth of a methionine auxotroph in S. enterica. Growth of an S. enterica strain auxotrophic for methionine (metE mddA+) was tested in NCE minimal medium (glycerol, 22 mM) in the absence of methionine or with methionine (100 μM), MSX (100 μM), or methionine and MSX (100 μM each). Growth curves were determined using a microplate reader (Bio-Tek Instruments) as described in Materials and Methods. The strain analyzed was JE6583 (metE205 mddA+). The error bars represent standard deviations.
An mddA+ strain displays biphasic growth at higher MSX concentrations with the addition of glutamine.
Growth of the mddA+ strain (JE10079) exposed to MSX with the addition of glutamine (200 μM) was examined at various concentrations of MSX (50 to 200 μM). Under these conditions, cultures of the mddA+ strain exhibited biphasic growth, with the initial growth onset occurring at the same time, followed by a plateau with an increasing lag phase. The observed lag in growth correlated with higher concentrations of MSX (greater than 5 μM MSX) (Fig. 8A). Interestingly, after the onset of growth, the growth rates for all conditions were similar (doubling times [h]: 1.4 [5 μM], 2.1 [50 μM], 2.4 [100 μM], and 3.2 [200 μM]).
FIG 8.

An mddA+ strain exhibits biphasic growth in minimal medium containing glutamine and MSX. (A) Growth of the S. enterica mddA+ strain in NCE minimal medium supplemented with glycerol (22 mM) was examined in the presence of glutamine (200 μM) and increasing concentrations of MSX (5 to 200 μM), as indicated. The strain analyzed was JE10079 (mddA+). (B) Growth of the S. enterica mddA+/pMDD8 mddA+ strain was examined in the same medium in the presence of MSX (50 μM) and increasing concentrations of inducer [l-(+)-arabinose; 50 to 500 μM], as indicated. The strain analyzed was JE18955 (mddA+/pMDD8 mddA+). Growth curves were determined using a microplate reader (Bio-Tek Instruments) as described in Materials and Methods. The error bars represent standard deviations.
Ectopic overexpression of mddA+ provides resistance to higher concentrations of MSX.
An mddA+ strain carrying mddA+ on a plasmid under the control of an inducible promoter (JE18961) was grown in minimal medium with MSX (50 μM) and various levels of inducer [50 to 500 μM l-(+)-arabinose]. At higher concentrations of the inducer, cultures of strain JE18961 grew with lag times shorter than those of the noninduced control (Fig. 8B). These data were consistent with the idea that increased SeMddA protein levels provided greater protection against the toxic effects of MSX.
Identification of genetic loci whose functions are required for MSX toxicity.
We took a genetic approach to find loss-of-function derivatives of an mddA1::cat+ strain (JE18333) that grew in the presence of MSX (10 μM). For this purpose, a P22 phage lysate grown on a pool of S. enterica mddA1::cat+ strains carrying Tn10d(tet+) elements randomly inserted in the chromosome was used as the donor to transduce the mddA1::cat+ strain to tetracycline resistance (for details, see Materials and Methods). Tetracycline-resistant (Tetr) colonies (∼50,000) were replica printed onto minimal medium supplemented with glycerol (22 mM), tetracycline (20 μg ml−1), and MSX (10 μM). A total of 22 mutant strains that grew under these conditions were analyzed further. To confirm that growth of the mddA1::cat+ strain was due to the inheritance of a Tn10d(tet+) element, P22 phage was grown on these strains to generate a phage lysate that was used as the donor to transduce the original mddA1::cat+ recipient strain to Tetr. Only one of the 22 MSXr mutants grew on medium containing MSX after reconstruction. The other mutants contained a mutation(s) not linked to the Tn10d(tet+) element. After freeing the reconstructed MSXr strain from phage, the location of the transposon insertion was identified by arbitrary PCR and subsequent DNA sequencing (46).
The transposon insertion in this strain was located within the glnP gene (insertion at bp 384 out of 660), which encodes the membrane component subunit of the glutamine ABC transporter GlnHPQ (Fig. 9A). The reconstructed mddA1::cat+ glnP1561::Tn10d(tet+) strain (JE20027) grew on minimal medium containing MSX (10 μM) (Fig. 9B, triangles). Identification of this mutant was not surprising, as the amino acid transporters MetNIQ (methionine permease) and GlnHPQ (glutamine permease) are responsible for transporting MSX in S. enterica. To our knowledge, transport of MSO has not been investigated (55).
