

Abstract
Over-expression of Ribosomal RNA Processing 1 Homolog B (RRP1B) induces a transcriptional profile that accurately predicts patient outcome in breast cancer. However, the mechanism by which RRP1B modulates transcription is unclear. Here, the chromatin-binding properties of RRP1B were examined to define how it regulates metastasis-associated transcription.
To identify genome-wide RRP1B binding sites, high-throughput ChIP-seq was performed in the human breast cancer cell line MDA-MB-231 and HeLa cells using antibodies against endogenous RRP1B. Global changes in repressive marks such as histone H3 lysine 9 trimethylation (H3K9me3) were also examined by ChIP-seq. Analysis of these samples identified 339 binding regions in MDA-MB-231 cells and 689 RRP1B binding regions in HeLa cells. Among these, 136 regions were common to both cell lines. Gene expression analyses of these RRP1B-binding regions revealed that transcriptional repression is the primary result of RRP1B binding to chromatin. ChIP-reChIP assays demonstrated that RRP1B co-occupies loci with decreased gene expression with the heterochromatin-associated proteins, tripartite motif-containing protein 28 (TRIM28/KAP1) and heterochromatin protein 1-α (CBX5/HP1α). RRP1B occupancy at these loci was also associated with higher H3K9me3 levels, indicative of heterochromatinization mediated by the TRIM28/HP1α complex. In addition, RRP1B up-regulation, which is associated with metastasis suppression, induced global changes in histone methylation.
Implications
RRP1B, a breast cancer metastasis suppressor, regulates gene expression through heterochromatinization and transcriptional repression, which helps our understanding of mechanisms that drive prognostic gene expression in human breast cancer.
Keywords: Breast cancer, metastasis, histone methylation, gene expression, chromatin immunoprecipitation (ChIP)
Introduction
Breast carcinoma is the most commonly diagnosed malignancy in women in the United States, with over 235,000 new cases and over 40,000 deaths in 2012 (1). Metastasis is the main cause of death in breast cancer and metastatic disease is considered incurable (2). Tumorigenesis and metastasis form a spectrum of progression that are induced by genetic alterations and disruption of epigenetic modifications (3). The sequential acquisition of random somatic mutations within the primary tumor is the most widely accepted model of metastasis at the molecular level. However, it is becoming increasingly clear that although somatic mutation is the primary driver of metastasis, the propensity of a primary tumor to metastasize is strongly influenced by inter-individual germline variation (reviewed in (4)).
The consideration of metastasis susceptibility as a heritable trait with a complex inheritance pattern has led to the identification of several germline-encoded metastasis modifier genes, including Ribosomal RNA Processing 1 Homolog B (RRP1B). RRP1B was identified as a germline susceptibility gene for breast cancer metastasis using expression quantitative trait locus mapping in the FVB/N-Tg(MMTV-PyVT)634Mul/J mouse mammary tumorigenesis model (5). Specifically, two concurrent and independent experimental results identified Rrp1b as a novel germline modifier of metastasis: first, RRP1B is a binding partner of the metastasis modifier SIPA1; second, RRP1B is a germline regulator of extracellular matrix gene expression, which are a class of genes frequently dysregulated in tumors prone to metastasizing (5). Following its identification using modifier locus mapping, the properties of Rrp1b were investigated by ectopic expression in the highly metastatic Mvt-1 mouse mammary tumor cell line. These experiments demonstrated that Rrp1b dysregulation induced a gene expression signature that predicts survival in multiple human breast cancer datasets with a high degree of reproducibility (5, 6). The relevance of RRP1B was further highlighted by the finding that a coding germline polymorphism within human RRP1B is consistently associated with clinical outcome in multiple breast cancer cohorts representing over 2,000 breast cancer patients (5, 7).
Subsequent protein-protein interaction analyses revealed several probable mechanisms by which Rrp1b dysregulation has such profound and clinically relevant effects on global gene expression (6). Most notably, these initial studies suggested that RRP1B is most likely a facultative heterochromatin protein since it co-localizes with several heterochromatin-associated histone marks. Concomitantly, RRP1B was shown to physically interact with a number of heterochromatin-associated proteins including, tripartite motif-containing protein 28 (TRIM28; KAP1) and heterochromatin protein 1-α (HP1α) (8-11), which are potent inducers of gene silencing. TRIM28 interacts with and recruits SETDB1 (SET domain, bifurcated 1) (9), a histone methyltransferase which has been shown to co-localize with and mediate trimethylation of histone H3 at lysine 9 (H3K9me3) (12). This creates high-affinity binding sites for the TRIM28/HP1α complex (13). H3K9me3 is a well-known marker of heterochromatin (14) and strong association between TRIM28 and H3K9me3 has been reported (15, 16).
In our current study, we aim to elucidate the mechanism by which RRP1B regulates metastasis-associated gene expression. We utilized a variety of approaches to define the mechanism by which RRP1B suppresses gene expression. We have used chromatin immunoprecipitation (ChIP) assays to identify chromatin regions bound by RRP1B, and demonstrated that these interactions are often associated with binding of the transcriptional repressors HP1α and TRIM28. Further, we demonstrate an association of these proteins at discrete genomic loci is concurrent with H3K9me3 and down-regulation of gene expression. Taken together, these data demonstrate that the clinically relevant changes in gene expression induced by RRP1B dysregulation and subsequent effects upon metastasis in breast cancer are, at least in part, due to regulation of epigenetic mechanisms.
