

Applications of nanomaterials, including nanoparticles, in analytical chemistry continue to grow in recent years. Consequently, many excellent reviews have been published, covering different aspects of nanomaterials for chemical analysis.1–6 The present review focuses on applications of single nanoparticles in analytical chemistry. Detecting, counting, imaging, and tracking of single nanoparticles represent a critically important capability in analytical chemistry. In addition to revealing basic properties of nanoparticle that could be otherwise washed out in the ensemble analysis of nanoparticles, the capability of single nanoparticle analysis offers many unique applications. Due to limited space, this review includes mainly works published from 2011 to 2013.
Nanoparticles have been used in many analytical assays because of their extraordinary physical and chemical properties. Studies have shown that the physical (e.g., light emission and absorption7) and chemical (e.g., catalytic reactions) properties of a nanoparticle depend not only on its chemical composition, but also on its size and shape. For example, the optical responses of 10 different gold nanorods (AuNRs) modified with aptamers were found to vary by 3–4 times.8 In another study, highly anisotropic nanoparticles were found to be more sensitive to the binding to Hg2+ compared to the isotropic nanoparticles.9 By analyzing over 100 nanoparticles, Kim et al.10 discovered a correlation between the maximum extinction wavelength of a single AuNR and its sensitivity to a change in the surrounding refractive index. Yi et al.11 studied the evolution of optical scattering spectrum of Au nanoparticle-catalyzed reduction of 4-nitrophenol, and observed faster electron transfer rates in high-index elongated tetrahexahedral Au nanoparticles compared with those of low-index AuNRs. The size of a nanoparticle also affects its properties. For example, in a study of antibody binding to prostate-specific antigen-modified Au nanoparticles, smaller nanoparticles were found to be more sensitive than the larger nanoparticles.12 These results were consistent with the study of polarized nanoparticles, such as nanoplates13 and nanorods,14 whose corners and ends exhibited higher responses to ligand binding. These findings demonstrate a need for single nanoparticle analysis.
The present review contains four sections, covering sensing, counting, imaging and tracking of single nanoparticles, respectively. In the section of single nanoparticle-based sensors, we discuss chemical sensing applications, where the signal transduction or readout is based on detecting a physical property, such as optical absorption, of each individual nanoparticles. In contrast, in the section of counting single nanoparticles, signals from different nanoparticles are detected as a function of time, which are identified individually because they are separated in time domain. In the section of imaging chemical processes of single nanoparticles, individual nanoparticles are resolved spatially and analyzed by different imaging techniques. Finally, the last section is devoted to single nanoparticle tracking, which is based on the dynamic movements of single nanoparticles revealed by time resolved images.
CHEMICAL SENSING WITH SINGLE NANOPARTICLES
Single nanoparticle spectroscopy
Single nanoparticle spectroscopy measures the optical absorption of single nanoparticles originated from local surface plasmon resonance. The spectrum is sensitive to the chemical composition, morphology and size of each nanoparticle, and also sensitive to the refractive index of the medium near the nanoparticle surface. Because of the surface sensitivity, molecular recognition and chemical reactions taking place on the surface of the nanoparticle can be detected, which is one of the basic principles of nanoparticle-based chemical sensing. The optical absorption can be analyzed from the RGB values of the colored digital cameras,15, 16 or more accurately, with a spectrometer. Progress of this rapidly growing field has been summarized in several excellent reviews. For example, Van Duyne et al.17 reviewed single nanoparticle spectroscopy based-biosensing applications, and efforts towards single molecule detection and single cell imaging, as well as coupling of the plasmonic-based optical spectroscopy with other molecular identification technologies, such as Raman spectroscopy18 and mass spectroscopy.19 Sannomiya et al.20 focused specifically on biosensing applications using single plasmonic nanoparticles. Long et al.21 summarized in vitro and in vivo biological imaging applications of plasmonic nanoparticles.
Monitor molecular binding and chemical reactions
When a molecule binds to a single nanoparticle, the optical spectrum shifts, which is measured to detect the molecule. The selectivity relies on the modification of the nanoparticle with molecular receptors that can specifically recognize the target molecule, a principle that has been widely used in other chemical sensors. The sensitivity of the nanoparticle sensing platform arises from the localization of the evanescent field to the nanoparticle surface, and sensitive dependence of the optical spectrum on refractive index change within the evanescent field. Despite the high sensitivity, reaching single molecule detection is still challenging because of the small shift in the optical spectrum and relative broad spectral band. Single nanoparticles often exhibit much narrower spectral bands than the nanoparticle ensembles,7 which helps improve the sensitivity for the detection of molecules.
Sönnichsen et al.22 recently attempted to detect single protein molecule binding to nanorods by improving both optics and data processing. In their setup, the nanorods were illuminated by intense light with a total internal reflection configuration, and the scattered light was captured by an intensified CCD camera (Fig. 1a). The optical spectrum was fit with a model to determine the shift in the absorption band with a precision of 0.07 nm (Fig. 1b). Using this approach, discrete wavelength shift of 0.3 nm was reported, which was attributed to the individual binding events of single fibronectin molecules with a molecular weight of 450 kDa onto the single nanorod. The time resolution was a few milliseconds, allowing for continuous recording of the dynamic process of single fibronectin binding events. The same group took a step further to achieve simultaneous detection of multiple analytes in one microfluidic flow cell.23 In that work, individual AuNRs modified with corresponding analyte-targeting aptamers served as multiplexed nanosensors for 4 analytes. This single nanoparticle spectroscopy approach also enabled the sensitive detection of DNA molecules.24
Fig. 1.

Single molecule binding event resolved as a wavelength shift in single nanoparticle spectroscopy. (a) Single AuNRs were illuminated by white light and the scattered spectrum was analyzed by a dark-field microscopy coupled with a spectrometer. (b) The binding of a single protein molecule onto a AuNR was resolved as the wavelength shift in the scattered spectrum. Reprinted with permission from Ref. 22. Copyright (2012) American Chemical Society.
In addition to specific binding between a receptor and a target molecule, chemical reaction of a nanoparticle with a ligand is another way to achieve chemical selectivity. For example, the formation of Ag2S coating in the surface of Au-Ag core-shell nanoparticles was found to induce wavelength shifts in their scattering spectra, leading to selective and sensitive mapping of sulphide concentrations in single live cells.25
In order to amplify the spectral response with the geometry-dependent optical properties, several strategies have been used to change the nanoparticle morphology, such as seeded growth and the formation of satellite nanoparticles. For example, reduced nicotinamide adenine dinucleotide (NADH) was found to facilitate the deposition of a copper shell on Au nanoparticles in the presence of Cu2+.26 By detecting the shift in the maximum scattering wavelength, NADH concentration near a single Au nanoparticle was determined. In another work, the interaction of adenosine triphosphate (ATP) to a Au nanoparticle functionalized with ATP-binding aptamer induced the release of the aptamer molecules from the Au nanoparticle surface due to the formation of a rigid quadrupled structure.27 The resulted un-protected Au nanoparticle could catalyze its self-growth in the presence of certain reactants, leading to an enhanced wavelength shift by as large as 75 nm. Furthermore, Cu+ catalyzed click reaction between alkyne and azide was used to link alkyne-modified 14-nm Au nanoparticles to azide-modified 60-nm Au nanoparticles.28 The formation of satellite nanoparticles generated a significant shift in the maximum scattering wavelength, allowing for sensitive detection of Cu2+.
While single molecule detection limit is impressive, the number of detectable molecules per unit area is less impressive compared to other detection methods, such as the planar surface plasmon resonance. This is because the surface area of a nanoparticle is small. For example, the detection limit per unit area for single proteins22 achieved with a 40 nm-nanoparticle is about 100 pg/mm2, while the planar surface plasmon resonance can achieve a detection limit of <1 pg/mm2 (e.g., www.biosensingusa.com). The detection limit per unit area reflects the lowest detectable concentration target molecules in the solution, which is often more important than the total number of detectable molecules. However, single nanoparticle detection can provide localized information of analytes in a complex samples, such as cells.
Hydrogen gas sensing
Several methods have been proposed for hydrogen sensing based on the changes in the dielectric properties of metal nanoparticles upon hydrogen adsorption. The real-time monitoring on the scattering spectrum of single metal nanoparticle reveals the mechanisms of hydrogen adsorption and transition, and enables the study of the adsorption efficiency on the nanoparticle geometry. Because Pd adsorbs hydrogen strongly while Au nanoparticles exhibit the best plasmonic scattering features in the visible range, a Pd nanoparticle was placed in the near field of Au nanostructure so that hydrogen adsorption on Pd nanoparticle could be detected from the scattering spectrum of Au nanostructures (Fig. 2a).29 Using this strategy, the presence of 33 torr hydrogen gas induced a 10 nm shift in the maximum scattering wavelength of Au nanostructure (Fig. 2b) and the sensitivity was highly dependent on the Pd-Au distance as well as the geometry of the Au nanostructures.29 This is an example of using two materials to achieve a desired function, where one material serves as a recognition interface and another material provides signal transduction.
Fig. 2.
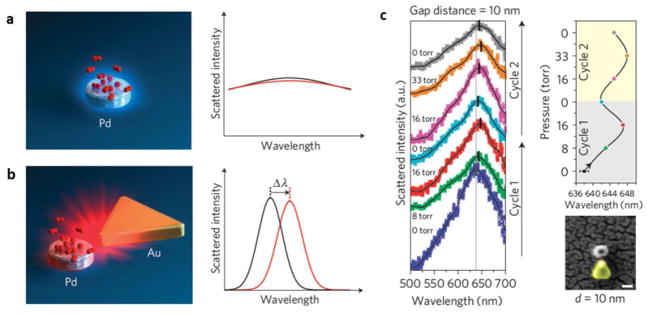
Hydrogen sensing with a single Pd nanoparticle coupled with a plasmonic Au nanostructure. (a) Hydrogen adsorption on Pd nanoparticle induces minimal wavelength shift in its own spectrum. (b) The wavelength shift is amplified in the presence of a Au nanoplate, which serves as a nano-antenna to detect the tiny change in its near field. (c) The maximum scattering wavelength in the single nanoparticle spectrum depends on hydrogen concentration. Adapted by permission from Macmillan Publishers Ltd:[Nature Materials] (Ref. 29), copyright (2011).