FIG 9.
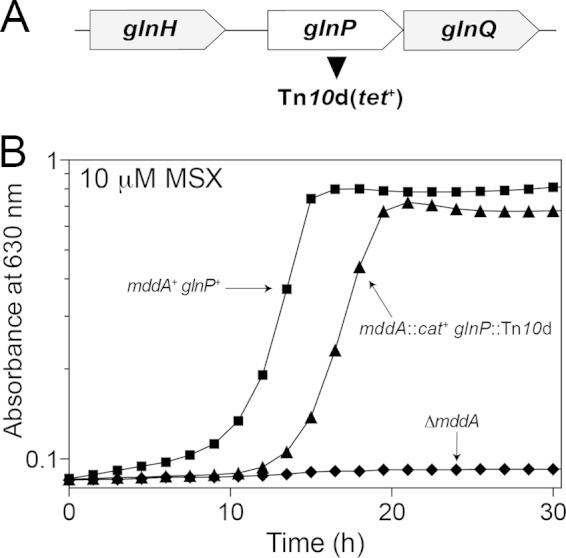
Growth of the mddA1::cat+ glnP1561::Tn10d(tet+) strain in the presence of MSX. (A) Transposon mutagenesis [Tn10d(tet+)] of an mddA1::cat+ strain determined that inactivation of glnHPQ (glutamine permease) restored growth in the presence of MSX (10 μM). (B) Growth of the reconstructed S. enterica mddA1::cat+ glnP1561::Tn10d(tet+) strain identified in the mutagenesis screen was examined in NCE minimal medium supplemented with glycerol (22 mM) and MSX (10 μM). Growth curves were determined using a microplate reader (Bio-Tek Instruments) as described in Materials and Methods. The following strains were analyzed: JE10079 (mddA+ glnP+), JE18622 (ΔmddA2), and JE20027 [mddA1::cat+ glnP1561::Tn10d(tet+)].
Deletion of two amino acid transport systems relieves MSX toxicity.
A deletion of glnP in combination with glnQ (ATP-binding subunit, strain JE20064) was constructed to ensure that the cell was devoid of transporter. Growth of the mddA+ and ΔmddA2 strains in the absence of GlnPQ was examined in medium containing MSX (20 μM) (Fig. 10A). A glnPQ1562::cat+ strain (JE20064) exposed to MSX reached stationary phase faster than the wild-type strain (∼10 h) (Fig. 10A, inverted triangles). A ΔmddA2 glnPQ1562::cat+ strain (JE20065) displayed an increased lag phase compared to the wild-type strain, but once the culture started growing, it grew at a rate similar to that of the glnPQ mddA+ strain (Fig. 10A, diamonds). Ectopic synthesis of glnPQ in the ΔmddA2 glnPQ1562::cat+ strain (JE20073) restored the apparent transport of MSX and abolished growth in medium containing MSX (Fig. 10A, circles).
FIG 10.
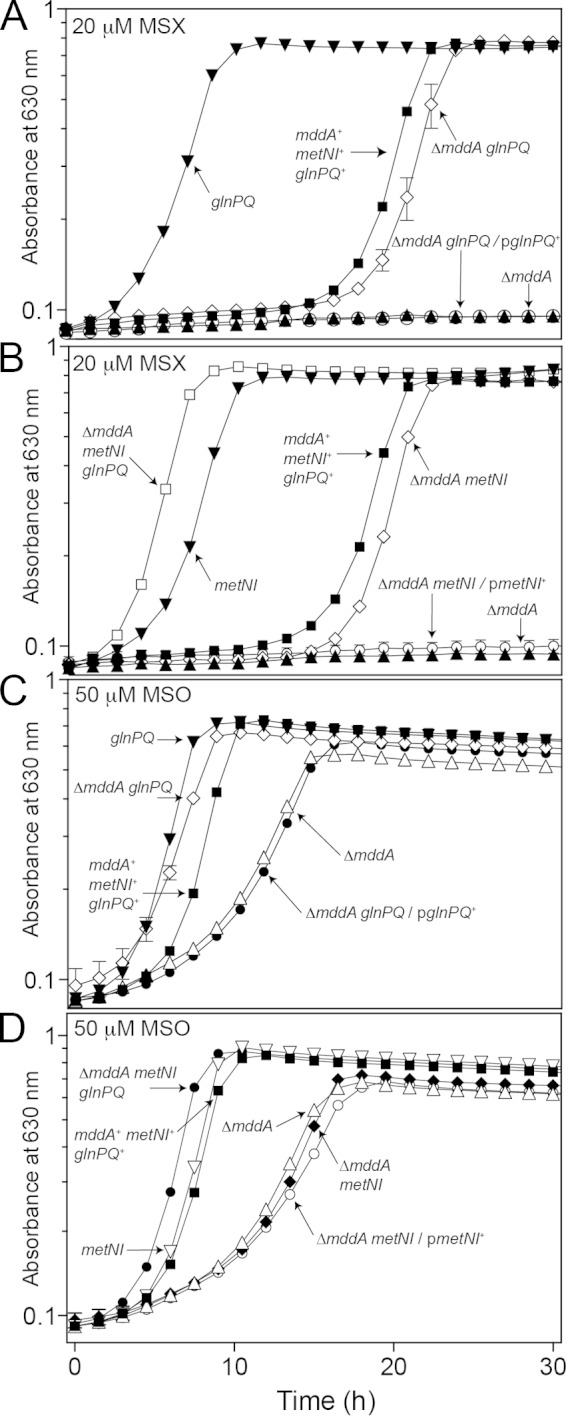
Deletion of two amino acid transporters (GlnHPQ and MetNIQ) restores growth of a ΔmddA2 strain exposed to MSX and MSO. (A to D) Growth of S. enterica mddA+ and ΔmddA2 strains with or without glutamine permease (glnPQ) or methionine permease (metNI) was examined in the presence of MSX (20 μM) or MSO (50 μM) in NCE minimal medium with glycerol (22 mM). Plasmids were induced with 10 μM l-(+)-arabinose. Growth curves were determined using a microplate