Materials and Methods
Cell culture
MDA-MB-231 and HeLa cells were acquired from ATCC (Manassas, VA). All cell lines were maintained in DMEM with 10% fetal bovine serum and 1% penicillin-streptomycin (Gibco, Grand Island, NY) and incubated in 5% CO2 at 37 °C.
Lentiviral transduction
Lentiviral particles were generated as previously described (17) using the pDest-688 vector containing either human hemagglutinin (HA)-tagged RRP1B or no insert as a control. After viral infection, cells were selected in normal growth medium containing 10 μg/ml puromycin (Sigma Aldrich, St. Louis, MO), transferred to 96-well plates, and individual clones were selected by limiting dilution. Ectopic expression of RRP1B in single clonal isolate colonies was confirmed by qPCR as previously described (6).
siRNA transfection
Two different sequences of RRP1B Silencer Select siRNA (Life Technologies, Grand Island, NY, ID s22978 and s225927) were transfected in MDA-MB-231 cells using Lipofectamine RNAiMAX Transfection Reagent (Life Technologies) according to manufacturer's reverse transfection protocol. For RNA isolation, cells were plated at 1 × 105 per 24-well and collected 48 hr after transfection. For protein analysis and ChIP assays, cells were plated in 6-well and 150 mm plates at 2 × 105 per ml.
Cell growth and invasion assays
MDA-MB-231 clonal isolates infected with either HA-RRP1B over-expression or control lentiviral particles were seeded in triplicate on 24 mm plates at a density of 105 cells per plate and incubated in 5% CO2 at 37 °C for the indicated periods of time. After incubation, the cells were harvested and the number of cells was counted. For soft agar growth assays, cells were plated in triplicate in 0.3% soft agar and allowed to grow for 7 days. Colonies were quantified after staining with 0.005% crystal violet. Cell invasion assays were performed as previously described (18) using the MDA-MB-231 clonal isolate cell lines described above.
Lung colonization assays
One million MDA-MB-231 RRP1B or control cells were intravenously injected via the tail vein of NU/J female mice (Jackson Laboratory, Bar Harbor, ME). Twenty-six mice were injected with MDA-MB-231 RRP1B cells and 11 mice were injected with control cells. Lungs were harvested 90 days post-injection and pulmonary surface metastases were counted. All animal experiments were performed in compliance with the National Human Genome Research Institute Animal Care and Use Committee's guidelines.
Total RNA isolation
Total RNA was isolated from cells using RNeasy Mini Kit (QIAGEN, Valencia, CA). All samples were subjected to on-column DNase I digestion, and RNA quality and quantity were determined using NanoDrop 2000 (Thermo Scientific, Wilmington, DE). Only samples with A260/A280 ratios between 1.8 and 2.1 were used for downstream analysis.
Microarray analysis
Microarray was performed with MDA-MB-231 over-expressing RRP1B and control clonal isolates as previously described (5). Briefly, total RNA was extracted from MDA-MB-231 clonal isolates and processed using Affymetrix GeneChIP Human Gene 1.0 ST array (Santa Clara, CA) according to manufacturer's protocol. Data was analyzed using Partek Genomics Suite (Saint Louis, MO) and heatmaps was generated using R (19) based on the most significant gene expression data. Data was submitted to Gene Expression Omnibus (accession number GSE53979).
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed using a modified protocol (20). Briefly, cells were fixed with 1% formaldehyde and lysed with lysis buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 0.1% sodium deoxycholate, and protease inhibitors). Cell lysates were sonicated with a Daigger Ultrasonic Processor and Sonicator Rod (Vernon Hills, IL) for a total of 3 min with 30 sec pulse and 20 sec off cycles to shear the DNA to a final size of 200–500 base pairs. After pre-clearing with Protein G Sepharose beads (GE Healthcare, Piscataway, NJ), antibodies targeting either RRP1B (sc-83327, Santa Cruz Biotechnology, Dallas, TX), H3K9me3 (ab8898, Abcam, Cambridge, UK), or IgG (12-370, Millipore, Billerica, MA) was added to the lysate and incubated at 4 °C for 3 hours. Protein G Sepharose beads were added and incubated overnight. The complex was washed twice with lysis buffer, once with high salt buffer (50 mM HEPES-KOH pH 7.5, 500 mM NaCl, 1 mM EDTA, 0.1% Triton X-100, 0.1% sodium deoxycholate), twice with LiCl buffer (10 mM Tris-HCl pH 8.0, 0.25 M LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA) and once with TE buffer, followed by elution in TE buffer containing 1% SDS. Crosslinks were then reversed and the DNA was purified with QIAquick PCR purification kit (QIAGEN). The DNA obtained was analyzed either by high-throughput sequencing or qPCR.