Similar hybrid nanostructures (e.g., Au/Pd core/shell nanoparticles) could be synthesized via convenient wet-chemistry.30 Another example is to coat Au nanostructures with a thin SiO2 shell (10 nm), and place the Au/SiO2 nanostructures on top of a Pd thin layer.31 Hydrogen adsorption onto the Pd layer near the Au/SiO2 nanostructure was detected from the plasmonic signals of the Au nanostructures. Au nanostructures were also capsulated by a platinized CdS shell.32 The CdS shell served as a photocatalyst to induce the decomposition of lactic acid to produce hydrogen, which was subsequently detected by single nanoparticle spectroscopy. Langhammer et al.33 used a different fabrication procedure to deposit a single Pd nanoparticle on the top of a Au nanocone with a thin SiO2 layer as spacer. Aizpurua et al.34 simulated light scattering during the formation of Pd hydride in single Pd nanodisk, which provided insights into the plasmonic sensing of hydrogen absorption. Schmidt et al.35 placed a plasmonic nanoparticle on top of a Fabry-Perot microcavity, which increased the sensitivity by ~36 times.
Electrochemistry of single nanoparticles
The impact of the electrode size on electrochemistry has been widely recognized,3 and electrochemistry of single nanoparticles has been carried out. One approach is to immobilize single nanoparticles on an ultramicroelectrode (UME). Chen and Kucernak36 developed a method to electrochemically deposit single Pt nanoparticles on a carbon UME with an electroactive area of 1 nm2. The area was small such that only one nucleation center could formed on the UME, which allowed synthesis of a single Pt nanoparticle on the electrode. They further investigated electrocatalytic reactions (e.g., hydrogen reduction) of the single nanoparticle.37,38 Zhang et al. immobilized pre-synthesized nanoparticles on nanoelectrodes.39,40,41 Although this approach involved an extra step, the size of the nanoparticles was better defined than that of the electrodeposited nanoparticles. Sun et al. studied the electrochemistry of single Au nanoparticles electrostatically adsorbed on a Pt nanoelectrode, and found that the energy required to achieve efficient oxidation of the Au nanoparticles increased when the size of the nanoparticle decreased.42 More recently, they synthesized a single Au nanoparticle on a Pt nanoelectrode under open circuit conditions.43 This approach allowed the immobilization of a single nanoparticle on an electrode without an organic protection layer, which reduced the influence of the protection layer on the electrochemical activities of the nanoparticle.
COUNTING SINGLE NANOPARTICLES
Colorimetric approaches
Plasmonic coupling between two or more metal nanoparticles changes the optical spectrum, thus the color, of the nanoparticle assembly, which has been used as a signal transduction mechanism to detect molecular binding events.44 This sensing mechanism has been studied at the single nanoparticle level by counting the individual nanoparticles. For example, using dark-field microscopy, 18-nm Au nanoparticles modified with single stranded DNA were resolved as green dots. Upon exposure to a DNA with complimentary sequence, nanoparticle dimers and oligomers formed, which changed the nanoparticles from green to yellow. The number of yellow dots indicated the number of hybridization events, allowing for the quantification of DNA molecules by counting the yellow emitters in the dark-field images.45 In a reversed approach, the nanoparticle-assemblies were cleaved into separated nanoparticles with an analyte, leading to blue-shift of the optical spectrum.46 A flash-lamp dark-field imaging was used to capture the transient image of Au nanoparticles, allowing analysis of molecular binding processes in free solution phase.47
Light scattering from individual polystyrene particles (100 nm in diameter) was detected in in a flow cytometer configuration using a hydrodynamic focusing technique.48 The size distribution of different particles was quantified by statistically analyzing the burst of light scattering from the particles. The technique was later applied to detect Au nanoparticles as small as 24 nm by enhanced light scattering.49 In order to distinguish Au and Ag nanoparticles in a mixed solution, incident light with wavelengths corresponding to the maximum extinction wavelengths of the Au and Ag nanoparticles was used in the detection.50 To increase the detection limit of single nanoparticles, dark-field scattering was combined with heterodyne interferometry, which allowed the detection of individual human viruses and bacteriophages as small as 24 nm.51
Non-optical methods have also been applied to detect and count nanoparticles. For example, inductively coupled plasma mass spectroscopy (ICP-MS) was used to detect single Au nanoparticles. When the Au nanoparticles passed the mass detection chamber, it resulted in burst signals associated with the individual nanoparticles. For DNA coated nanoparticles, hybridization induced aggregation of the nanoparticles. Consequently, the burst frequency decreased and the burst intensity increased. This DNA assay could reach a detection limit of 1 pM.52 The ICP-MS method was combined with hydrodynamic chromatography to further improve the capability of counting and characterizing the nanoparticles.53
Fluorescence resonance energy transfer (FRET) on single nanoparticles
Fluorescence-based detection is a major technique that can achieve single molecule detection capability. In order to perform the fluorescence measurements, the target molecules are either intrinsically fluorescent, or more commonly, labeled with fluorescent tags, such as organic dyes and fluorescent semiconductor nanoparticles, i.e., quantum dots (QDs). Due to its narrow emission band, tunable optical properties, high quantum efficiency, as well as the excellent photo-stability, QDs have been widely used as the fluorescent tags in many chemical and biological applications, such as ultrasensitive detection and molecular imaging. Single QD analysis of individual QDs was carried out by detecting transient light emission from the QDs.54, 55 In that work, a single QD was modified with a probe DNA via the biotin-streptavidin interaction, which were then linked to a Cy5-modified reporter DNA in the presence of target DNA (Fig. 3a).54 The close proximity between fluorescent Cy5 and QD in the resulted Cy5-reporter-target-probe-QD structure led to FRET, where QD served as a donor and Cy5 as an acceptor (Fig. 3b). The emission of each QD was simultaneously measured at the donor and acceptor emission wavelengths by two detectors (Fig. 3c). In order to resolve the hybridization of each individual QD and to minimize the photobleaching of Cy5 acceptor, the QD-containing solution was flown through a micro-capillary so that each QD was illuminated once for a short period of time. In such a configuration, the simultaneous appearance of discrete Cy5 and QD emission bursts served as an indicator of FRET and thus the DNA hybridization, while the sole QD emission represented the individual QDs without DNA hybridization (Figs. 3d and 3e). The subsequent counting of QDs with FRET allowed the determination of target DNA concentration with an improved sensitivity by 100 times.54
Fig. 3.
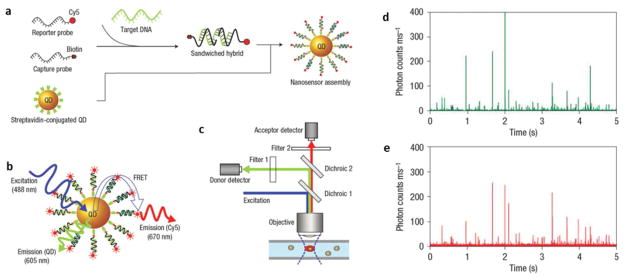
Fluorescence resonance energy transfer (FRET) on single QDs for quantitative DNA assay. (a) Schematic demonstration of the assembly of QDs. (b) QDs involved in the FRET process as a donor. (c) The existence of effective FRET on a single QD is shown as fluorescent bursts at both donor (d) and acceptor (e) emission wavelengths at the same time. Adapted by permission from Macmillan Publishers Ltd:[Nature Materials] (Ref. 54), copyright (2005).
The above approach was applied to the detection of various analytes. For example, using QDs that were modified with a RNA fragment, their specific binding to a peptide labeled with Cy5 was studied from the FRET signals.56 Another FRET-based application was to monitor the conformational change of cocaine-binding aptamer.57 Simultaneous detection of multiple analytes was also achieved by co-modifying two single stranded DNA probes on QDs, which could recognize different target DNA with respective dye labeling.58 In another report, miRNA was amplified and subsequently converted to the target oligonucleotides, which could be detected by the single QD-based FRET nanosensor, allowing a miRNA detection limit of 0.1 aM.59
More quantitative information can be obtained by analyzing the frequency and intensity of the discrete QDs’ fluorescent bursts. By counting the bursts during a certain period of time, one can estimate the concentration of QDs, leading to interesting applications.60 For example, by counting QDs in the presence of cation ions, Ca2+ and Mg2 at the physiological concentrations were found to induce aggregation of QDs.61 In another study, QD-encapsulated liposome was modified with probe DNA, and then immobilized on magnetic beads in the presence of target DNA. After magnetic separation, the release of QDs and subsequent single QD counting allowed detection of target DNA concentration as low as a few aM.62 Mattoussi et al.63 analyzed the ratio between donor and acceptor emissions for each QD and discovered two QD populations with different emission ratios. They modified QDs with maltose binding protein (MBP) labeled with rhodamine red as a FRET acceptor, and observed decreased fluorescence quantum yield of rhodamine red in the presence of maltose due to the MBP conformational change upon maltose binding. In another work by Opperwall et al.,64 tens of MBP-modified QDs immobilized on a glass substrate were monitored by total internal reflection fluorescence (TIRF) microscopy, where each QD served as a nanosized maltose sensor. Detection of maltose was achieved by analyzing the concentration-dependent emission from the individual QDs. Different QDs exhibited different sensitivities to maltose with a variation of up to seven orders of magnitudes, which was attributed to heterogeneous protein orientation and conformation after modification.
In addition to FRET, the coincidence emission of two QDs has also been utilized as an indicator of molecular interactions when the two QDs are modified with respective molecules in a recognition pair.65, 66 Each free QD exhibited its characteristic emission when it entered the detection volume in a micro-capillary. However, when two QDs were brought together by the molecular interactions and entered the detection volume, two bursts were simultaneously detected. This coincidence analysis was used to study molecular interactions and used in detecting mutations in DNA.65, 66 Note that QDs are ideal fluorescent tags in this method because of their narrow emission bands and broad excitation bands, allowing one to use one excitation wavelength to create emission at two distinct wavelengths without overlapping.