reader (Bio-Tek Instruments) as described in Materials and Methods. The following were analyzed: mddA+ glnPQ+ metNI+ (JE10079), ΔmddA2 (JE18622), glnPQ1562::cat+ (JE20064), ΔmddA2 glnPQ1562::cat+ (JE20065), metNI2703::kan+ (JE19583), ΔmddA2 metNI2703::kan+ (JE19730), ΔmddA2 metNI2703::kan+ glnPQ1562::cat+ (JE20067), ΔmddA2 glnPQ1562::cat+/pGLN2 glnPQ+ (JE20073), and ΔmddA2 metNI2703::kan+/pMETN1 metNI+ (JE20329) strains. The error bars represent standard deviations.
Transport of MSX has been examined in S. enterica in relation to the transport of amino acids (Met) (56, 57). These studies demonstrated that the uptake of MSX and methionine sulfoxide was inhibited when both glutamine permease (GlnHPQ) and methionine permease (MetNIQ) were blocked. As seen in Fig. 10B, growth of a ΔmddA2 glnPQ1562::cat+ metNI2703::kan+ strain (JE20067) lacking both transporters was examined in the presence of MSX. Strain JE20067 grew better than the wild-type strain, with no observed lag when MSX was added to the medium (Fig. 10B, open squares). The ΔmddA2 metNI2703::kan+ strain behaved similarly to the ΔmddA2 glnPQ1562::cat+ strain (Fig. 10B, diamonds).
Ectopic synthesis of MetNI in the ΔmddA2 metNI2703::kan+ strain background also failed to support growth in the presence of MSX, confirming that GlnHPQ and MetNIQ transport MSX into the cell and that deletion of these two systems supports growth of an mddA strain on medium containing MSX (Fig. 10B, circles). It is interesting that in either the ΔmddA2 glnPQ1562::cat+ or the ΔmddA2 metNI2703::cat+ strain, growth occurred at a rate similar to that in the mddA+ strain, but with a longer lag phase.
MSO is transported by GlnHPQ but not by MetNIQ.
Similar growth experiments using the above-mentioned strains were carried out with the addition of MSO (50 μM) to investigate the transport of MSO (Fig. 10C and D). Deletion of glnPQ in a ΔmddA2 strain background (JE20065) restored growth to wild-type levels (Fig. 10C, diamonds), suggesting that a decreased amount of MSO was being transported into the cell. This effect was reversed when glnPQ+ were expressed in trans (Fig. 10C, solid circles).