For ChIP-reChIP, MDA-MB-231 lysates were collected as described above and incubated overnight incubation with the first antibody, either against TRIM28 (ab22553, Abcam) or HP1α (05-689, Millipore), and Protein G Sepharose beads. Immuno-complexes were washed twice with lysis buffer, once with high salt buffer, and once with TE buffer, followed by elution. The eluted samples were diluted with lysis buffer and incubated with the second antibody, anti-RRP1B, for 3 hours at 4 °C. Protein G Sepharose beads were then added and incubated overnight with rotation at 4 °C. The following day, samples were processed according to the ChIP protocol. All immunoprecipitations were performed in triplicate.
High-throughput sequencing
Approximately 100 ng of ChIP DNA with a fragment size of 200 bp was used to produce a library compatible with Illumina sequencing on a GAiix. Single-end adapters and sample DNA were input into a Beckman SPRI-TE robot along with SPRI-TE reagents for automated library construction. Post-construction size selection was performed using a 2% agarose gel stained with SYBR-gold and imaged on a Dark Reader. A band representing the range 250-350 bp was cut out and gel extracted using QIAquick Gel Extraction Kit (QIAGEN). To ensure minimal amplification, test PCR amplifications were set up for each library. Aliquots were removed at every two cycles from 6 through 16 cycles and analyzed for yield on a 2% agarose gel. Based on these results, large scale amplification was performed on the remainder of the library. Typically 12-14 cycles were required. The PCR reaction was cleaned up using Agencourt Ampure Xp beads (Beckman Coulter, Indianapolis IN). Libraries were quantitated by qPCR (KAPA Biosystems, Woburn, MA) and sequenced on an Illumina GAiix on single read flow cells. Early data was generated using version 4 chemistry and later data was generated using version 5 chemistry. Read lengths were 35 bases.
ChIP-sequencing (ChIP-seq) analysis
Read Mapping: ChIP DNAs were sequenced in duplicate. ChIP-seq reads were mapped to the hg18 human genome build and analyzed using the samtool suite. Using a database of repetitive elements (21) and the UCSC Genome Browser track characterizing ribosomal RNAs (22), a bowtie library (23) was generated for the characterization of unmapped reads. The set of unmapped reads was then aligned to the repetitive regions of the human genome using the bowtie tool. H3K9me3 ChIP-seq reads were mapped to the hg19 human genome build. Peak Calling: Using the Input reads as background for HeLa and MDA-MB-231 cells respectively, MACS (model-based analysis of ChIP-seq, version 1.4.1) was used to determine peaks under a statistical significance range of P = 10-3 to 10-7. For H3K9me3 ChIP-seq data, Sole-Search (Version v2) was used. Visual inspection of the different peak profiles allowed for the identification of relevant replicates and p-value settings. Peak Partitioning: Peaks were partitioned using the RefSeq gene annotations available for the UCSC Genome Browser. The resulting genomic partitions include proximal promoters (1 kb upstream to 50 bp downstream of TSS), distal promoters (10 kb upstream to 1 kb upstream of TSS), downstream regions (10 kb directly downstream of the transcription end site) as well as coding regions, exons, introns, untranslated, and intergenic regions. In addition to using genomic partitions, peaks were categorized using UCSC Genome Browser track annotations for noncoding and ribosomal RNAs, alternative events, the top scoring 1% and 5% conserved elements and DNaseI hypersensitive sites, as well as track data derived for the location of CpG Islands (24) and repetitive elements (21). Venn diagrams were generated using R (19).
Quantitative real-time PCR gene expression analysis
cDNA was synthesized from RNA isolated from MDA-MB-231 cells using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) following the manufacturer's protocol. Real-time qPCR was performed to detect cDNA levels of RRP1B and a variety of genes (Supplementary Table S1) using an ABI 7900 Sequence Detection System (Life Technologies). Reactions were performed using ABI SYBR Green Master Mix per the manufacturer's protocol. The cDNA level of each gene was normalized to GAPDH cDNA levels using custom-designed primers for SYBR green-amplified target genes. All ChIP-qPCR samples were amplified in duplicate. Statistical significance was calculated by Student's t-test.
Results
Ectopic expression of RRP1B in a human breast cancer cell line significantly affects cell growth, invasiveness, motility, and lung colonization
Previous studies have shown that knockdown of Rrp1b increases the metastatic capacity of Mvt-1 and 4T1 mouse mammary tumor cell lines (25). To gain a more comprehensive understanding of the role of this gene in metastasis, we used lentiviral-mediated ectopic expression of RRP1B in the human breast cancer cell line, MDA-MB-231. Over-expression of RRP1B was confirmed through Western blotting and real-time quantitative PCR (qPCR;Supplementary Fig. S1). Ectopic expression of RRP1B significantly decreased cell growth rates compared to control cell lines (Fig. 1A). In soft agar assays, fewer colonies were observed with the cells over-expressing RRP1B compared to the control cell lines (Fig. 1B). When seeded in transwells, cells over-expressing RRP1B displayed decreased invasiveness compared to the control cell lines (Fig. 1C). In addition, decreased pulmonary metastases were observed with the over-expression of RRP1B when intravenously injected into the tail vein of NU/J female mice (Fig. 1D). These data, which complement our earlier studies (25), confirm that RRP1B is a metastasis suppressor, and that the less metastatic phenotype induced by over-expression of RRP1B is due to changes in cellular growth rate, invasiveness, and migratory capacity.