Examples described above are mainly based on the counting of single QDs in time domain as each QD passes through a detection volume confined by micro-capillary or evanescent wave. Using fluorescence imaging techniques, it is also possible to spatially count individual nanoparticles immobilized on a surface. For example, Zhang et al.67 immobilized a Cy5-labeled peptide to QDs, and observed FRET on each individual QD by dual-color fluorescence imaging. When renin was present in the solution, it cleaved the peptide and led to the release of Cy5 labels, which was observed as a decrease in the emission of FRET acceptor, Cy5. Wang et al.68 found that co-localization of two QDs in the coincidence emission method exhibited different combinations of colors, depending on the original colors and the assembly status, which allowed them to detect single molecule hybridization using a color camera.
Counting nanoparticle-electrode collision events electrochemically
In electrochemistry, the electrode current was typically recorded as a result of the Faradaic (e.g., electrochemical reactions) and non-Faradaic processes (e.g., electrical charging). Interactions or collisions of individual nanoparticles with the electrode surface result in transient changes in the current, which can be analyzed sequentially, leading to electrochemical counting and analysis of single nanoparticles.
Blockade of current by non-conductive nanoparticles
One straightforward attempt to observe single nanoparticle collision events on an electrode is to detect the blockade of the electrochemical reactions by non-conductive particles. This effect was demonstrated more than 10 years ago using sub-micron-sized non-conductive liposomes,69 where each collision event corresponded to a transient reduction in the current, leading to a negative peak in the current vs. time plot. However, since the area affected by each particle is small compared to the surface area of the entire electrode, the signal-to-noise ratio of the method is rather poor. By decreasing the size of the electrode, one could improve the signal-to-noise ratio. This was demonstrated using an UME with a diameter of 1 micron, where irreversible adsorption of individual carboxylated latex beads with a diameter of 300 nm led to large drops in the electrochemical current of ferrocenemethanol redox process.70 Recently, a systematic study with different types of non-conductive nanoparticles (silica and polystyrene) further confirmed that the blockade of the diffusion of redox molecules was responsible for the observed decrease in current.71 Experiments and numerical simulations also suggested that the nanoparticles tended to hit the electrode edges more frequently because of the enhanced electrical field at the edges. Such edge effect was directly visualized in a recent study by observing the individual nanoparticles with an optical microscope.72 Although useful, this approach faces difficulty for small nanoparticles (e.g., < ~100 nm).
Boosted current by catalytic nanoparticles
A new strategy to count single nanoparticles was reported by Bard et al.73, 74 In their approach, a single nanoparticle collision led to a well-resolved increase in the electrode current, which is in contrast to the decreased electrochemical current due to blocked mass diffusion by insulating nanoparticles. The increase in the current was due to electro-catalytic reaction of the metal nanoparticles. In the first demonstration of this strategy, the collision events of individual Pt nanoparticles onto a carbon microelectrode exhibited stepwise increases in the electro-oxidation current of hydrazine.73 Detailed analysis of the current profiles revealed a correlation between collision frequency and strength with the properties of nanoparticles, such as concentration and diffusion coefficients.74 This single nanoparticle electrochemistry method was further expanded to different electrochemical reactions for various nanoparticles and electrodes.3, 75, 76 In addition to recording the current response under a constant electrode potential, single nanoparticle collision was also observed under a potentiometric design by measuring the open-circuit potential of an inert electrode.77
It has been realized that the ways of the nanoparticles interact with electrode greatly affect the associated current profiles, leading to either spikes (transient change) or stepwise increases (permanent change) as shown in Fig. 4a. Hit-n-run nanoparticles would contribute a current spike due to the limited time of residence78 and the permanent adsorption of nanoparticles would generate stepwise current increases.74 However, more complicated mechanisms have been identified recently. For example, deactivation of adsorbed nanoparticles could distort the current profile, resulting in a fast current change followed by gradual decay.79, 80 The deactivation was attributed to the oxidation of nanoparticles by oxygen, a product of the catalytic reaction. In contrast, the collision of Au nanoparticle-decorated single wall carbon nanotubes onto a Pt nanoelectrode led to well-defined stepwise current responses without decay because of the absence of deactivation mechanisms.81 Deactivation of the adsorbed nanoparticles could maintain the background current for continuous counting of nanoparticle collision with the electrode.80 This was shown with an Hg modified Pt UME (Hg/Pt UME), on which Pt nanoparticles catalyzed the oxidation of hydrazine after the collision. Once the Pt nanoparticles hit and adsorbed on the Hg/Pt UME, Hg atoms interacted with the nanoparticles to form a Hg-Pt alloy, which deactivated the Pt nanoparticles and quenched the catalytic current. Consequently, the current returned to the level before Pt nanoparticle collision, leading to a well-defined peak with stable baseline (Fig. 4b).
Fig. 4.

Single nanoparticle collision-based electrochemistry. (a) A transient “hit-n-run” collision usually generates a peak in the electrochemical current profile, while the permanent “hit-n-stand” collision results in stair-wise features. (b) The collision of Pt nanoparticle on an Hg/Pt electrode leads to the rapid formation of Hg/Pt alloy and effectively regenerates the electrode surface. Adapted with permission from 80. Copyright (2013) American Chemical Society.
In order to achieve sensitive recognition of single nanoparticle collision events, the size of the electrode in general must be small enough to reduce the background current. Unwin et al. 82 described a different approach to reduce background current. They used a micropipette to form a micro-scale contact between the solution-containing micropipette and a carbon electrode surface. The small electro-active area at the contact helped reduce the background current, and allowed them to observe catalytic current due to the collision of individual nanoparticles. Using a carbon-coated copper grid electrode, they were able to examine the nanoparticles after collision with transmission electron microscopy (TEM), allowing the study of size-activity relationship.
Further investigations were devoted to the driving forces behind the nanoparticle collisions. In a recent scanning electrochemical microscopy study,79 the random walk model was found to work well to describe the nanoparticle collisions with the electrode at high ionic strengths. Analysis on the random walk model was also applied to explain the diffusion-controlled movement of nanoparticles in an earlier study.83 However, directional migration of charged nanoparticles under an electrical field was found to play a role at low ionic strengths, affecting the collision efficiency and strength of both insulating71 and conductive nanoparticles.84 Specific molecular interactions, such as DNA hybridization, were introduced to induce the nanoparticle collision in a more controllable way.85 In this approach, Pt nanoparticles were functionalized with single-stranded DNA and the electrode was modified with a DNA with complementary sequence. The specific DNA hybridization led to the binding of the nanoparticles to the electrode surface, which was detected by the catalytic current of the Pt nanoparticles. Analysis of the current profiles revealed information regarding the frequency and kinetics of single DNA hybridization events.85 Similar approach was utilized in a recent study to achieve the sensitive detection of single DNA molecules.86
Direct electro-oxidation current from electro-active nanoparticles
In addition to detecting catalytic current associated with single nanoparticle collisions, Compton’s group focused more on the direct electrochemistry of single electro-active nanoparticles as summarized in the review paper published by this group.76 The initial attempt was made by measuring the Faradaic current from individual Ag nanoparticles as they collided with the electrode.87 The systematic studies of metal nanoparticles, such as Ag,87 Cu88, and Ni89, revealed quantitative relationship between nanoparticle concentration and the collision frequency. In order to avoid the oxidative destruction of nanoparticles, an electro-active tag, which was usually redox molecule with a lower formal potential such as 1,4-nitrothiophenol, was immobilized on the Ag nanoparticles to act as a sacrificial reagent.90 By analyzing the potential-dependent collision frequency and strength, the electron transfer kinetics of metal nanoparticles could be studied on single nanoparticle basis,91 which is useful for the development of catalytic materials.92 This method was recently expanded to organic nanoparticles.93 For example, the random collision of single electro-active indigo nanoparticle onto a carbon microelectrode was found to generate a transient reductive Faradaic current that depended on the size of the nanoparticles, allowing the measurements of the size distribution of organic nanoparticles.93
One of the goals of such collision-based electrochemistry is to study the electro-catalytic or electrochemical activities on single nanoparticle level. Although promising, analysis based on the current profile alone cannot easily answer some of the important questions. For example, what is the reason for the observed variations in the current response of the individual collisions? The variations in the current were generally larger than the variations in geometrical parameters (size and morphology, aggregation status) and optical properties (such as optical absorption and scattering). The collision of nanoparticles with the electrode was stochastic, and multiple factors, such as the speed and the orientation of each nanoparticle, could affect the charge transfer during the collision.72 More well-defined surface chemistry80 and controllable nanoparticle-electrode interactions85, 86 would minimize the variations and provide clear interpretations to the electrochemical current profiles.
IMAGING CHEMICAL PROCESSES OF SINGLE NANOPARTICLES
Surface plasmon resonance (SPR) microscopy
SPR imaging technique has been developed long time ago to study molecular binding in a microarray format.94 Traditional SPR imaging systems are mainly prism-based, which has limited spatial resolution. Using a high numerical aperture objective-based SPR microscope,95 we recently demonstrated the imaging of single viruses and silica nanoparticles attached on a Au film.96 Each individual virus or nanoparticle was imaged as a V-shape pattern, which was attributed to the scattering of the surface plasmon waves by the virus or nanoparticle. The high spatial resolution capability was combined with a plasmonic-based electrochemical imaging technique97 to image the electrocatalytic reactions of single nanoparticles.98 The principle of the plasmonic-based electrochemical current microscope (P-ECM) is that the Faradaic current is determined by the generation rate of reaction products (or the consumption rate of reactants) in an electrochemical reaction process, which can be imaged by SPR because of the sensitive dependence of SPR signal to the local concentration changes of reactants or products.97 Based on this principle, the local Faradaic current was expressed in terms of the SPR signal, from which local cyclic voltammogram of the electrode surface was obtained. Such capability was utilized to study the electro-reduction of hydrogen on single Pt nanoparticle.98 The production of hydrogen molecules under the appropriate reduction potential resulted in an obvious change in the local SPR intensities (Figs. 5a–f). Based on a theoretical model between SPR intensity and current density, the voltammogram of hydrogen evolution on a single Pt nanoparticle was resolved (Fig. 5g). This method allowed measurement of cyclic voltammograms of multiple Pt nanoparticles individually, providing an unprecedented capability of studying single nanoparticle electrochemistry.98
Fig. 5.