Surprisingly, the kinetics of growth of the ΔmddA2 metNI2703::kan+ strain (JE19730) (Fig. 10D, diamonds) and the ΔmddA2 strain (JE18622) (Fig. 10D, triangles) in the presence of MSO were similar, demonstrating that deletion of the methionine transporter did not prevent inhibitory levels of MSO from entering the cell. Expression of metNI+ in trans in the ΔmddA2 metNI2703::kan+ strain (JE20329) (Fig. 10D, open circles) resulted in growth that was similar to that of the ΔmddA2 strain (Fig. 10D, triangles). Taken together, these results implied that, unlike MSX and methionine sulfoxide, MSO was not transported through the methionine permease.
DISCUSSION
SeMddA is necessary for cell survival in the presence of MSX in minimal but not in rich medium.
On minimal medium, S. enterica strains lacking MddA cannot grow in the presence of MSX; however, in rich medium, a higher concentration (5×) of MSX is needed to observe even a delay in growth (Fig. 2). This raises the question as to what enzymes or metabolites are present under rich medium conditions that help prevent MSX toxicity in the absence of MddA. One explanation could be differences in the availability of glutamine or methionine in rich versus minimal medium, since the addition of these compounds restored growth of a ΔmddA strain in the presence of MSX (Fig. 5). Other possibilities include increased expression of glnA (the gene encoding the enzyme known to be affected by MSX) and the presence of antioxidant compounds in rich medium.
SeMddA does not acetylate PHO.
SeMddA and its homologues have been annotated as PHO acetyltransferases (like the Bar protein of Streptomyces spp.), an activity that MddA homologues in several organisms do not posses (12, 16). Although PHO is inhibitory to S. enterica when present at 100 μM, it is clear that the deleterious effect of PHO is affected by the absence or presence of MddA (Fig. 4). In contrast, our data support the conclusion that a physiological role of MddA in S. enterica is to block the harmful effects of oxidized methionine derivatives, such as MSX and MSO (Fig. 2 and 8 and Table 1).
The fact that SeMddA can acetylate MSX and MSO but not PHO suggests a relatively high degree of specificity of the enzyme for structural analogues of its bona fide substrates (Fig. 2 and 4). There are, however, examples of homologues of the Streptomyces Bar protein that efficiently acetylate PHO and MSX. Such homologues are found in Streptomyces hygroscopicus and Rhodococcus spp. (24, 58). Whether PHO is acetylated by another S. enterica GNAT remains an open question.
Comparison of enzyme activities between MddA homologues.
The activity of an MddA-related homologue in P. aeruginosa, PITA (PA4866), has been investigated (12). The authors of the study observed a Km(app) of 1.3 mM for both MSX and MSO, while we observed much lower Km(app) values (MSX, 576 μM; MSO, 230 μM), with MSX having a 2-fold-higher Km(app) than MSO (Table 1). The activity of PITA versus MddA also differs in the turnover number. The kcat of PITA ranged from 505 s−1 (MSX) to 610 s−1 (MSO), while the kcat of MddA varied from 34.5 s−1 (MSX) to 30.6 s−1 (MSO), indicating that the PITA enzyme has a faster turnover than MddA. It is also interesting that the authors saw no inhibition of enzyme activity when using ∼20 mM substrate. We observed inhibition of MddA activity when the concentration of acetyl-CoA was >750 μM and when the concentration of MSO was >1,500 μM. Comparison of the catalytic efficiencies (M−1 s−1) of MddA when MSO (1.3 × 105) or MSX (6.0 × 104) was the substrate also suggests that MSO is a better substrate than MSX.
Expression of mddA is induced in response to MSX.
In the presence of glutamine with increasing concentrations of MSX, an mddA+ strain grows biphasically. Clearly, the addition of glutamine allows the initial onset of growth, but at a certain point, the cell either runs out of available glutamine or GlnA is inhibited by MSX, leading to growth arrest (Fig. 8). Not surprisingly, the higher the concentration of MSX, the longer growth arrest persists. We hypothesize that during growth arrest there may be an increase in the expression of mddA+ in response to the level of MSX. Once the MSX is acetylated and rendered innocuous, the cell can resume growth, a conclusion that is supported by the fact that growth rates remain similar when growth is restored.
How does a strain devoid of MddA and either transporter grow in the presence of MSX?