Figure 1. RRP1B suppresses metastasis in vivo and decreases cell invasiveness in vitro.

A cell proliferation assay. B, soft agar assay. C, trans-well invasion assays. D, pulmonary surface metastases from intravenous injection of RRP1B over-expressing cells and control cells in NU/J mice. All data represent the average of three biological replicates.
Microarray analyses of these cells depicted a significant change in gene expression due to the over-expression of RRP1B with 219 down-regulated and 73 up-regulated genes compared to that of the control (Fig. 2). These data support the conclusion that the decrease in metastasis and changes in cell behavior were associated with changes in gene expression elicited by RRP1B. Analysis of the associated functions of these genes using Ingenuity Pathway Analysis (IPA, www.ingenuity.com) indicated that cellular growth and proliferation were among the top associated networks (Supplementary Table S2).
Figure 2. Over-expression of RRP1B in MDA-MB-231 cells causes a significant change in gene expression.

Global gene expression was quantified in five clonal isolates of MDA-MB-231 cells ectopically expressing RRP1B and control. Heatmap represents relative gene expression levels in the five clonal isolates of MDA-MB-231 cells ectopically expressing RRP1B and control detected by microarray analysis (WT; RRP1B, ctrl; control).
RRP1B binds to various genic regions within the genome
RRP1B was previously shown to interact with multiple nucleosome binding proteins, implying that RRP1B may be involved in chromatin modification, which in turn could account for a proportion of the transcriptional changes seen with dysregulation of RRP1B (6). However, the consequence of these interactions, and the precise role of RRP1B binding is unclear. The aim of these experiments was to use ChIP-sequencing (ChIP-seq) to define regions of chromatin bound by endogenous RRP1B. We hypothesized that RRP1B induces prognostic changes in gene expression and suppresses metastasis by interacting with these chromatin-associated factors at target gene sites. We performed genome-wide ChIP-seq in the MDA-MB-231 breast cancer cell line and confirmed in HeLa cells using an antibody targeting endogenous RRP1B. Although not a breast cancer cell line, we deemed ChIP-seq analysis in HeLa cells to be of importance since earlier protein-protein interaction studies in this cell line defined RRP1B as an interactor of chromatin-associated factors (6). A total of 339 peaks binding RRP1B were identified in MDA-MB-231 cells and 680 peaks were identified in HeLa cells. Among these, 203 and 544 peak regions were unique to MDA-MB-231 and HeLa, respectively, and 136 peak regions were common to both cell lines (Fig. 3A). To define RRP1B occupancy in relation to gene position, the locations of RRP1B binding peaks were partitioned into four bins relative to RefSeq gene coordinates; distal promoter, proximal promoter, downstream, and genic regions (Fig. 3B). The locations of RRP1B occupancy peaks relative to gene position in MDA-MB-231 and HeLa cells are shown in Table 1. These data demonstrate RRP1B occupancy peaks were comparatively evenly distributed relative to genic position in both cell lines.
Figure 3. RRP1B interacts with multiple chromatin regions.
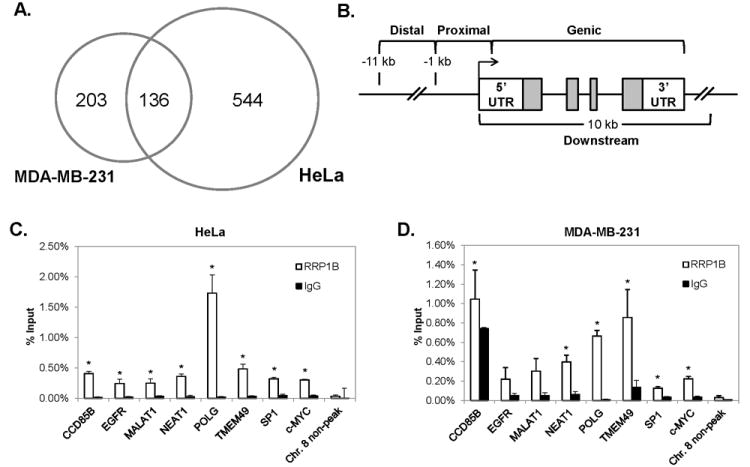
A, Venn diagram of unique and common binding regions detected by ChIP-seq in MDA-MB-231 and HeLa cells. B, division of chromatin regions based on relative genic locations. C, verification of common RRP1B binding regions via ChIP-qPCR in HeLa cells, and D, MDA-MB-231 cells. *; P ≤ 0.05 compared to IgG controls.
Table 1.