Plasmonic-based electrochemical current images of a single 80-nm Pt nanoparticle at different potentials during a cyclic voltammetry scan. (a–f) Current density images of a single 80 nm Pt nanoparticle at potentials of − 0.05, −0.36, −0.4, −0.5, −0.4 and −0.05 V, respectively. (g) Typical cyclic voltammograms of three different single Pt nanoparticles resolved by P-ECM. Scale bars (a–f): 3 μm. Adapted by permission from Macmillan Publishers Ltd:[Nature Nanotechnology] (Ref. 98), copyright (2012).
Single molecule fluorescence on single nanoparticles
Chen and co-workers99, 100 developed a single molecule fluorescence imaging method to map catalytic activity distribution and its temporal evolution on single or sub-nanoparticle level. Individual nanoparticles were sparsely immobilized on a glass surface to catalyze a fluorogenic chemical reaction that can convert a non-fluorescent reactant molecule to a fluorescent product molecule. This process was recorded as the sudden appearance of fluorescence emission by using a single molecule fluorescence microscopy (Fig. 6a). A continuous flow was applied to accelerate the release of product molecules from the nanoparticles after a short period of stay, leading to a sudden disappearance of the fluorescence emission and an un-occupied nanoparticle for the next chemical reactions. In the first work studying the Au nanoparticle-catalyzed fluorogenic reactions from non-fluorescent resazurin to highly fluorescent resorufin by NH2OH, quantized fluorescence bursts were observed from the fluorescence intensity trajectory around the location of the Au nanoparticles (Fig. 6b).99 Several control experiments confirmed that each fluorescent burst represented the generation of one resorufin molecule, and thus one chemical reaction, indicating the real time monitoring of single chemical reaction events on different individual catalyst in parallel. Further data analysis on the intensity trajectories over time revealed interesting reaction kinetics on each of the single nanoparticles.101 This method provided valuable information of the heterogeneous catalytic activity of different nanoparticles and complex reaction kinetics of the same nanoparticle.102, 103
Fig. 6.
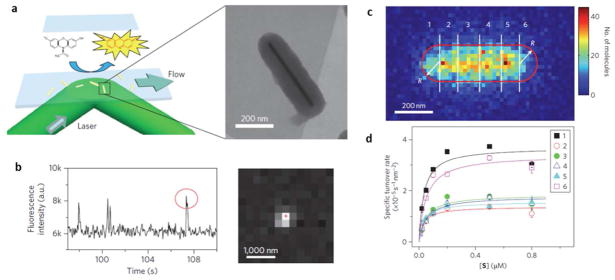
Super-localization of single fluorescent molecules reveals the reactivity of single nanoparticles. (a) The fluorogenic reaction converts a non-fluorescent molecule (reactant) to a fluorescent molecule (product). (b) Such single molecule reaction results in the appearance of a single molecule fluorescence pattern and a burst in the fluorescence emission at certain location. (c) By counting the number of single molecule reaction events across the Au nanorod, one can map the sub-particle distribution of reactivity. (d) Substrate concentration-dependent turn-over rate in six sub-particle regions as defined in (c) exhibited quite different reaction kinetics. Adapted by permission from Macmillan Publishers Ltd:[Nature Nanotechnology] (Ref. 104), copyright (2012).
Gaussian fitting was applied to precisely locate each single product molecule, e.g., the catalytic site, with a spatial resolution better than the optical diffraction limit, allowing super-resolution imaging of catalytic reactions within single nanoparticles.104–106 A spatial resolution of ~40 nm was achieved on a single mesoporus silica-coated Au nanorod, which exhibited catalytic activity to the oxidation of Amplex Red.104 Significantly different catalytic activities were found between the ends and the center of the nanorod. It was believed that heterogeneous surface defects mainly contributed to the non-uniform catalytic activity distribution. A subsequent study on single Au nanoplate further discovered that the corner regions exhibited higher catalytic reactivity than edges and center regions.107 Mapping of the reactive sites on a single TiO2 nanoparticle decorated by 14-nm Au nanoparticle was achieved by tracking fluorescence bursts with accuracy of 20 nm.105 A comparative study on the plain and decorated TiO2 nanoparticles revealed that the catalytic reactions tended to occur around the deposited Au nanoparticle on the decorated TiO2 nanoparticle. Recently, the localization accuracy was further improved to 10 nm by modifying the chemical structure of the fluorescent probe, which enhanced the fluorescence quantum efficiency as well as the lifetime, leading to an improved signal-to-noise ratio even under lower fluorophore concentration.108
Size-dependent catalytic activity of nanoparticles is always of interest for clarifying catalysis mechanisms and for developing novel catalysts. In earlier studies, three sets of Au nanoparticles with different sizes pre-determined by TEM were immobilized on the substrate respectively to explore the relationship between catalytic kinetics and nanoparticle size.109 Although useful, this method did not take full advantage of its capability to resolve individual nanoparticles. In a recent work, scanning electron microscopy was applied to characterize the very same nanoparticles on the substrate after performing the catalytic reactions.110 A clear relationship was established between the size and catalytic activity by statistically analyzing over 1000 nanoparticles. The sub-population in a mixture of different catalysts could be easily determined and mapped by this method, leading to an excellent screening platform to select the specific catalyst of interest.
One of the challenges of this method is the restriction on the types of chemical reactions that can be studied. First, the reactions must be fluorogenic. Second, the fluorescent products must dissociate from the nanoparticle surface after a short period of stay. Third, the catalyst should neither enhance nor quench the fluorophore to avoid the complications in the data interpretation. The last point is a concern, particularly for metal nanoparticles, which are known to affect fluorescence emission. Orrit et al.111 recently reported that the single molecule fluorescence near a single nanorod could be enhanced by hundreds of times if the fluorophore spectrum matched the surface plasmon of the nanorod. In order to generalize this method, Majima explored the versatile chemical synthesis to combine a fluorophore moiety and a redox moiety such as catechol112 and dinitrophenyl,105, 108 in which the fluorescence quantum efficiency of the fluorophore was regulated by redox reaction via intra-molecular electron transfer. Chen et al.110 studied the activity correlation between the classical fluorogenic reactions and commonly-used model redox reactions, such as the reduction of 4-nitrophenol and the oxidation of hydroquinone. If a correlation between these model reactions were established, one could then investigate many kinds of catalysts using the classical fluorogenic reactions as standards.
TRACKING SINGLE NANOPARTICLES
Techniques for single molecule tracking have been applied to the study of single nanoparticles.113–115 By labeling specific membrane components, such as membrane proteins or phospholipids, with nanoparticle tags, one can obtain important mechanical and biochemical information of the cell membrane and its components (Fig. 7). Another application of nanoparticle tracking is related to the molecular imaging and clinical therapy where the nanoparticles act as contrast enhancers and drug carriers. By accurately tracking the nanoparticles, nanoparticle transport in and out of the cells, and its interactions with the cellular components can be monitored and studied. In these applications, fluorescent QDs and plasmonic nanoparticles are two of the most widely used nanoparticles.
Fig. 7.
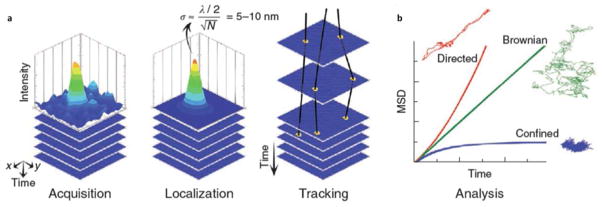
Single nanoparticle tracking reveals the mechanical and biochemical properties of cell membranes and membrane components. (a) The tracking starts with image acquisition of a nanoparticle, localization of the nanoparticle by fitting of the image profile with a Gaussian function, and tracking of the nanoparticle over time. (b) Different types of movement revealed by the tracking method. Reprinted by permission from Macmillan Publishers Ltd:[Nature Methods] (Ref. 115), copyright (2010).
Tracking single fluorescent nanoparticles
The classic fluid-mosaic model of cell membrane considers that the plasma membrane of animal cells consists of membrane components, such as proteins and cholesterols, embedded in a uniform and fluidic lipid bilayer. However, evidence has shown that the proteins and lipids distribute heterogeneously in the membrane, as protein microdomains116, 117 and lipid rafts.118 By labeling the Glycosyl-phosphatidylinositol (GPI)-anchored protein in a cell membrane with QDs and then analyzing the trajectory of a single QD, it was observed that the QDs in some sub-micron size membrane regions diffused much slower than in other regions.118 The regions with slow QD diffusion overlapped with the locations of glycosphingolipid GM1-rich microdomains by staining the microdomains. Direct labeling of glycosphingolipid GM1 with QDs was recently achieved using streptavidin-conjugated QDs bound to biotinylated protein cholera toxin B subunits, which were then selectively attached to glycosphingolipid GM1.119 This allowed continuous observation of confined diffusion of QDs in the microdomains, and the results suggested a persisting time as long as tens of seconds for the individual lipid rafts, which could be important for cellular signaling processes.