On the basis of the growth data presented (Fig. 9 and 10), we conclude that a deletion of only one of the MSX transport systems still allows inhibitory concentrations of MSX into the cell, which raises the question of how the mddA glnPQ and mddA metNI strains can survive the toxic effects of MSX. A plausible explanation is the existence of an enzyme with SeMddA-like activity whose kcat for MSX is slower than that of SeMddA but sufficient to support growth under such conditions. Examples of redundant functions in cells are not rare (e.g., MetE and MetH [methionine synthases] and PurN and PurT [glycinamide ribonucleotide transformylase]). It should also be noted that mutations in GlnA that prevent MSX binding and inhibition while retaining activity have been characterized (59, 60). It is also interesting that while both MSX and methionine sulfoxide are transported through the methionine permease MetNIQ, our data indicate that the structural analogue MSO is not (Fig. 10), shedding light on the specificity of the MetNIQ transporter.
Why does SeMddA acetylate oxidized methionine derivatives?
There is precedent for the role of GNATs in detoxifying toxic compounds (5, 11) by using acetylation as a means to inactivate antibiotics. While our data clearly demonstrate that MddA of S. enterica can prevent the deleterious effects of MSX and MSO (Fig. 2 and 8), it is less clear what environmental conditions expose S. enterica to MSX and MSO.
MSX has been identified in processed foods (in the 1940s) and occurs naturally in the roots and stems of some plant species (25). It is also possible that these compounds are produced as a consequence of the host response and reactive nitrogen or oxygen species (61) or are synthesized endogenously as a by-product of a normal metabolic process (62), which would not be unprecedented for S. enterica. Oxidation of methionine residues of proteins can generate methionine sulfoxide, but no studies looking at protein oxidation have detected the production of MSO (63–65). Although the sources of MSX and MSO are currently unknown, MddA function is necessary for S. enterica growth when these compounds are present. Future studies aim to understand the role of acetylation with respect to environmental stressors.
Supplementary Material
ACKNOWLEDGMENTS
We declare that no competing interests exist.
This work was supported by USPHS grant R01 GM062203 to J.C.E.-S.
We thank James Workman and Rachel Burckhardt for technical assistance.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02311-14.
REFERENCES
- 1.Thao S, Escalante-Semerena JC. 2011. Control of protein function by reversible N(epsilon)-lysine acetylation in bacteria. Curr Opin Microbiol 14:200–204. doi: 10.1016/j.mib.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernal V, Castano-Cerezo S, Gallego-Jara J, Ecija-Conesa A, de Diego T, Iborra JL, Canovas M. 2014. Regulation of bacterial physiology by lysine acetylation of proteins. N Biotechnol 31:586–595. doi: 10.1016/j.nbt.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka S, Matsushita Y, Yoshikawa A, Isono K. 1989. Cloning and molecular characterization of the gene rimL which encodes an enzyme acetylating ribosomal protein L12 of Escherichia coli K12. Mol Gen Genet 217:289–293. doi: 10.1007/BF02464895. [DOI] [PubMed] [Google Scholar]
- 4.Yoshikawa A, Isono S, Sheback A, Isono K. 1987. Cloning and nucleotide sequencing of the genes rimI and rimJ which encode enzymes acetylating ribosomal proteins S18 and S5 of Escherichia coli K12. Mol Gen Genet 209:481–488. doi: 10.1007/BF00331153. [DOI] [PubMed] [Google Scholar]
- 5.Wright GD, Ladak P. 1997. Overexpression and characterization of the chromosomal aminoglycoside 6′-N-acetyltransferase from Enterococcus faecium. Antimicrob Agents Chemother 41:956–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marvil DK, Leisinger T. 1977. N-Acetylglutamate synthase of Escherichia coli: purification, characterization, and molecular properties. J Biol Chem 252:3295–3303. [PubMed] [Google Scholar]
- 7.Fukuchi J, Kashiwagi K, Takio K, Igarashi K. 1994. Properties and structure of spermidine acetyltransferase in Escherichia coli. J Biol Chem 269:22581–22585. [PubMed] [Google Scholar]