Analysis of RRP1B-binding regions detected through ChIP-seq. Regions bound by endogenous RRP1B in MDA-MB-231 and HeLa cells were defined with MACS. Genomic sequences were partitioned into four bins relative to RefSeq genes; distal promoter, proximal promoter, downstream, and genic. The genic bin was further divided by 5′ UTR, exons, introns, and 3′ UTR. The number of peaks for each bin is shown here
| Peak Position Relative to Gene | HeLa | MDA-MB-231 | |
|---|---|---|---|
| Distal promoter | 150 | 64 | |
| Proximal promoter | 156 | 54 | |
| Downstream | 263 | 102 | |
| Genic | 205 | 90 | |
|
| |||
| Genic | 5′ UTR | 61 | 21 |
| Exons | 60 | 13 | |
| Introns | 129 | 74 | |
| 3′ UTR | 33 | 19 | |
Of the peaks identified by ChIP-seq, eight common RRP1B binding regions were randomly selected for validation by ChIP-qPCR. A genomic region on chromosome 8 with no evidence of RRP1B binding was selected as a negative control. In both MDA-MB-231 and HeLa cells, we confirmed significant enrichment of RRP1B at binding regions compared to the IgG control, and no significant enrichment of RRP1B was detected at the negative control region (Fig. 3C, D). To confirm the specificity of the antibody used for ChIP-seq, we performed ChIP-qPCR assays using an alternative RRP1B antibody, which was generated using a different epitope of this protein. RRP1B binding was confirmed at the same eight peak regions using this alternative RRP1B antibody, thus demonstrating the binding specificity of the antibody used to generate global ChIP-seq data (Supplementary Fig. S2).
RRP1B associates with potent inducers of heterochromatinization to suppress gene expression
Next, we examined how RRP1B binding to chromatin influences gene expression at these loci. In MDA-MB-231 clonal isolates ectopically expressing RRP1B, the expression of 40 genes with RRP1B occupancy peaks either within 5 kb up- or downstream were quantified by qPCR (Supplementary Table S3-1). Transcriptional repression was observed for the majority of genes that were dysregulated in RRP1B over–expressing cells compared to that of the control.Genes that displayed a significant decrease in comparison to the control were selected forfurther investigation (Fig. 4; Supplementary Table S3-2). To examine how changes in RRP1B expression levels influenced the transcription of genes with nearby RRP1B occupancy peaks, MDA-MB-231 cells were transfected with one of two different siRNA sequences targeting human RRP1B. Knockdown of RRP1B was confirmed by qPCR and Western Blotting(Supplementary Fig. S1). Quantitative real-time PCR analysis demonstrated that the expressionof the majority of these genes, with the exception of SYNJ2 and c-MYC, was rescued andincreased significantly compared to that of the RRP1B over-expressing cells (Fig. 4).Expression of SYNJ2 and c-MYC decreased in comparison to the control with both the overexpression and knockdown of RRP1B. This may be due to a change in an upstream regulator of these genes as a result of silencing RRP1B.
Figure 4. RRP1B-binding regions are frequently associated with decreased gene expression.

The consequences of RRP1B binding were examined in RRP1B over-expressing cells and compared to that of RRP1B knockdown cells. Fold change was calculated using the control for each treatment. RRP1B siRNA represents the average fold change of cells transfected with either one of the two different RRP1B siRNA sequences. *; P ≤ 0.05, **; P ≤ 0.005 compared to RRP1B over-expressed.
Given that RRP1B binding is frequently associated with reduced gene expression (Fig. 2), we chose to focus on the mechanism underlying these observations. We hypothesized that the decreased gene expression observed with RRP1B over-expression is due to increased association of RRP1B with the heterochromatin-associated proteins, TRIM28 and HP1α, at the chromatin region of these genes. We performed ChIP-reChIP-qPCR assays to examine whether RRP1B shares common occupancy with either TRIM28 or HP1α at binding peaks. Two consecutive immunoprecipitations were performed; first using either a TRIM28 or HP1α antibody, followed by a second immunoprecipitation using an RRP1B antibody. The majority of transcriptionally-repressed genes with a nearby RRP1B binding peak had an associated region of TRIM28 and/or HP1α binding. For TRIM28, co-occupancy with RRP1B was detected at significant levels at six of the twelve binding regions within 5 kb of genes that displayed a decreased expression in RRP1B over-expressing cells; c-MYC, DNM2, EGFR, NCOR2, RAB5B, SP1 (Fig. 5A). For HP1α, co-occupancy with RRP1B was determined to be significant at all binding regions that were examined, other than SYNJ2 (Fig. 5B). Both TRIM28 and HP1 have been shown to co-localize with markers of gene silencing and recruit transcriptional co-repressors (8-11, 16). These data demonstrate that a prominent mechanism by which RRP1B reduces gene expression is through an association with the TRIM28/HP1α heterochromatin complex.
Figure 5. RRP1B interacts with TRIM28 and HP1α at regions associated with H3K9me3 and decreased gene expression.

A, ChIP-reChIP with TRIM28 and RRP1B antibodies in MDA-MB-231 cells. *; P ≤ 0.05 compared to both IgG/TRIM28 and IgG/RRP1B controls. B, ChIP-reChIP with HP1α and RRP1B antibodies in MDA-MB-231 cells. *; P ≤ 0.05 compared to both IgG/HP1α and IgG/RRP1B controls. C, H3K9me3 ChIP-qPCR in MDA-MB-231 clonal isolates over-expressing RRP1B and control cell lines. D, H3K9me3 ChIP-qPCR in MDA-MB-231 cells transfected with either RRP1B siRNA or negative control siRNA. E, Venn diagram of unique and common H3K9me3-enriched regions identified by ChIP-seq in MDA-MB-231 clonal isolates over-expressing RRP1B and control cell lines.