QDs have been used to track individual membrane receptors and their dynamics via antibodies that bind to the receptors.120 By analyzing the diffusion pattern of the glycine receptors over time, some receptors were found to change between a free diffusion mode (larger diffusion coefficient) and a space-confined diffusion mode (smaller diffusion coefficient), which were subsequently attributed to the perisynaptic and synaptic locations, respectively. In the study of individual GABAA receptors on the membrane of a spinal neuron, the QD trajectory was found to consist of a Brownian diffusion and a directed movement with a velocity of 0.29 μm/sec,121 which was associated with the elongation of microtubule. A recent study compared the different diffusion patterns of nicotinic acetylcholine receptor (nAChR)-bound QDs at different subcellular locations in a single neuron, and discovered the local confinement of receptors and the reduced mobility and longer dwell time at perisynaptic locations.122 QDs were functionalized with small molecule antagonists targeting the serotonin transporter.123 A population of serotonin transporters was found to display restricted mobility in the cholesterol and ganglioside GM1-enriched microdomains, supporting the model of serotonin transporter-containing lipid rafts. Dumas et al. further studied the restricted mobility of CD4 receptors in the lipid rafts under different temperatures.124 Three types of motions including free diffusion, directional transportation and confined diffusion were found when analyzing the trajectory of a QD-bound prion protein, depending on the distinct phases during prion protein endocytosis.125
In addition to membrane proteins and lipids, intracellular proteins and organelles have also been studied with QDs. The direct labeling of nerve growth factor (NGF) with QDs and subsequent analysis of the movement enabled the study of NGF retrograde transport from axon terminal to the soma.126 Xie et al. used QDs as probes to track endocytic vesicles, and observed 8-nm discrete steps during the active transport of the vesicles along microtubule, revealing the underlying actions of the motor proteins, such as kinesin and dynein.127 Labeling a QD to the head or tail of a dynein was achieved in a recent work, which allowed the observation of 16-nm stepwise movement of single dynein molecules along the microtubule.128 The simultaneous labeling of both the head and the tail in the work further allowed the analysis on the interhead separation during the movement, leading to new insights into the stepping of the motor proteins. The rotational movement of a motor protein could also be studied with a polarized rod-shaped QD by analyzing the fluorescence intensities along four distinct polarization directions.129 This approach led to the observation of a 90-degree rotation of myosin V around its own axis in addition to the translation stepping movement. Another approach to simultaneously track the position and orientation of a single virus with QDs was to combine scattering interferometry130 with single QD fluorescence microscopy.131 Statistical analysis of the tracking data to extract the step size and dwell time is critical in these studies. A computational algorithm was proposed recently for the analysis based on finding patterns in trajectory compatible with expected behavior of motor proteins.132
Other fluorescent nanoparticles, such as fluorescent polymer nanoparticles, have also been used to track the movement of membrane receptors. 15-nm polymer nanoparticles composed of highly fluorescent conjugated polymers was utilized to stain fixed cells, and the trajectory of single nanoparticles revealed different diffusion patterns on the cell membrane and inside the cytoplasm.133 A recent work used hydroxyl-terminated conjugated polymer nanoparticles to specifically label the insulin-like growth factor 1 receptor. The restricted diffusion was observed on cholesterol-depleted cells treated by cyclodextran, demonstrating the great impact of cholesterol on the receptor mobility in the cell membrane.134
A two-circle scan method was reported to provide the feedback for 3-Dimensional (3-D) tracking of single fluorescent polystyrene nanoparticle in a well-defined oil-in-water 3-D interface.135 The 3-D trajectory reproduced the surface profile of an oil droplet. This method was subsequently used to track the 3-D diffusional patterns of a single streptavidin-conjugated QD before and after its binding to biotinylated DNA origami.136 Although the interaction with origami did not affect the fluorescence intensity of single QDs, the trajectory analysis exhibited significantly slower diffusion for the origami-bound QD. Further combination with single-photon statistics allowed the determination of the stoichiometry of the QD binding on origami with multiple QD binding sites.
TIRF imaging offered another opportunity to precisely determine the vertical distance of a single nanoparticle, because the fluorescence intensity is a function of its vertical distance to the substrate. Except for the intrinsic limitation on the detection depth of ~200 nm, the main challenge is that the absolute emission intensity was affected by several factors, such as quantum yield, surrounding medium, photobleaching and light source fluctuations. A recent work used the relative intensity rather than the absolute intensity to obtain vertical distance by scanning the incident angle.137 Based on the dependence of evanescent field on incident angles, vertical position of a single QD was determined with sub-10 nm precision, allowing for 3-D tracking of the QDs.137
Tracking single plasmonic nanoparticles
Dark-field microscopy has been used to track single plasmonic Au138, 139 and Ag140 nanoparticles. Compared to QDs, the plasmonic nanoparticles avoid the intrinsic blinking effect and cytotoxicity. By attaching Au nanoparticles to solid supported lipid bilayer via electrostatic interactions, 90% nanoparticles was found to freely diffuse on the lipid layer in the absence of ganglioside GM1.138 However, increasing GM1 concentration in the lipid layer led to a linear drop in the percentage of free Au nanoparticles. The results suggested the possible roles of GM1 in the formation of lipid rafts. In another work, decreased Au nanoparticles mobility was found in the presence of cholera toxin B subunit (CTB) because CTB was capable to bind to multiple GM1 molecules and led to a confined Au nanoparticle movement.139 Xu et al.140 monitored the uptake and transport of single Ag nanoparticles in embryos. From the analysis on trajectory, they concluded that random Brownian motion was responsible for the passive endocytosis of single Ag nanoparticles. A follow-up work on Au nanoparticles by the same group discovered that Au nanoparticles exhibited less toxicity than Ag nanoparticles with the same size.141
By taking advantage of the shape-tunable anisotropic optical properties of AuNRs, multiple microscopic techniques, including dark-field polarization microscopy142–144, photothermal imaging,145 multifocal two-photon microscopy146 and planar illumination microscopy147 were developed to study the movement patterns of single AuNRs. Using differential interference contrast (DIC) microscopy, Fang et al.148 recently developed a single particle orientation and rotational tracking method to measure and track the in-plane orientation and out-plane rotation of AuNR in artificial membrane and live cells. The conjugation of AuNR with certain proteins allows the investigation on the rotational behaviors of single proteins during many biological processes, such as the rotational walking of motor proteins. The DIC image of a single AuNR illuminated by light with wavelength that matched its longitudinal absorption exhibited bright and dark patterns depending on the orientation of the rod. AuNRs functionalized with cell-penetrating peptide allowed internalization of the AuNRs by live cells and formation of nanorod-containing vesicles. Real time measurement of the AuNR orientation revealed the rotational movement of motor proteins when the vesicle walked along microtubule.149 Fang et al.150 further discovered the influence of surface modifications of AuNRs on their rotational movement patterns, and concluded that although positive surface charge facilitated the initial adsorption of nanorod onto the cell membrane, the presence of transferrin protein specifically targeting membrane receptors was mostly responsible for the endocytosis process.
Several efforts have been made to further improve the performance of DIC imaging of AuNRs. Since the absolute bright/dark intensity in the DIC images could be easily affected by the unstable light source and vertical vibrations of AuNR, polarized DIC imaging was proposed, where intensity ratio between two orthogonally polarized images of each AuNR was used.151 Accurate determination of polar angle and analysis of polarized images enabled the identification between clockwise and counterclockwise rotations.152 Light with wavelength tuned to the longitudinal absorption band of AuNR is mostly used in the DIC measurements because it gives stronger DIC signals. However, longitudinal absorption band is also very sensitive to the morphology of the AuNR and the refractive index of the surround medium, resulting in fluctuating signals in complex biological media. For this reason, light with wavelength tuned to the transverse absorption band was used to track AuNRs a live cell membrane, which showed much less sensitivity to the environment.153 The effect of polarization on the quantitative analysis of AuNR orientation was discussed under both plasmonic and non-plasmonic wavelengths.154 The orientation analysis of the AuNRs with DIC microscopy described has been combined with position tracking with a lateral precision of better than 10 nm.155,156 These approaches have been applied to track both rotational and translation dynamics of AuNR-containing cargos along axons157, and membrane receptors.158,159
PERSPECTIVE
Nanoparticles are interesting because they possess many unique properties, such as electronic, optical, plasmonic, magnetic, mechanical and catalytic properties. They also serve as a bridge for one to investigate the transition in the fundamental physical properties of materials from bulk materials to single molecules and atoms. Many exciting discoveries have been made over the past decades, and more will likely to come in the coming years. Important new discoveries will benefit from coordinated efforts, including synthesis, structural determination, property characterization, and theoretical modeling. One example is catalytic activity of nanoparticles. It has been recognized the catalytic activity of a nanoparticle depends not only on its chemical composition and size, but also atomic scale structure, including edges (confined electromagnetic fields) 14, 30, 104, 107 and facets (reduced activation energy).11 Tools that could perform in situ characterization of both the atomic scale structure and catalytic activity of each individual nanoparticles would significantly advance the field.
Nanoparticles are interesting also because their unique properties have led to many important applications. A particularly rapid growing application is the use of nanoparticles as nano-probes to detect molecules, to report spatial distribution (imaging enhancers) and to track movement of biological materials (cells, membrane components, organelles and proteins). Fluorescent QDs and plasmonic metal nanoparticles represent the two major nano-probes. Most of the previous applications use nanoparticles as passive optical probes, which will continue to advance and make significant contributions to biology and medicine, especially when combined with other technologies. However, more sophisticated and multiple functions could be created with hybrid nano-objects. One example in this direction is the Pd-Au hybrid nanostructures for hydrogen detections, in which Pd provides specificity for hydrogen adsorption and Au reports the adsorption.29 Other efforts include synthesis of core-shell structures,25, 31 and coating of nanoparticles with functional organic molecules,64, 120 in order to achieve more sophisticated functions. Silicon transistors in commercial electronics have decreased to a couple of tens of nm. Further miniaturization of transistor size down to 5 nm has been projected. Integrating sophisticated functions, such as logic functions, into a nanoscale object, is, at least in principle, possible, which dramatically extend the current capability of nanoparticles as passive probes.
Finally, we note that single nanoparticle analysis as emphasized in the present review indeed provides new and detailed insights into the intrinsic properties of the nanoparticles, and biological processes probed by the nanoparticles. Most studies reported to date analyzed only a few numbers of nanoparticles. Because of the large variability both in the size, shape and surface chemistry of nanoparticles, and the processes probed by the nanoparticles,63,110 measurements on many individual nanoparticles will allow for statistical analysis and provide a more complete picture. This effort will also provide a better understanding of the connection between ensemble measurements involving many nanoparticles, and analysis based on single nanoparticles.
Acknowledgments
This work was supported by the Gordon and Betty Moore Foundation, National Science Foundation (NSF, CHE-1105588, BIO-1151005), and National Institutes of Health (NIH, 8R21GM103396).
Biographies
Wei Wang is currently an assistant research professor in Biodesign Institute at Arizona State University. He received his B.S. degree in 2004 and Ph.D. degree in 2009 from Department of Chemistry at University of Science and Technology of China (USTC) with Prof. Hua Cui. Since 2009, he was a postdoc in the group of Professor Nongjian Tao at Arizona State University before moving to his current position in 2013. His current researches involve single cell optical imaging and electroanalytical chemistry.