- 8.Errey JC, Blanchard JS. 2006. Functional annotation and kinetic characterization of PhnO from Salmonella enterica. Biochemistry 45:3033–3039. doi: 10.1021/bi052297p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hung MN, Rangarajan E, Munger C, Nadeau G, Sulea T, Matte A. 2006. Crystal structure of TDP-fucosamine acetyltransferase (WecD) from Escherichia coli, an enzyme required for enterobacterial common antigen synthesis. J Bacteriol 188:5606–5617. doi: 10.1128/JB.00306-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikeuchi Y, Kitahara K, Suzuki T. 2008. The RNA acetyltransferase driven by ATP hydrolysis synthesizes N4-acetylcytidine of tRNA anticodon. EMBO J 27:2194–2203. doi: 10.1038/emboj.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolf E, Vassilev A, Makino Y, Sali A, Nakatani Y, Burley SK. 1998. Crystal structure of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase. Cell 94:439–449. doi: 10.1016/S0092-8674(00)81585-8. [DOI] [PubMed] [Google Scholar]
- 12.Davies AM, Tata R, Beavil RL, Sutton BJ, Brown PR. 2007. l-Methionine sulfoximine, but not phosphinothricin, is a substrate for an acetyltransferase (gene PA4866) from Pseudomonas aeruginosa: structural and functional studies. Biochemistry 46:1829–1839. doi: 10.1021/bi0615238. [DOI] [PubMed] [Google Scholar]
- 13.McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK. 2001. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- 14.Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 15.Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O'Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 16.Davies AM, Tata R, Snape A, Sutton BJ, Brown PR. 2009. Structure and substrate specificity of acetyltransferase ACIAD1637 from Acinetobacter baylyi ADP1. Biochimie 91:484–489. doi: 10.1016/j.biochi.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Vetting MW, de Carvalho LPS, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. 2005. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys 433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Deblock M, Botterman J, Vandewiele M, Dockx J, Thoen C, Gossele V, Movva NR, Thompson C, Vanmontagu M, Leemans J. 1987. Engineering herbicide resistance in plants by expression of a detoxifying enzyme. EMBO J 6:2513–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogawa Y, Tsuruoka T, Inoue S, Niida T. 1973. Studies on a new antibiotic SF-1293. II. Chemical structure of antibiotic SF-1293. Sci Rep Meijii Kaisha 13:42–48. [Google Scholar]
- 20.Murakami T, Anzai H, Imai S, Satoh A, Nagaoka K, Thompson CJ. 1986. The bialaphos biosynthetic genes of Streptomyces-Hygroscopicus—molecular-cloning and characterization of the gene-cluster. Mol Gen Genet 205:42–50. doi: 10.1007/BF02428031. [DOI] [Google Scholar]
- 21.De Robertis E, Sellinger OZ, Rodriguez DL, Alberici LM, Zieher LM. 1967. Nerve endings in methionine sulphoximine convulsant rats, a neurochemical and ultrastructural study. J Neurochem 14:81–89. doi: 10.1111/j.1471-4159.1967.tb09496.x. [DOI] [PubMed] [Google Scholar]
- 22.Rowe WB, Meister A. 1970. Identification of L-methionine-S-sulfoximine as the convulsant isomer of methionine sulfoximine. Proc Natl Acad Sci U S A 66:500–506. doi: 10.1073/pnas.66.2.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Circello BT, Eliot AC, Lee JH, van der Donk WA, Metcalf WW. 2010. Molecular cloning and heterologous expression of the dehydrophos biosynthetic gene cluster. Chem Biol 17:402–411. doi: 10.1016/j.chembiol.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maughan SC, Cobbett CS. 2003. Methionine sulfoximine, an alternative selection for the bar marker in plants. J Biotechnol 102:125–128. doi: 10.1016/S0168-1656(03)00028-2. [DOI] [PubMed] [Google Scholar]
- 25.Jeannoda VL, Rakoto-Ranoromalala DA, Valisolalao J, Creppy EE, Dirheimer G. 1985. Natural occurrence of methionine sulfoximine in the Connaraceae family. J Ethnopharmacol 14:11–17. doi: 10.1016/0378-8741(85)90023-6. [DOI] [PubMed] [Google Scholar]
- 26.Bentley HR, Booth RG, Greer EN, Heathcote JG, Hutchinson JB, Mohan T. 1948. Action of nitrogen trichloride on proteins; production of toxic derivative. Nature 161:126. [DOI] [PubMed] [Google Scholar]