RRP1B binding is associated with increased H3K9 trimethylation
Given that both TRIM28 and HP1α bind to regions of RRP1B occupancy, and that TRIM28/HP1α binding is associated with an increase in H3K9me3 levels (15, 26), we hypothesized that the association of these proteins to the common chromatin regions would be accompanied by a concomitant increase in this repressive, heterochromatin-associated histone mark. To examine this, we performed ChIP-qPCR with an antibody against H3K9me3 with MDA-MB-231 cells stably over-expressing RRP1B and control cells. At RRP1B binding regions where reduced gene expression was observed in RRP1B over-expressing cells, nine of the twelve genes examined displayed significantly increased levels of H3K9me3 in the cells over-expressing RRP1B compared to the control cells (Fig. 5C). At these same regions, with the loss of RRP1B expression via siRNA, there was a significant decrease in H3K9me3 levels compared to that of the control (Fig. 5D). These data indicate that RRP1B-chromatin interactions are not only associated with decreased gene expression, but are also accompanied by transcriptionally-repressive changes in histone methylation typically associated with binding of the TRIM28/HP1α heterochromatinization complex.
Based on these observations, we chose to examine the global effects of RRP1B dysregulation on levels of H3K9me3. We performed ChIP-seq with H3K9me3-immunoprecipitated samples in MDA-MB-231 cells stably over-expressing RRP1B and control cell lines. A global increase in the number of H3K9me3-enriched regions was observed in cells over-expressing RRP1B (Table 2). Analysis using Sole-Search identified 78 unique peaks for control cells, 364 unique peaks for RRP1B over-expressing cells, and 190 common regions between the two groups (Fig. 5E; Supplementary Table S4). It is expected that some regions, such as the 78 regions identified here, will have decreased levels of H3K9me3 with RRP1B over-expression. This is supported by our microarray data and gene expression data, which demonstrates that although the predominant effect of RRP1B over-expression is transcriptional repression, there is a subset of genes that exhibit increased expression.
Table 2. H3K9me3-enriched regions detected through ChIP-seq.
ChIP-seq analysis of H3K9me3-enriched regions in MDA-MB-231 cells stably over-expressing RRP1B and control cell lines were identified using Sole-Search
| Peak Position Relative to Gene | Control | RRP1B |
|---|---|---|
| Distal promoter | 74 | 167 |
| Proximal promoter | 17 | 70 |
| Downstream | 159 | 355 |
| Genic | 336 | 663 |
Discussion
Over the last decade various multi-gene ‘poor-outcome’ breast cancer signatures have been identified (27-29). The prognostic power of such signatures has proven so great that they have been deployed in the clinic for risk stratification in newly diagnosed breast cancer patients. Examples of such signature-based clinical tests include the 21-gene Oncotype Dx and 70-gene MammaPrint (reviewed in (30)). However, the origins of these signatures and the specific mechanisms regulating gene expression during tumorigenesis are not fully understood.
It is becoming increasingly clear that germline variation has a significant influence upon gene expression within the primary tumor (31). It is also apparent that germline genetic variation strongly modulates the propensity of primary breast carcinomas to metastasize (reviewed in (4)), which is of significance given that more than 90% of deaths in breast cancer are a direct consequence of metastasis (2). RRP1B was previously identified as a germline metastasis suppressor that regulates gene expression in a manner that accurately predicts survival in human breast cancer (5, 6). In this report, we used a combination of ChIP analyses to define interactions between endogenous RRP1B and chromatin, coupled with expression analyses. This approach has defined the manner in which RRP1B interacts with chromatin, and has revealed one possible mechanism by which RRP1B regulates metastasis.
ChIP-seq experiments in MDA-MB-231 and HeLa cells revealed that genomic regions occupied by RRP1B are highly diverse with RRP1B binding to a wide variety of elements in relation to genic position. Given that these cell lines were derived from two different tissues, confirmation of our ChIP-seq findings in additional breast cancer cell lines may be of importance. However, given the degree of RRP1B occupancy peak overlap between these two different cell lines, as shown in Figure 3A, we predict that a similar degree of peak overlap would be observed with other breast cancer cell lines. Yet, such experiments are beyond the scope of our current work.
Our interest in the TRIM28/HP1α heterochromatin-associated complex stems from an earlier observation showing that both of these proteins physically interacts with RRP1B, and co-localizes with H3K9me3 (6). Both HP1α and H3K9me3 have been purified with several transcriptionally repressive protein complexes (13, 26, 32, 33). HP1α, a component of the HP1 tetramer, interacts with TRIM28 (8, 16), which was also identified as a RRP1B binding partner, primarily in the perinucleolar region (6). TRIM28 assembles a macromolecular complex that contains proteins such as Mi2α, SETDB1 (SET domain, bifurcated 1), and HP1, which acts to create a stable heterochromatic microenvironment (8,10,13,16). We found that RRP1B is located near genes with decreased expression that also bound TRIM28 and/or HP1α. In addition, higher levels of H3K9me3 were seen at most RRP1B binding regions with reduced expression in adjacent genes. Previous studies have shown that TRIM28 and H3K9me3 are specifically enriched at the 3′ end regions of zinc finger (ZNF) genes (15, 26). Consistent with these findings we found that 38 of the H3K9me3-enriched regions unique to RRP1B over-expressing MDA-MB-231 cells were ZNF genes (Supplementary Table S4). Based on the data presented here, we conclude that RRP1B serves not only to localize TRIM28 and HP1α to specific genomic loci, but also increases H3K9me3 at these same loci, which leads to transcriptional repression. These conclusions are supported by our RRP1B knockdown experiments, which demonstrated that there was a decrease in H3K9me3 levels (Fig. 5D) coupled with no significant changes in TRIM28 or HP1α levels at these loci compared to the control (Supplementary Fig. S3). More specifically, a lower level of RRP1B expression lessened its ability to guide the TRIM28/HP1α heterochromatinization complex to target loci.