Nongjian “NJ” Tao, Ph.D., is the director of the Center for Bioelectronics and Biosensors at the Biodesign Institute and is a professor in the Ira A. Fulton Schools of Engineering at Arizona State University. Tao joined ASU faculty as a professor of electrical engineering and an affiliated professor of chemistry and biochemistry in August 2001. His expertise lies in molecular electronics and nanoelectronics, chemical and biological sensors, and wireless devices for mobile health and environmental applications.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Mayer KM, Hafner JH. Chem Rev. 2011;111:3828–3857. doi: 10.1021/cr100313v. [DOI] [PubMed] [Google Scholar]
- 2.Chen KI, Li BR, Chen YT. Nano Today. 2011;6:131–154. [Google Scholar]
- 3.Oja SM, Wood M, Zhang B. Anal Chem. 2013;85:473–486. doi: 10.1021/ac3031702. [DOI] [PubMed] [Google Scholar]
- 4.Scida K, Stege PW, Haby G, Messina GA, Garcia CD. Analytica Chimica Acta. 2011;691:6–17. doi: 10.1016/j.aca.2011.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sapsford KE, Tyner KM, Dair BJ, Deschamps JR, Medintz IL. Anal Chem. 2011;83:4453–4488. doi: 10.1021/ac200853a. [DOI] [PubMed] [Google Scholar]
- 6.Chiang CK, Chen WT, Chang HT. Chem Soc Rev. 2011;40:1269–1281. doi: 10.1039/c0cs00050g. [DOI] [PubMed] [Google Scholar]
- 7.Cui J, Beyler AP, Marshall LF, Chen O, Harris DK, Wanger DD, Brokmann X, Bawendi MG. Nature Chemistry. 2013;5:602–606. doi: 10.1038/nchem.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo LH, Kim DH. Chem Commun. 2011;47:7125–7127. doi: 10.1039/c1cc11865j. [DOI] [PubMed] [Google Scholar]
- 9.Bootharaju MS, Chaudhari K, Pradeep T. RSC Advances. 2012;2:10048–10056. [Google Scholar]
- 10.Guo LH, Ferhan AR, Lee K, Kim DH. Anal Chem. 2011;83:2605–2612. doi: 10.1021/ac200432c. [DOI] [PubMed] [Google Scholar]
- 11.Eo M, Baek J, Song HD, Lee S, Yi J. Chem Commun. 2013;49:5204–5206. doi: 10.1039/c3cc41627e. [DOI] [PubMed] [Google Scholar]
- 12.Hwang WS, Truong PL, Sim SJ. Anal Biochem. 2012;421:213–218. doi: 10.1016/j.ab.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Beeram SR, Zamborini FP. J Phys Chem C. 2011;115:7364–7371. [Google Scholar]
- 14.Zijlstra P, Paulo PMR, Orrit M. Nat Nanotechnol. 2012;7:379–382. doi: 10.1038/nnano.2012.51. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Ling J, Huang CZ. Chem Commun. 2011;47:8121–8123. doi: 10.1039/c1cc11503k. [DOI] [PubMed] [Google Scholar]
- 16.Jing C, Gu Z, Ying YL, Li DW, Zhang L, Long YT. Anal Chem. 2012;84:4284–4291. doi: 10.1021/ac203118g. [DOI] [PubMed] [Google Scholar]
- 17.Ringe E, Sharma B, Henry AI, Marks LD, Van Duyne RP. Phys Chem Chem Phys. 2013;15:4110–4129. doi: 10.1039/c3cp44574g. [DOI] [PubMed] [Google Scholar]
- 18.Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP. Nat Mater. 2008;7:442–453. doi: 10.1038/nmat2162. [DOI] [PubMed] [Google Scholar]
- 19.Sagle LB, Ruvuna LK, Ruemmele JA, Van Duyne RP. Nanomedicine. 2011;6:1447–1462. doi: 10.2217/nnm.11.117. [DOI] [PubMed] [Google Scholar]
- 20.Sannomiya T, Voros J. Trends Biotechnol. 2011;29:343–351. doi: 10.1016/j.tibtech.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Jing C, Zhang L, Long YT. Chem Soc Rev. 2012;41:632–642. doi: 10.1039/c1cs15143f. [DOI] [PubMed] [Google Scholar]
- 22.Ament I, Prasad J, Henkel A, Schmachtel S, Sonnichsen C. Nano Lett. 2012;12:1092–1095. doi: 10.1021/nl204496g. [DOI] [PubMed] [Google Scholar]
- 23.Rosman C, Prasad J, Neiser A, Henkel A, Edgar J, Sonnichsen C. Nano Lett. 2013;13:3243–3247. doi: 10.1021/nl401354f. [DOI] [PubMed] [Google Scholar]
- 24.Schneider T, Jahr N, Jatschka J, Csaki A, Stranik O, Fritzsche W. J Nanopart Res. 2013;15 [Google Scholar]
- 25.Xiong B, Zhou R, Hao JR, Jia YH, He Y, Yeung ES. Nature Communications. 2013;4:1708. doi: 10.1038/ncomms2722. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, Li Y, Li DW, Jing C, Chen XY, Lv M, Huang Q, Long YT, Willner I. Angew Chem-Int Edit. 2011;50:6789–6792. doi: 10.1002/anie.201102151. [DOI] [PubMed] [Google Scholar]
- 27.Liu Q, Jing C, Zheng XX, Gu Z, Li D, Li DW, Huang Q, Long YT, Fan CH. Chem Commun. 2012;48:9574–9576. doi: 10.1039/c2cc34632j. [DOI] [PubMed] [Google Scholar]
- 28.Shi L, Jing C, Ma W, Li DW, Halls JE, Marken F, Long YT. Angew Chem-Int Edit. 2013;52:6011–6014. doi: 10.1002/anie.201301930. [DOI] [PubMed] [Google Scholar]
- 29.Liu N, Tang ML, Hentschel M, Giessen H, Alivisatos AP. Nat Mater. 2011;10:631–636. doi: 10.1038/nmat3029. [DOI] [PubMed] [Google Scholar]
- 30.Tang ML, Liu N, Dionne JA, Alivisatos AP. J Am Chem Soc. 2011;133:13220–13223. doi: 10.1021/ja203215b. [DOI] [PubMed] [Google Scholar]
- 31.Tittl A, Yin XH, Giessen H, Tian XD, Tian ZQ, Kremers C, Chigrin DN, Liu N. Nano Lett. 2013;13:1816–1821. doi: 10.1021/nl4005089. [DOI] [PubMed] [Google Scholar]
- 32.Seo D, Park G, Song H. J Am Chem Soc. 2012;134:1221–1227. doi: 10.1021/ja2093663. [DOI] [PubMed] [Google Scholar]
- 33.Shegai T, Langhammer C. Adv Mater. 2011;23:4409–4414. doi: 10.1002/adma.201101976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poyli MA, Silkin VM, Chernov IP, Echenique PM, Muino RD, Aizpurua J. J Phys Chem Lett. 2012;3:2556–2561. doi: 10.1021/jz3007723. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt MA, Lei DY, Wondraczek L, Nazabal V, Maier SA. Nature Communications. 2012;3 doi: 10.1038/ncomms2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen SL, Kucernak A. J Phys Chem B. 2003;107:8392–8402. [Google Scholar]
- 37.Chen SL, Kucernak A. J Phys Chem B. 2004;108:13984–13994. [Google Scholar]
- 38.Chen SL, Kucernak A. J Phys Chem B. 2004;108:3262–3276. [Google Scholar]
- 39.Li YX, Cox JT, Zhang B. J Am Chem Soc. 2010;132:3047–3054. doi: 10.1021/ja909408q. [DOI] [PubMed] [Google Scholar]
- 40.Li YX, Bergman D, Zhang B. Anal Chem. 2009;81:5496–5502. doi: 10.1021/ac900777n. [DOI] [PubMed] [Google Scholar]
- 41.Jena BK, Percival SJ, Zhang B. Anal Chem. 2010;82:6737–6743. doi: 10.1021/ac101261m. [DOI] [PubMed] [Google Scholar]
- 42.Lakbub J, Pouliwe A, Kamasah A, Yang C, Sun P. Electroanalysis. 2011;23:2270–2274. [Google Scholar]
- 43.Sun P, Li F, Yang C, Sun T, Kady I, Hunt B, Zhuang J. J Phys Chem C. 2013;117:6120–6125. [Google Scholar]
- 44.Lee JS, Han MS, Mirkin CA. Angew Chem-Int Edit. 2007;46:4093–4096. doi: 10.1002/anie.200700269. [DOI] [PubMed] [Google Scholar]
- 45.Xiao LH, Wei L, He Y, Yeung ES. Anal Chem. 2010;82:6308–6314. doi: 10.1021/ac101018v. [DOI] [PubMed] [Google Scholar]
- 46.Waldeisen JR, Wang T, Ross BM, Lee LP. ACS Nano. 2011;5:5383–5389. doi: 10.1021/nn2002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan ZQ, Cheng J, Cheng XD, He Y, Yeung ES. Analyst. 2012;137:2930–2932. doi: 10.1039/c2an16171k. [DOI] [PubMed] [Google Scholar]
- 48.Yang LL, Zhu SB, Hang W, Wu L, Yan XM. Anal Chem. 2009;81:2555–2563. doi: 10.1021/ac802464a. [DOI] [PubMed] [Google Scholar]
- 49.Zhu SB, Yang LL, Long Y, Gao M, Huang TX, Hang W, Yan XM. J Am Chem Soc. 2010;132:12176–12178. doi: 10.1021/ja104052c. [DOI] [PubMed] [Google Scholar]
- 50.Person S, Deutsch B, Mitra A, Novotny L. Nano Lett. 2011;11:257–261. doi: 10.1021/nl103656y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitra A, Ignatovich F, Novotny L. Biosens Bioelectron. 2012;31:499–504. doi: 10.1016/j.bios.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han GJ, Xing Z, Dong YH, Zhang SC, Zhang XR. Angew Chem-Int Edit. 2011;50:3462–3465. doi: 10.1002/anie.201006838. [DOI] [PubMed] [Google Scholar]