- 27.Bentley HR, McDermott EE, Pace J, Whitehead JK, Moran T. 1949. Action of nitrogen trichloride (agene) on proteins; isolation of crystalline toxic factor. Nature 164:438. doi: 10.1038/164438a0. [DOI] [PubMed] [Google Scholar]
- 28.Bentley HR, Pace J, Whitehead JK, Moran T. 1949. Action of nitrogen trichloride on proteins; progress in the isolation of the toxic factor. Nature 163:675. doi: 10.1038/163675a0. [DOI] [PubMed] [Google Scholar]
- 29.Shaw CA, Bains JS. 1998. Did consumption of flour bleached by the agene process contribute to the incidence of neurological disease? Med Hypotheses 51:477–481. doi: 10.1016/S0306-9877(98)90067-6. [DOI] [PubMed] [Google Scholar]
- 30.Blin M, Crusio WE, Hevor T, Cloix JF. 2002. Chronic inhibition of glutamine synthetase is not associated with impairment of learning and memory in mice. Brain Res Bull 57:11–15. doi: 10.1016/S0361-9230(01)00631-1. [DOI] [PubMed] [Google Scholar]
- 31.Berkowitz D, Hushon JM, Whitfield HJ Jr, Roth J, Ames BN. 1968. Procedure for identifying nonsense mutations. J Bacteriol 96:215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balch WE, Wolfe RS. 1976. New approach to the cultivation of methanogenic bacteria: 2-mercaptoethanesulfonic acid (HS-CoM)-dependent growth of Methanobacterium ruminantium in a pressurized atmosphere. Appl Environ Microbiol 32:781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Way JC, Davis MA, Morisato D, Roberts DE, Kleckner N. 1984. New Tn10 derivatives for transposon mutagenesis and for construction of lacZ operon fusions by transposition. Gene 32:369–379. doi: 10.1016/0378-1119(84)90012-X. [DOI] [PubMed] [Google Scholar]
- 34.Davis RW, Botstein D, Roth JR. 1980. A manual for genetic engineering: advanced bacterial genetics, p 78–79. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 35.Schmieger H. 1971. The fate of the bacterial chromosome in P22-infected cells of Salmonella typhimurium. Mol Gen Genet 110:238–244. doi: 10.1007/BF00337836. [DOI] [PubMed] [Google Scholar]
- 36.Schmieger H, Backhaus H. 1973. The origin of DNA in transducing particles in P22-mutants with increased transduction-frequencies (HT-mutants). Mol Gen Genet 120:181–190. doi: 10.1007/BF00267246. [DOI] [PubMed] [Google Scholar]
- 37.Chan RK, Botstein D, Watanabe T, Ogata Y. 1972. Specialized transduction of tetracycline resistance by phage P22 in Salmonella typhimurium. II. Properties of a high-frequency-transducing lysate. Virology 50:883–898. [DOI] [PubMed] [Google Scholar]
- 38.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galloway NR, Toutkoushian H, Nune M, Bose N, Momany C. 2013. Rapid cloning for protein crystallography using type IIS restriction enzymes. Crystal Growth Design 13:2833–2839. doi: 10.1021/cg400171z. [DOI] [Google Scholar]
- 40.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rocco CJ, Dennison KL, Klenchin VA, Rayment I, Escalante-Semerena JC. 2008. Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid 59:231–237. doi: 10.1016/j.plasmid.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blommel PG, Fox BG. 2007. A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr Purif 55:53–68. doi: 10.1016/j.pep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM. 1961. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 44.Griffith KL, Wolf RE Jr. 2002. Measuring beta-galactosidase activity in bacteria: cell growth, permeabilization, and enzyme assays in 96-well arrays. Biochem Biophys Res Commun 290:397–402. doi: 10.1006/bbrc.2001.6152. [DOI] [PubMed] [Google Scholar]
- 45.Riddles PW, Blakeley RL, Zerner B. 1983. Reassessment of Ellman's reagent. Methods Enzymol 91:49–60. doi: 10.1016/S0076-6879(83)91010-8. [DOI] [PubMed] [Google Scholar]
- 46.Caetano-Anolles G. 1993. Amplifying DNA with arbitrary oligonucleotide primers. PCR Methods Appl 3:85–94. doi: 10.1101/gr.3.2.85. [DOI] [PubMed] [Google Scholar]
- 47.Ronzio RA, Rowe WB, Meister A. 1969. Studies on mechanism of inhibition of glutamine synthetase by methionine sulfoximine. Biochemistry 8:1066–1075. doi: 10.1021/bi00831a038. [DOI] [PubMed] [Google Scholar]