RRP1B may be influencing both gene expression and metastasis through additional mechanisms as well. For example, it was apparent that activation of RRP1B suppresses the expression of oncogenes (e.g., c-MYC) and transcription factors with wide-ranging effects (e.g., SP1). Also, our work demonstrates that three genes exhibiting transcriptional repression (DNM2, PHLDA and PTCH2) do not exhibit a significant increase in H3K9me3 levels, as shown in Fig. 5C. This suggests that additional, and as yet uncharacterized, H3K9me3-independent mechanisms are in part responsible for RRP1B-mediated transcriptional repression. Further, although the majority of genes adjacent to RRP1B occupancy peaks did display reduced expression upon RRP1B over-expression, some genes did display increased expression (Fig. 2) and a reduction in H3K9me3 levels (Fig. 5E). These complex effects upon gene expression are a reflection of the highly promiscuous manner in which RRP1B interacts with other proteins (34). For example, it has been previously demonstrated that RRP1B interacts with PARP1 (6), which is a known positive regulator of transcription (35). Additionally, RRP1B also co-localizes with markers of euchromatin (6). This suggests that RRP1B likely regulates transcription by interacting with multiple transcriptional complexes, in addition to regulating other cellular processes, such as ribosome biogenesis through its interaction with Protein Phosphatase 1 (34). Supporting its role in rRNA processing, several nucleolar proteins (e.g. Nucleophosmin, Nucleolin) have been shown to interact with RRP1B. Interestingly, previous studies reported that these nucleolar proteins also regulate transcription by directly binding to chromatin (36, 37). RRP1B, which is primarily located in nucleoli, most likely interacts indirectly with chromatin, as it does not possess any known DNA binding motifs, and recruits chromatin-binding proteins as a means of transcriptional regulation. Given that heterochromatin is concentrated in the nucleus in perinucleolar clusters, it is not surprising that the primarily nucleolar protein RRP1B acts as a regulator of heterochromatinization.
Tumor expression profiling has become a useful tool for assessing prognosis in breast cancer; however, the origin of these prognostic signatures remains somewhat unclear. In this study, we demonstrate that RRP1B regulates metastasis-associated gene expression by interacting with the transcriptional co-repressors TRIM28 and HP1α, which act by recruiting chromatin-modifying enzymes such as SETDB1. Based on previous studies and our data presented here, the manner in which RRP1B regulates both transcription and metastasis proves to be highly complex, and its impact upon both of these processes are not the result of a simplistic, linear effect on one signaling pathway. Rather, RRP1B dysregulation influences both metastasis and prognostic gene expression through a net effect on multiple pathways and biological processes.
Supplementary Material
Acknowledgments
We thank Dr. Kent Hunter for his critical review of this manuscript. We also thank Dr. Abdel Elkahloun at the NHGRI Microarray Core Facility and Alice Young at the NIH Intramural Sequencing Center for their technical assistance.
Notes: This research was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, USA.