- 53.Rakcheev D, Philippe A, Schaumann GE. Anal Chem. 2013;85:10643–10647. doi: 10.1021/ac4019395. [DOI] [PubMed] [Google Scholar]
- 54.Zhang CY, Yeh HC, Kuroki MT, Wang TH. Nat Mater. 2005;4:826–831. doi: 10.1038/nmat1508. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Y, Wang TH. Theranostics. 2012;2:631–654. doi: 10.7150/thno.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang CY, Johnson LW. Anal Chem. 2007;79:7775–7781. doi: 10.1021/ac071225w. [DOI] [PubMed] [Google Scholar]
- 57.Zhang CY, Johnson LW. Anal Chem. 2009;81:3051–3055. doi: 10.1021/ac802737b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang CY, Hu J. Anal Chem. 2010;82:1921–1927. doi: 10.1021/ac9026675. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, Zhang CY. Anal Chem. 2012;84:224–231. doi: 10.1021/ac202405q. [DOI] [PubMed] [Google Scholar]
- 60.Zhang CY, Johnson LW. J Am Chem Soc. 2008;130:3750-+. doi: 10.1021/ja711493q. [DOI] [PubMed] [Google Scholar]
- 61.Zhang CY, Li DR. Analyst. 2010;135:2355–2359. doi: 10.1039/c0an00248h. [DOI] [PubMed] [Google Scholar]
- 62.Zhou J, Wang QX, Zhang CY. J Am Chem Soc. 2013;135:2056–2059. doi: 10.1021/ja3110329. [DOI] [PubMed] [Google Scholar]
- 63.Pons T, Medintz IL, Wang X, English DS, Mattoussi H. J Am Chem Soc. 2006;128:15324–15331. doi: 10.1021/ja0657253. [DOI] [PubMed] [Google Scholar]
- 64.Opperwall SR, Divakaran A, Porter EG, Christians JA, DenHartigh AJ, Benson DE. ACS Nano. 2012;6:8078–8086. doi: 10.1021/nn303347k. [DOI] [PubMed] [Google Scholar]
- 65.Yeh HC, Ho YP, Shih IM, Wang TH. Nucleic Acids Research. 2006;34 doi: 10.1093/nar/gkl021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Song YK, Zhang Y, Wang TH. Small. 2013;9:1096–1105. doi: 10.1002/smll.201202242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Long Y, Zhang LF, Zhang Y, Zhang CY. Anal Chem. 2012;84:8846–8852. doi: 10.1021/ac302284s. [DOI] [PubMed] [Google Scholar]
- 68.Ho YP, Kung MC, Yang S, Wang TH. Nano Lett. 2005;5:1693–1697. doi: 10.1021/nl050888v. [DOI] [PubMed] [Google Scholar]
- 69.Hellberg D, Scholz F, Schauer F, Weitschies W. Electrochemistry Communications. 2002;4:305–309. [Google Scholar]
- 70.Quinn BM, van ’t Hof P, Lemay SG. J Am Chem Soc. 2004;126:8360–8361. doi: 10.1021/ja0478577. [DOI] [PubMed] [Google Scholar]
- 71.Boika A, Thorgaard SN, Bard AJ. J Phys Chem B. 2013;117:4371–4380. doi: 10.1021/jp306934g. [DOI] [PubMed] [Google Scholar]
- 72.Fosdick SE, Anderson MJ, Nettleton EG, Crooks RM. J Am Chem Soc. 2013;135:5994–5997. doi: 10.1021/ja401864k. [DOI] [PubMed] [Google Scholar]
- 73.Xiao XY, Bard AJ. J Am Chem Soc. 2007;129:9610–9612. doi: 10.1021/ja072344w. [DOI] [PubMed] [Google Scholar]
- 74.Xiao XY, Fan FRF, Zhou JP, Bard AJ. J Am Chem Soc. 2008;130:16669–16677. doi: 10.1021/ja8051393. [DOI] [PubMed] [Google Scholar]
- 75.Batchelor-McAuley C, Dickinson EJF, Rees NV, Toghill KE, Compton RG. Anal Chem. 2012;84:669–684. doi: 10.1021/ac2026767. [DOI] [PubMed] [Google Scholar]
- 76.Rees NV, Zhou YG, Compton RG. RSC Advances. 2012;2:379–384. [Google Scholar]
- 77.Zhou HJ, Park JH, Fan FRF, Bard AJ. J Am Chem Soc. 2012;134:13212–13215. doi: 10.1021/ja305573g. [DOI] [PubMed] [Google Scholar]
- 78.Kwon SJ, Fan FRF, Bard AJ. J Am Chem Soc. 2010;132:13165–13167. doi: 10.1021/ja106054c. [DOI] [PubMed] [Google Scholar]
- 79.Kwon SJ, Bard AJ. J Am Chem Soc. 2012;134:7102–7108. doi: 10.1021/ja300894f. [DOI] [PubMed] [Google Scholar]
- 80.Dasari R, Robinson DA, Stevenson KJ. J Am Chem Soc. 2013;135:570–573. doi: 10.1021/ja310614x. [DOI] [PubMed] [Google Scholar]
- 81.Park JH, Thorgaard SN, Zhang B, Bard AJ. J Am Chem Soc. 2013;135:5258–5261. doi: 10.1021/ja4000917. [DOI] [PubMed] [Google Scholar]
- 82.Kleijn SEF, Lai SCS, Miller TS, Yanson AI, Koper MTM, Unwin PR. J Am Chem Soc. 2012;134:18558–18561. doi: 10.1021/ja309220m. [DOI] [PubMed] [Google Scholar]
- 83.Kwon SJ, Zhou HJ, Fan FRF, Vorobyev V, Zhang B, Bard AJ. Phys Chem Chem Phys. 2011;13:5394–5402. doi: 10.1039/c0cp02543g. [DOI] [PubMed] [Google Scholar]
- 84.Park JH, Boika A, Park HS, Lee HC, Bard AJ. J Phys Chem C. 2013;117:6651–6657. [Google Scholar]
- 85.Alligrant TM, Nettleton EG, Crooks RM. Lab Chip. 2013;13:349–354. doi: 10.1039/c2lc40993c. [DOI] [PubMed] [Google Scholar]
- 86.Kwon SJ, Bard AJ. J Am Chem Soc. 2012;134:10777–10779. doi: 10.1021/ja304074f. [DOI] [PubMed] [Google Scholar]
- 87.Zhou YG, Rees NV, Compton RG. Angew Chem-Int Edit. 2011;50:4219–4221. doi: 10.1002/anie.201100885. [DOI] [PubMed] [Google Scholar]
- 88.Haddou B, Rees NV, Compton RG. Phys Chem Chem Phys. 2012;14:13612–13617. doi: 10.1039/c2cp42585h. [DOI] [PubMed] [Google Scholar]
- 89.Zhou YG, Haddou B, Rees NV, Compton RG. Phys Chem Chem Phys. 2012;14:14354–14357. doi: 10.1039/c2cp42940c. [DOI] [PubMed] [Google Scholar]
- 90.Zhou YG, Rees NV, Compton RG. Chem Commun. 2012;48:2510–2512. doi: 10.1039/c2cc17481b. [DOI] [PubMed] [Google Scholar]
- 91.Kahk JM, Rees NV, Pillay J, Tshikhudo R, Vilakazi S, Compton RG. Nano Today. 2012;7:174–179. [Google Scholar]
- 92.Brainina KZ, Galperin LG, Kiryuhina TY, Galperin AL, Stozhko NY, Murzakaev AM, Timoshenkova OR. J Solid State Electrochem. 2012;16:2365–2372. [Google Scholar]
- 93.Cheng W, Zhou X-F, Compton RG. Angew Chem-Int Edit. 2013;52:12980–12982. doi: 10.1002/anie.201307653. [DOI] [PubMed] [Google Scholar]
- 94.Rothenhausler B, Knoll W. Nature. 1988;332:615–617. [Google Scholar]
- 95.Huang B, Yu F, Zare RN. Anal Chem. 2007;79:2979–2983. doi: 10.1021/ac062284x. [DOI] [PubMed] [Google Scholar]
- 96.Wang SP, Shan XN, Patel U, Huang XP, Lu J, Li JH, Tao NJ. Proc Natl Acad Sci U S A. 2010;107:16028–16032. doi: 10.1073/pnas.1005264107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shan XN, Patel U, Wang SP, Iglesias R, Tao NJ. Science. 2010;327:1363–1366. doi: 10.1126/science.1186476. [DOI] [PubMed] [Google Scholar]
- 98.Shan XN, Diez-Perez I, Wang LJ, Wiktor P, Gu Y, Zhang LH, Wang W, Lu J, Wang SP, Gong QH, Li JH, Tao NJ. Nat Nanotechnol. 2012;7:668–672. doi: 10.1038/nnano.2012.134. [DOI] [PubMed] [Google Scholar]
- 99.Xu WL, Kong JS, Yeh YTE, Chen P. Nat Mater. 2008;7:992–996. doi: 10.1038/nmat2319. [DOI] [PubMed] [Google Scholar]
- 100.Chen P, Zhou XC, Shen H, Andoy NM, Choudhary E, Han KS, Liu GK, Meng WL. Chem Soc Rev. 2010;39:4560–4570. doi: 10.1039/b909052p. [DOI] [PubMed] [Google Scholar]
- 101.Han KS, Liu GK, Zhou XC, Medina RE, Chen P. Nano Lett. 2012;12:1253–1259. doi: 10.1021/nl203677b. [DOI] [PubMed] [Google Scholar]
- 102.De Cremer G, Sels BF, De Vos DE, Hofkens J, Roeffaers MBJ. Chem Soc Rev. 2010;39:4703–4717. doi: 10.1039/c0cs00047g. [DOI] [PubMed] [Google Scholar]
- 103.Buurmans ILC, Weckhuysen BM. Nature Chemistry. 2012;4:873–886. doi: 10.1038/nchem.1478. [DOI] [PubMed] [Google Scholar]
- 104.Zhou XC, Andoy NM, Liu GK, Choudhary E, Han KS, Shen H, Chen P. Nat Nanotechnol. 2012;7:237–241. doi: 10.1038/nnano.2012.18. [DOI] [PubMed] [Google Scholar]
- 105.Wang N, Tachikawa T, Majima T. Chem Sci. 2011;2:891–900. [Google Scholar]
- 106.Tachikawa T, Majima T. Langmuir. 2012;28:8933–8943. doi: 10.1021/la300177h. [DOI] [PubMed] [Google Scholar]
- 107.Andoy NM, Zhou XC, Choudhary E, Shen H, Liu GK, Chen P. J Am Chem Soc. 2013;135:1845–1852. doi: 10.1021/ja309948y. [DOI] [PubMed] [Google Scholar]
- 108.Tachikawa T, Yonezawa T, Majima T. ACS Nano. 2013;7:263–275. doi: 10.1021/nn303964v. [DOI] [PubMed] [Google Scholar]