- 48.Liaw SH, Eisenberg D. 1994. Structural model for the reaction mechanism of glutamine synthetase, based on five crystal structures of enzyme-substrate complexes. Biochemistry 33:675–681. doi: 10.1021/bi00169a007. [DOI] [PubMed] [Google Scholar]
- 49.Steimer-Veale K, Brenchley J. 1974. Characterization of Salmonella typhimurium strains sensitive and resistant to methionine sulfoximine. J Bacteriol 119:848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wedler FC, Sugiyama Y, Fisher KE. 1982. Catalytic cooperativity and subunit interactions in Escherichia coli glutamine synthetase—binding and kinetics with methionine sulfoximine and related inhibitors. Biochemistry 21:2168–2177. doi: 10.1021/bi00538a028. [DOI] [PubMed] [Google Scholar]
- 51.Rowe WB, Ronzio RA, Meister A. 1969. Inhibition of glutamine synthetase by methionine sulfoximine. Studies on methionine sulfoximine phosphate. Biochemistry 8:2674–2680. [DOI] [PubMed] [Google Scholar]
- 52.Crespo JL, Guerrero MG, Florencio FJ. 1999. Mutational analysis of Asp51 of Anabaena azollae glutamine synthetase. D51E mutation confers resistance to the active site inhibitors L-methionine-DL-sulfoximine and phosphinothricin. Eur J Biochem 266:1202–1209. [DOI] [PubMed] [Google Scholar]
- 53.Brenchley J. 1973. Effect of methionine sulfoximine and methionine sulfone on glutamate synthesis in Klebsiella aerogenes. J Bacteriol 114:666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rowe WB, Meister A. 1973. Studies on inhibition of glutamine synthetase by methionine sulfone. Biochemistry 12:1578–1582. doi: 10.1021/bi00732a018. [DOI] [PubMed] [Google Scholar]
- 55.Betteridge PR, Ayling PD. 1975. The role of methionine transport-defective mutations in resistance to methionine sulphoximine in Salmonella typhimurium. Mol Gen Genet 138:41–52. doi: 10.1007/BF00268826. [DOI] [PubMed] [Google Scholar]
- 56.Kadner RJ. 1977. Transport and utilization of D-methionine and other methionine sources in Escherichia coli. J Bacteriol 129:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ayling PD. 1981. Methionine sulfoxide is transported by high-affinity methionine and glutamine transport systems in Salmonella typhimurium. J Bacteriol 148:514–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu GB, Yuan MR, Wei L, Zhang Y, Lin YJ, Zhang LL, Liu ZD. 2014. Characterization of a novel cold-adapted phosphinothricin N-acetyltransferase from the marine bacterium Rhodococcus sp strain YM12. J Mol Catal B Enzym 104:23–28. doi: 10.1016/j.molcatb.2014.03.001. [DOI] [Google Scholar]
- 59.Carroll P, Waddell SJ, Butcher PD, Parish T. 2011. Methionine sulfoximine resistance in Mycobacterium tuberculosis is due to a single nucleotide deletion resulting in increased expression of the major glutamine synthetase, GlnA1. Microb Drug Resist 17:351–355. doi: 10.1089/mdr.2010.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller ES, Brenchley JE. 1981. l-Methionine SR-sulfoximine-resistant glutamine-synthetase from mutants of Salmonella typhimurium. J Biol Chem 256:11307–11312. [PubMed] [Google Scholar]
- 61.Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, Roth JR, Baumler AJ. 2010. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467:426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lambrecht JA, Schmitz GE, Downs DM. 2013. RidA proteins prevent metabolic damage inflicted by PLP-dependent dehydratases in all domains of life. mBio 4:e00033–00013. doi: 10.1128/mBio.00033-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cabiscol E, Piulats E, Echave P, Herrero E, Ros J. 2000. Oxidative stress promotes specific protein damage in Saccharomyces cerevisiae. J Biol Chem 275:27393–27398. doi: 10.1074/jbc.M003140200. [DOI] [PubMed] [Google Scholar]
- 64.Drazic A, Winter J. 2014. The physiological role of reversible methionine oxidation. Biochim Biophys Acta 1844:1367–1382. doi: 10.1016/j.bbapap.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 65.Stadtman ER, Moskovitz J, Levine RL. 2003. Oxidation of methionine residues of proteins: biological consequences. Antioxid Redox Signal 5:577–582. doi: 10.1089/152308603770310239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.