Footnotes
The authors disclose no potential conflicts of interest.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Scully OJ, Bay BH, Yip G, Yu Y. Breast cancer metastasis. Cancer Genomics Proteomics. 2012;9:311–320. [PubMed] [Google Scholar]
- 3.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 4.Hunter KW, Crawford NP. The future of mouse QTL mapping to diagnose disease in mice in the age of whole-genome association studies. Annu Rev Genet. 2008;42:131–141. doi: 10.1146/annurev.genet.42.110807.091659. [DOI] [PubMed] [Google Scholar]
- 5.Crawford NP, Qian X, Ziogas A, Papageorge AG, Boersma BJ, Walker RC, et al. Rrp1b, a new candidate susceptibility gene for breast cancer progression and metastasis. PLoS genetics. 2007;3:e214. doi: 10.1371/journal.pgen.0030214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crawford NP, Yang H, Mattaini KR, Hunter KW. The metastasis efficiency modifier ribosomal RNA processing 1 homolog B (RRP1B) is a chromatin-associated factor. J Biol Chem. 2009;284:28660–28673. doi: 10.1074/jbc.M109.023457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh SM, Look MP, Sieuwerts AM, Foekens JA, Hunter KW. Distinct inherited metastasis susceptibility exists for different breast cancer subtypes: a prognosis study. Breast cancer research : BCR. 2009;11:R75. doi: 10.1186/bcr2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nielsen AL, Ortiz JA, You J, Oulad-Abdelghani M, Khechumian R, Gansmuller A, et al. Interaction with members of the heterochromatin protein 1 (HP1) family and histone deacetylation are differentially involved in transcriptional silencing by members of the TIF1 family. EMBO J. 1999;18:6385–6395. doi: 10.1093/emboj/18.22.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ., 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schultz DC, Friedman JR, Rauscher FJ., 3rd Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 2001;15:428–443. doi: 10.1101/gad.869501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryan RF, Schultz DC, Ayyanathan K, Singh PB, Friedman JR, Fredericks WJ, et al. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: a potential role for Kruppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol Cell Biol. 1999;19:4366–4378. doi: 10.1128/mcb.19.6.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frietze S, O'Geen H, Blahnik KR, Jin VX, Farnham PJ. ZNF274 recruits the histone methyltransferase SETDB1 to the 3′ ends of ZNF genes. PloS one. 2010;5:e15082. doi: 10.1371/journal.pone.0015082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iyengar S, Farnham PJ. KAP1 protein: an enigmatic master regulator of the genome. J Biol Chem. 2011;286:26267–26276. doi: 10.1074/jbc.R111.252569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hublitz P, Albert M, Peters AH. Mechanisms of transcriptional repression by histone lysine methylation. Int J Dev Biol. 2009;53:335–354. doi: 10.1387/ijdb.082717ph. [DOI] [PubMed] [Google Scholar]
- 15.O'Geen H, Squazzo SL, Iyengar S, Blahnik K, Rinn JL, Chang HY, et al. Genome-wide analysis of KAP1 binding suggests autoregulation of KRAB-ZNFs. PLoS genetics. 2007;3:e89. doi: 10.1371/journal.pgen.0030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sripathy SP, Stevens J, Schultz DC. The KAP1 corepressor functions to coordinate the assembly of de novo HP1-demarcated microenvironments of heterochromatin required for KRAB zinc finger protein-mediated transcriptional repression. Mol Cell Biol. 2006;26:8623–8638. doi: 10.1128/MCB.00487-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alsarraj J, Walker RC, Webster JD, Geiger TR, Crawford NP, Simpson RM, et al. Deletion of the proline-rich region of the murine metastasis susceptibility gene Brd4 promotes epithelial-to-mesenchymal transition- and stem cell-like conversion. Cancer Res. 2011;71:3121–3131. doi: 10.1158/0008-5472.CAN-10-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crawford NP, Alsarraj J, Lukes L, Walker RC, Officewala JS, Yang HH, et al. Bromodomain 4 activation predicts breast cancer survival. Proc Natl Acad Sci U S A. 2008;105:6380–6385. doi: 10.1073/pnas.0710331105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, et al. Various R programming tools for plotting data. R package version 2.11.3 2013 [Google Scholar]
- 20.Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, et al. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- 21.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 22.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, et al. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011;39:D876–882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ, et al. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS genetics. 2010:6. doi: 10.1371/journal.pgen.1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee M, Dworkin AM, Gildea D, Trivedi NS, Moorhead GB, Crawford NP. RRP1B is a metastasis modifier that regulates the expression of alternative mRNA isoforms through interactions with SRSF1. Oncogene. 2013 doi: 10.1038/onc.2013.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iyengar S, Ivanov AV, Jin VX, Rauscher FJ, 3rd, Farnham PJ. Functional analysis of KAP1 genomic recruitment. Mol Cell Biol. 2011;31:1833–1847. doi: 10.1128/MCB.01331-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 28.van't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 29.van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. The New England journal of medicine. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 30.Paik S. Is gene array testing to be considered routine now. Breast. 2011;20(Suppl 3):S87–91. doi: 10.1016/S0960-9776(11)70301-0. [DOI] [PubMed] [Google Scholar]
- 31.Li Q, Seo JH, Stranger B, McKenna A, Pe'er I, Laframboise T, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152:633–641. doi: 10.1016/j.cell.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, et al. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 33.Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science. 2002;296:1132–1136. doi: 10.1126/science.1069861. [DOI] [PubMed] [Google Scholar]
- 34.Chamousset D, De Wever V, Moorhead GB, Chen Y, Boisvert FM, Lamond AI, et al. RRP1B targets PP1 to mammalian cell nucleoli and is associated with Pre-60S ribosomal subunits. Mol Biol Cell. 2010;21:4212–4226. doi: 10.1091/mbc.E10-04-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnakumar R, Gamble MJ, Frizzell KM, Berrocal JG, Kininis M, Kraus WL. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science. 2008;319:819–821. doi: 10.1126/science.1149250. [DOI] [PubMed] [Google Scholar]
- 36.Gonzalez V, Guo K, Hurley L, Sun D. Identification and characterization of nucleolin as a c-myc G-quadruplex-binding protein. J Biol Chem. 2009;284:23622–23635. doi: 10.1074/jbc.M109.018028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z, Boone D, Hann SR. Nucleophosmin interacts directly with c-Myc and controls c-Myc-induced hyperproliferation and transformation. Proc Natl Acad Sci U S A. 2008;105:18794–18799. doi: 10.1073/pnas.0806879105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.