- 109.Zhou XC, Xu WL, Liu GK, Panda D, Chen P. J Am Chem Soc. 2010;132:138–146. doi: 10.1021/ja904307n. [DOI] [PubMed] [Google Scholar]
- 110.Zhou X, Choudhary E, Andoy NM, Zou N, Chen P. ACS Catalysis. 2013;3:1448–1453. [Google Scholar]
- 111.Yuan HF, Khatua S, Zijlstra P, Yorulmaz M, Orrit M. Angew Chem-Int Edit. 2013;52:1217–1221. doi: 10.1002/anie.201208125. [DOI] [PubMed] [Google Scholar]
- 112.Tachikawa T, Ohsaka T, Bian Z, Majima T. J Phys Chem C. 2013;117:11219–11228. [Google Scholar]
- 113.Baba K, Nishida K. Theranostics. 2012;2:655–667. doi: 10.7150/thno.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bruchez MP. Current Opinion in Chemical Biology. 2011;15:775–780. doi: 10.1016/j.cbpa.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pinaud F, Clarke S, Sittner A, Dahan M. Nat Methods. 2010;7:275–285. doi: 10.1038/nmeth.1444. [DOI] [PubMed] [Google Scholar]
- 116.Gudheti MV, Curthoys NM, Gould TJ, Kim D, Gunewardene MS, Gabor KA, Gosse JA, Kim CH, Zimmerberg J, Hess ST. Biophys J. 2013;104:2182–2192. doi: 10.1016/j.bpj.2013.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Winter PW, Van Orden AK, Roess DA, Barisas BG. Biochimica Et Biophysica Acta-Biomembranes. 2012;1818:467–473. doi: 10.1016/j.bbamem.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 118.Pinaud F, Michalet X, Iyer G, Margeat E, Moore HP, Weiss S. Traffic. 2009;10:691–712. doi: 10.1111/j.1600-0854.2009.00902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chang JC, Rosenthal SJ. ACS Chemical Neuroscience. 2012;3:737–743. doi: 10.1021/cn3000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dahan M, Levi S, Luccardini C, Rostaing P, Riveau B, Triller A. Science. 2003;302:442–445. doi: 10.1126/science.1088525. [DOI] [PubMed] [Google Scholar]
- 121.Bouzigues C, Morel M, Triller A, Dahan M. Proc Natl Acad Sci U S A. 2007;104:11251–11256. doi: 10.1073/pnas.0702536104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Burli T, Baer K, Ewers H, Sidler C, Fuhrer C, Fritschy JM. Plos One. 2010;5:e11507. doi: 10.1371/journal.pone.0011507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chang JC, Tomlinson ID, Warnement MR, Ustione A, Carneiro AMD, Piston DW, Blakely RD, Rosenthal SJ. Journal of Neuroscience. 2012;32:8919–8929. doi: 10.1523/JNEUROSCI.0048-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mascalchi P, Lamort AS, Salome L, Dumas F. Biochem Biophys Res Commun. 2012;417:409–413. doi: 10.1016/j.bbrc.2011.11.129. [DOI] [PubMed] [Google Scholar]
- 125.Chen LQ, Xiao SJ, Hu PP, Peng L, Ma J, Luo LF, Li YF, Huang CZ. Anal Chem. 2012;84:3099–3110. doi: 10.1021/ac202810b. [DOI] [PubMed] [Google Scholar]
- 126.Cui BX, Wu CB, Chen L, Ramirez A, Bearer EL, Li WP, Mobley WC, Chu S. Proc Natl Acad Sci U S A. 2007;104:13666–13671. doi: 10.1073/pnas.0706192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nan XL, Sims PA, Chen P, Xie XS. J Phys Chem B. 2005;109:24220–24224. doi: 10.1021/jp056360w. [DOI] [PubMed] [Google Scholar]
- 128.DeWitt MA, Chang AY, Combs PA, Yildiz A. Science. 2012;335:221–225. doi: 10.1126/science.1215804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ohmachi M, Komori Y, Iwane AH, Fujii F, Jin T, Yanagida T. Proc Natl Acad Sci U S A. 2012;109:5294–5298. doi: 10.1073/pnas.1118472109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ortega-Arroyo J, Kukura P. Phys Chem Chem Phys. 2012;14:15625–15636. doi: 10.1039/c2cp41013c. [DOI] [PubMed] [Google Scholar]
- 131.Kukura P, Ewers H, Muller C, Renn A, Helenius A, Sandoghdar V. Nat Methods. 2009;6:923-U85. doi: 10.1038/nmeth.1395. [DOI] [PubMed] [Google Scholar]
- 132.Bruno A, Bruno L, Levi V. Cell Biochemistry and Biophysics. 2013;65:1–11. doi: 10.1007/s12013-012-9397-3. [DOI] [PubMed] [Google Scholar]
- 133.Yu JB, Wu CF, Sahu SP, Fernando LP, Szymanski C, McNeill J. J Am Chem Soc. 2009;131:18410–18414. doi: 10.1021/ja907228q. [DOI] [PubMed] [Google Scholar]
- 134.Koner AL, Krndija D, Hou Q, Sherratt DJ, Howarth M. ACS Nano. 2013;7:1137–1144. doi: 10.1021/nn3042122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Du K, Liddle JA, Berglund AJ. Langmuir. 2012;28:9181–9188. doi: 10.1021/la300292r. [DOI] [PubMed] [Google Scholar]
- 136.Du K, Ko SH, Gallatin GM, Yoon HP, Liddle JA, Berglund AJ. Chem Commun. 2013;49:907–909. doi: 10.1039/c2cc37517f. [DOI] [PubMed] [Google Scholar]
- 137.Marchuk K, Guo YJ, Sun W, Vela J, Fang N. J Am Chem Soc. 2012;134:6108–6111. doi: 10.1021/ja301332t. [DOI] [PubMed] [Google Scholar]
- 138.Sagle LB, Ruvuna LK, Bingham JM, Liu CM, Cremer PS, Van Duyne RP. J Am Chem Soc. 2012;134:15832–15839. doi: 10.1021/ja3054095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yang YH, Nam JM. Anal Chem. 2009;81:2564–2568. doi: 10.1021/ac802477h. [DOI] [PubMed] [Google Scholar]
- 140.Lee KJ, Nallathamby PD, Browning LM, Desai T, Cherukuri PK, Xu XHN. Analyst. 2012;137:2973–2986. doi: 10.1039/c2an35293a. [DOI] [PubMed] [Google Scholar]
- 141.Browning LM, Huang T, Xu XHN. Interface Focus. 2013;3 doi: 10.1098/rsfs.2012.0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pierrat S, Hartinger E, Faiss S, Janshoff A, Sonnichsen C. J Phys Chem C. 2009;113:11179–11183. [Google Scholar]
- 143.Sonnichsen C, Alivisatos AP. Nano Lett. 2005;5:301–304. doi: 10.1021/nl048089k. [DOI] [PubMed] [Google Scholar]
- 144.Huang YJ, Kim DH. Nanoscale. 2011;3:3228–3232. doi: 10.1039/c1nr10336a. [DOI] [PubMed] [Google Scholar]
- 145.Chang WS, Ha JW, Slaughter LS, Link S. Proc Natl Acad Sci U S A. 2010;107:2781–2786. doi: 10.1073/pnas.0910127107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.van den Broek B, Ashcroft B, Oosterkamp TH, van Noort J. Nano Lett. 2013;13:980–986. doi: 10.1021/nl3040509. [DOI] [PubMed] [Google Scholar]
- 147.Xiao LH, Qiao YX, He Y, Yeung ES. J Am Chem Soc. 2011;133:10638–10645. doi: 10.1021/ja203289m. [DOI] [PubMed] [Google Scholar]
- 148.Wang GF, Sun W, Luo Y, Fang N. J Am Chem Soc. 2010;132:16417–16422. doi: 10.1021/ja106506k. [DOI] [PubMed] [Google Scholar]
- 149.Sun W, Gu Y, Wang GF, Fang N. Anal Chem. 2012;84:1134–1138. doi: 10.1021/ac202824v. [DOI] [PubMed] [Google Scholar]
- 150.Gu Y, Sun W, Wang GF, Fang N. J Am Chem Soc. 2011;133:5720–5723. doi: 10.1021/ja200603x. [DOI] [PubMed] [Google Scholar]
- 151.Ha JW, Sun W, Wang GF, Fang N. Chem Commun. 2011;47:7743–7745. doi: 10.1039/c1cc11679g. [DOI] [PubMed] [Google Scholar]
- 152.Xiao LH, Ha JW, Wei L, Wang GF, Fang N. Angew Chem-Int Edit. 2012;51:7734–7738. doi: 10.1002/anie.201202340. [DOI] [PubMed] [Google Scholar]
- 153.Ha JW, Sun W, Stender AS, Fang N. J Phys Chem C. 2012;116:2766–2771. [Google Scholar]
- 154.Stender AS, Augspurger AE, Wang GF, Fang N. Anal Chem. 2012;84:5210–5215. doi: 10.1021/ac301072f. [DOI] [PubMed] [Google Scholar]
- 155.Gu Y, Di XW, Sun W, Wang GF, Fang N. Anal Chem. 2012;84:4111–4117. doi: 10.1021/ac300249d. [DOI] [PubMed] [Google Scholar]
- 156.Gu Y, Wang GF, Fang N. ACS Nano. 2013;7:1658–1665. doi: 10.1021/nn305640y. [DOI] [PubMed] [Google Scholar]
- 157.Gu Y, Sun W, Wang GF, Jeftinija K, Jeftinija S, Fang N. Nature Communications. 2012;3 doi: 10.1038/ncomms2037. [DOI] [PubMed] [Google Scholar]
- 158.Gu Y, Sun W, Wang GF, Zimmermann MT, Jernigan RL, Fang N. Small. 2013;9:785–792. doi: 10.1002/smll.201201808. [DOI] [PubMed] [Google Scholar]
- 159.Xiao LH, Wei L, Liu C, He Y, Yeung ES. Angew Chem-Int Edit. 2012;51:4181–4184. doi: 10.1002/anie.201108647. [DOI] [PubMed] [Google Scholar]